
Synthesis of Yttrium Oxide Nanoneedles with Carbon Dioxide Carbonization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Procedures
2.1. Process for Synthesizing Yttrium Oxide Nanoneedles
2.2. Testing and Characterization
3. Results and Discussion
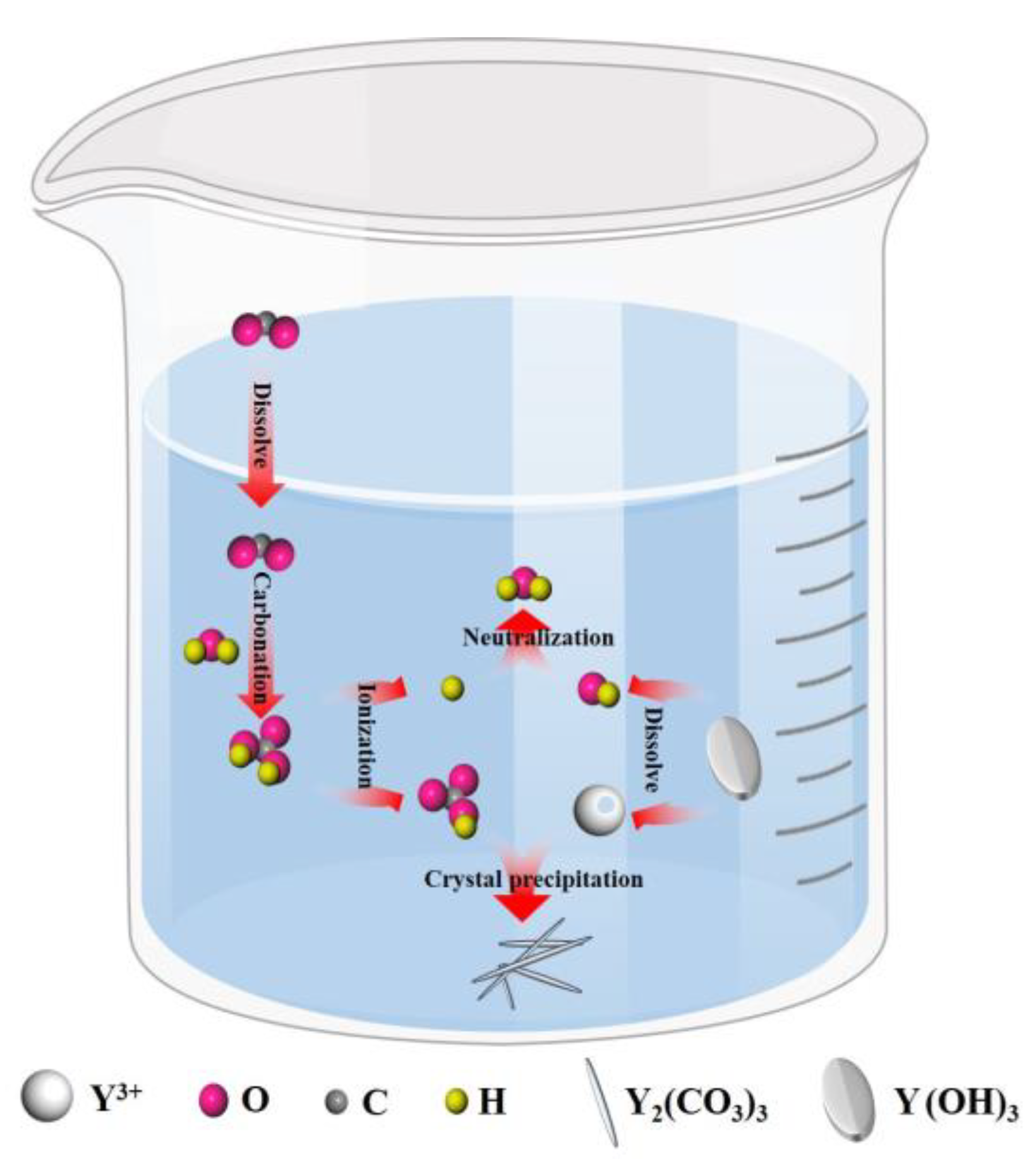
3.1. Carbonization Reaction Process
3.2. Research on the Process for Synthesizing Needle-like Yttrium Carbonate Crystals
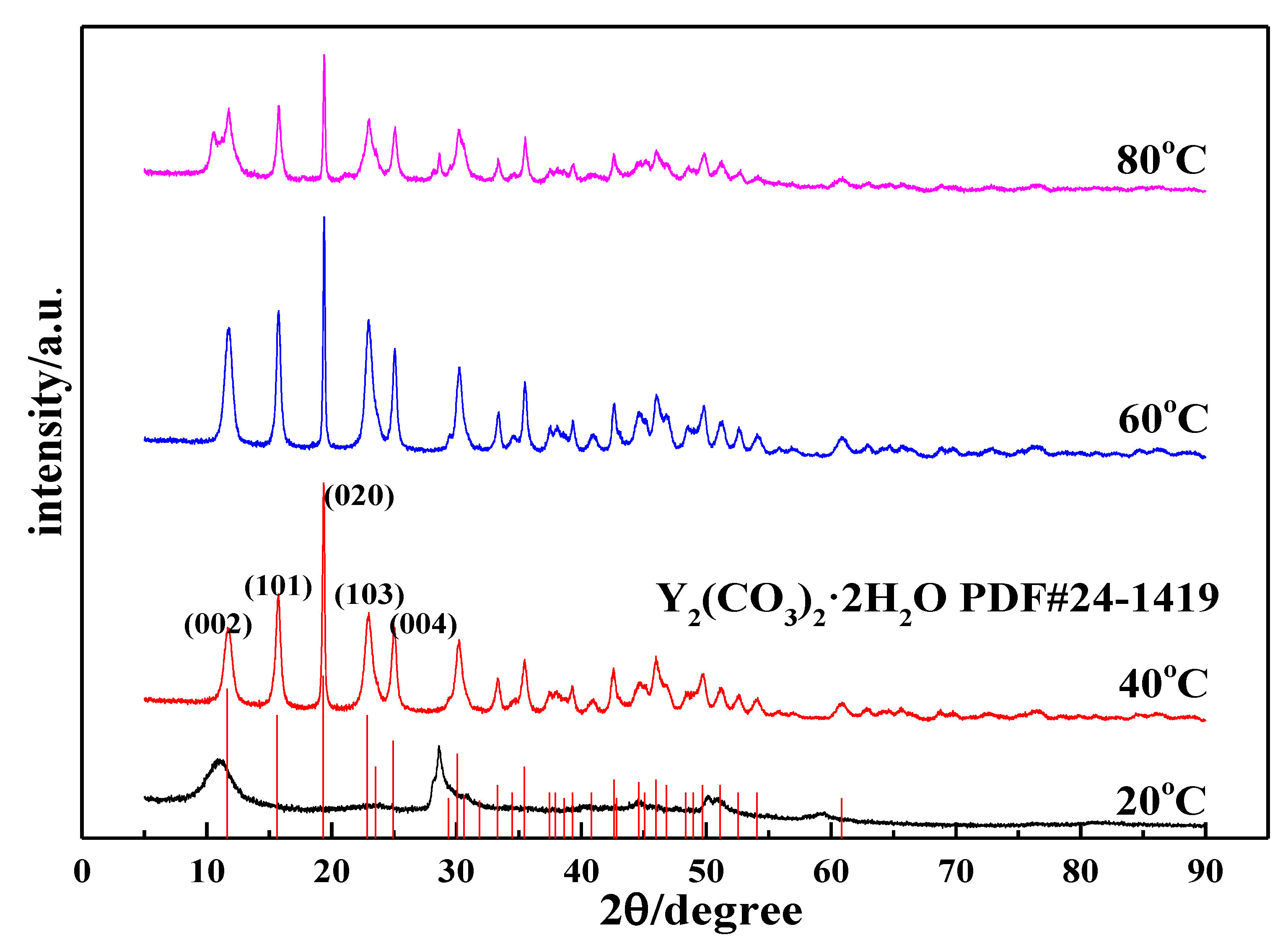
3.2.1. Effects of the Carbonization Temperature on the Morphology and Phase Type of Carbonization Products
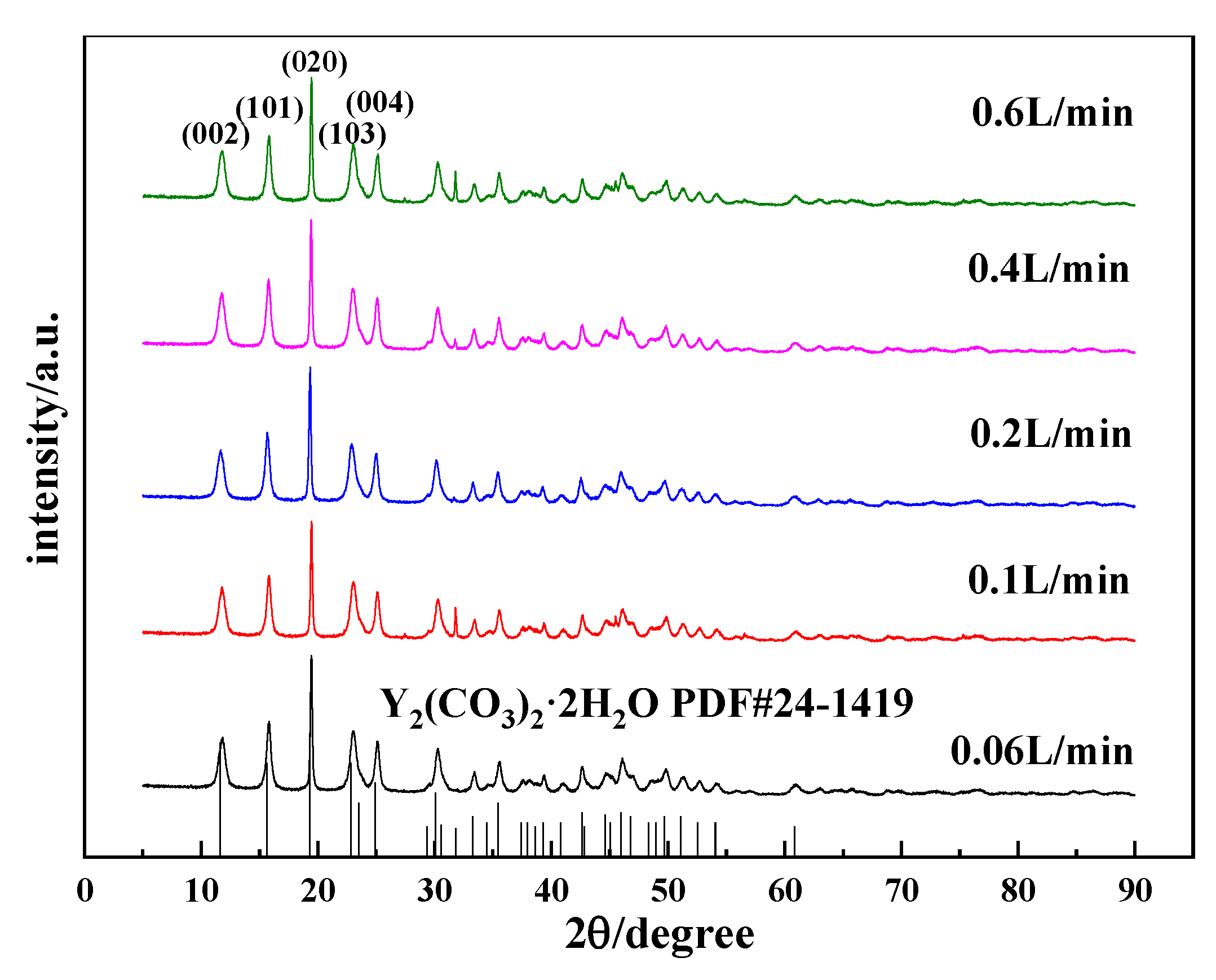
3.2.2. Effects of the CO2 Flow Rate on the Morphology and Phase Type of Carbonization Products
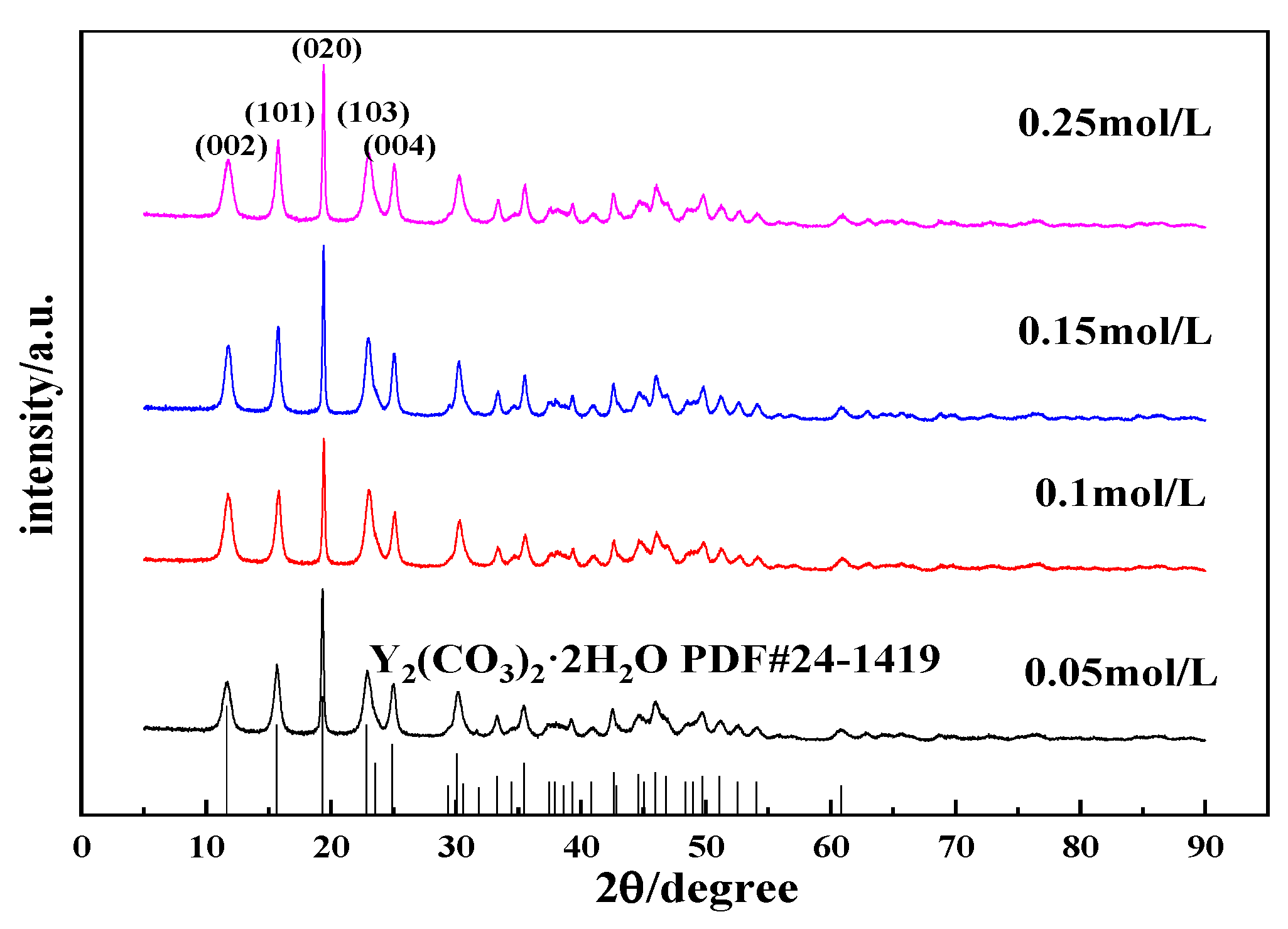
3.2.3. Effects of the Y(OH)3 Slurry Concentration on the Morphology and Phase Type of Carbonization Products
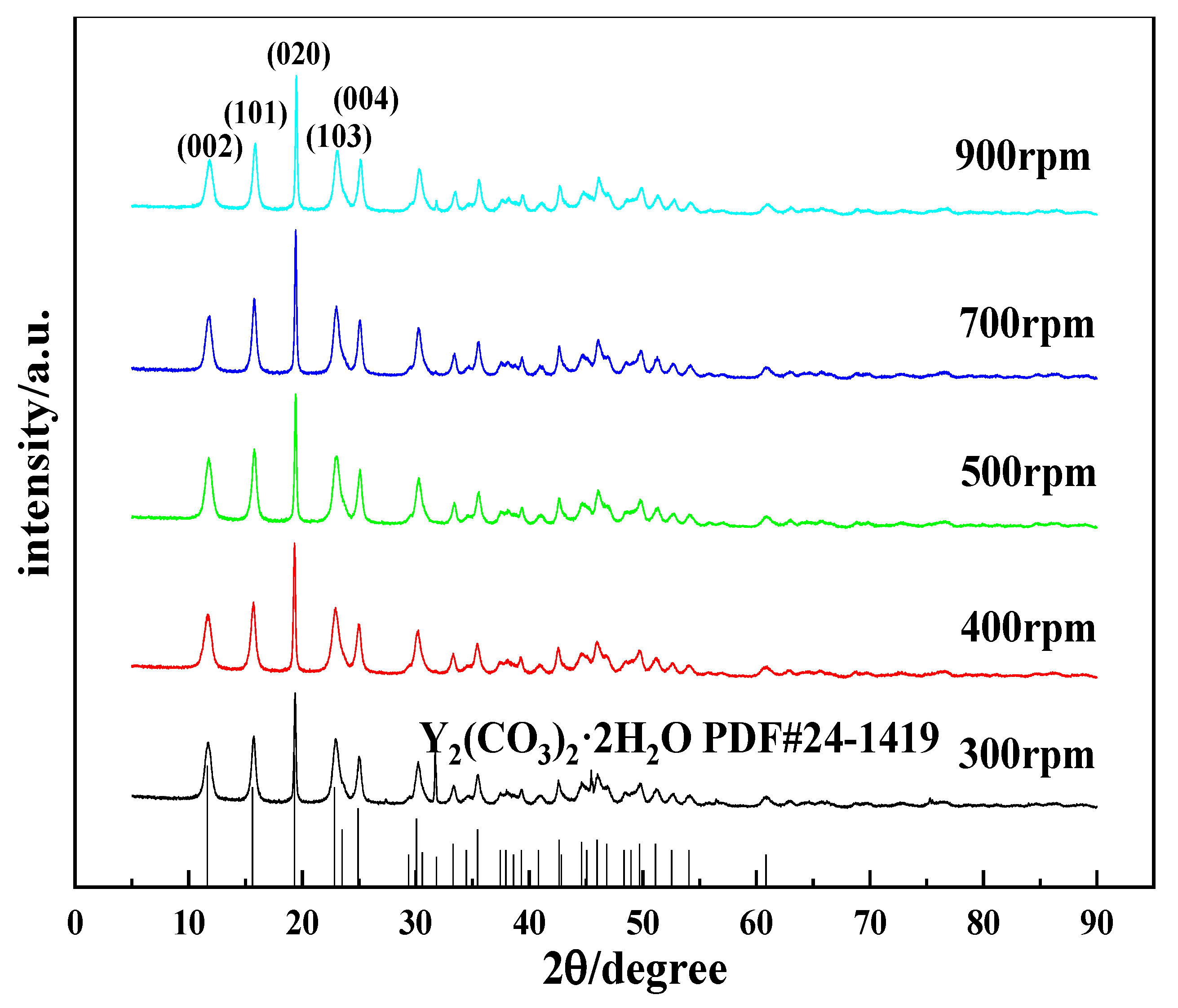
3.2.4. Effects of the Stirring Speed on the Morphology and Phase Type of Carbonization Products
3.3. Research on the Crystallization Mechanism of Needle-like Yttrium Carbonate
3.3.1. XRD, SEM, and TEM Analyses of Changes in the Carbonization Products Formed at Different Carbonization Times
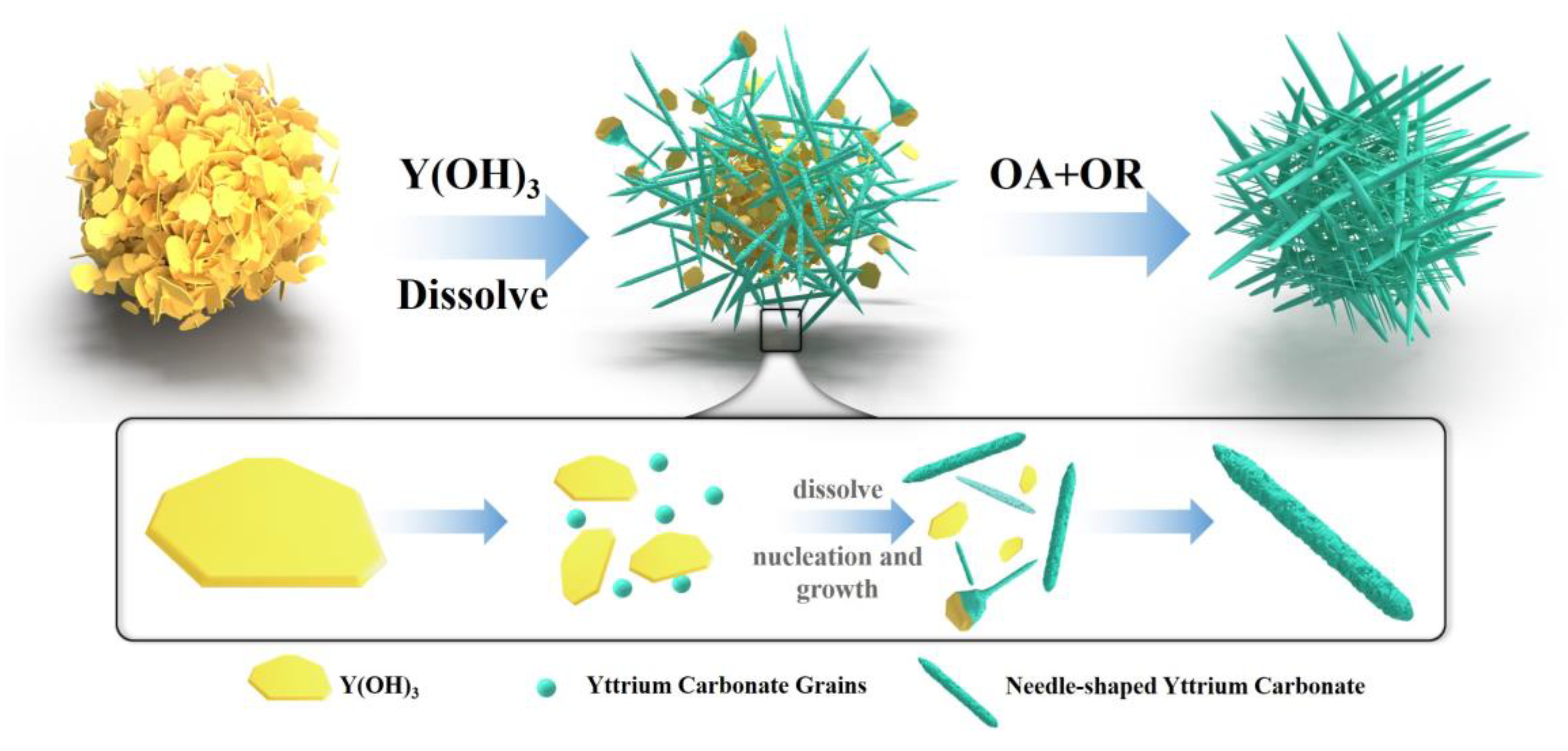
3.3.2. Crystal Growth Mechanism during Carbonization
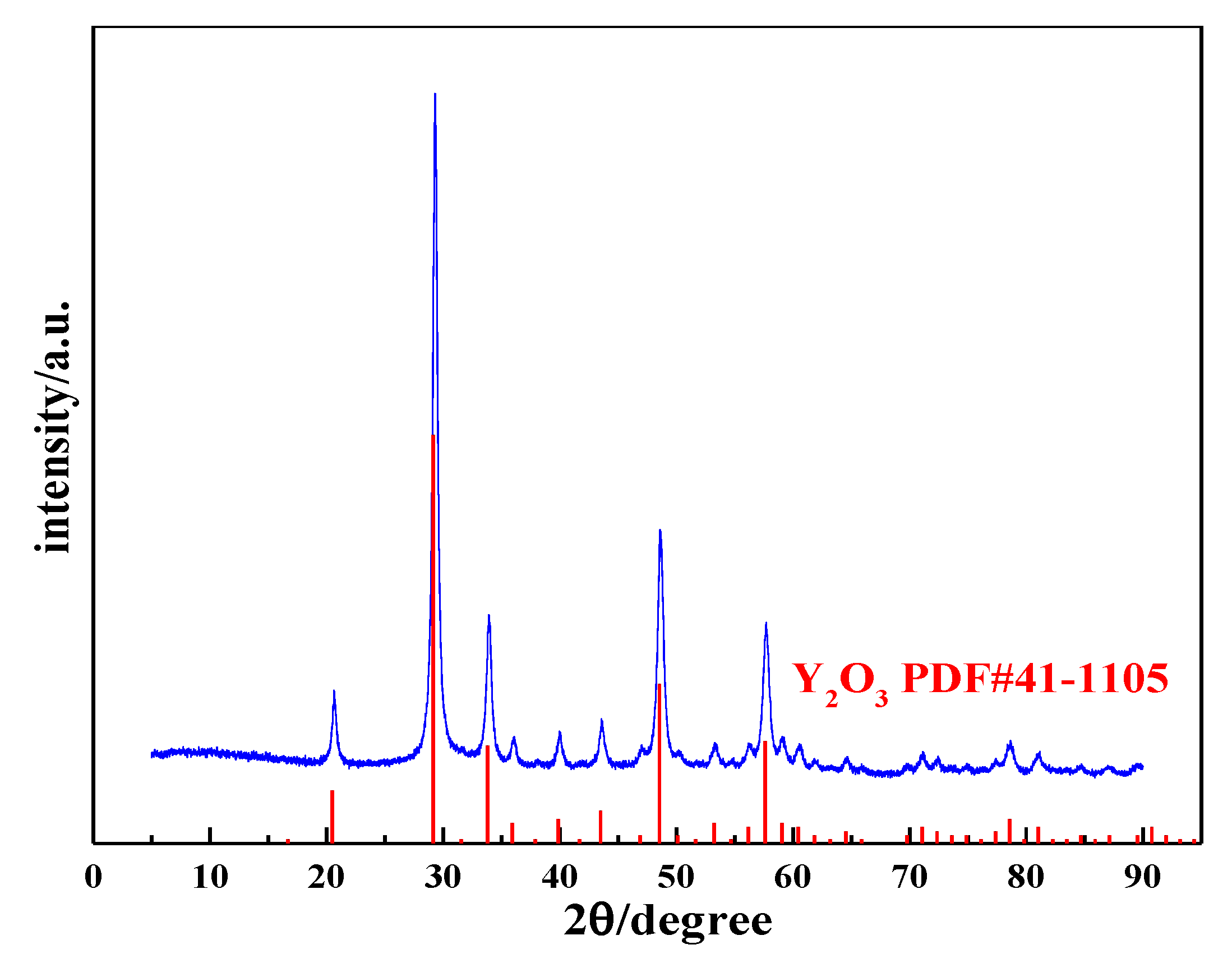
3.4. Characterization of Yttrium Oxide Nanoneedle Powder
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, S.; Zhong, S.; Ou-Yang, X.; Hu, N.; Chen, X.; Xu, R. Y(OH)3 and Y2O3 with novel structures: Formation and mechanism. Mater. Sci. Eng. B 2009, 162, 200–204. [Google Scholar] [CrossRef]
- Xing, K.; Yin, Y.; Zheng, H.; Wan, S.; Liu, P. Research progress on the preparation and morphology control of nano-yttrium oxide. Inf. Rec. Mater. 2019, 20, 3–5. [Google Scholar]
- Atuchin, V.V.; Aleksandrovsky, A.S.; Chimitova, O.D.; Diao, C.P.; Gavrilova, T.A.; Kesler, V.G.; Molokeev, M.S.; Krylov, A.S.; Bazarov, B.G.; Bazarova, J.G.; et al. Electronic structure of β-RbSm(MoO4)2 and chemical bonding in molybdates. Dalton Trans. 2015, 44, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Atuchin, V.V.; Beisel, N.F.; Galashov, E.N.; Mandrik, E.M.; Molokeev, M.S.; Yelisseyev, A.P.; Yusuf, A.A.; Xia, Z. Pressure-Stimulated Synthesis and Luminescence Properties of Microcrystalline (Lu,Y)3Al5O12:Ce3+ Garnet Phosphors. ACS Appl. Mater. Interfaces 2015, 7, 26235–26243. [Google Scholar] [CrossRef] [PubMed]
- Atuchin, V.V.; Gavrilova, T.A.; Grigorieva, T.I.; Kuratieva, N.V.; Okotrub, K.A.; Pervukhina, N.V.; Surovtsev, N.V. Sublimation growth and vibrational microspectrometry of α-MoO3 single crystals. J. Cryst. Growth 2011, 318, 987–990. [Google Scholar] [CrossRef]
- Ramana, C.; Carbajal-Franco, G.; Vemuri, R.; Troitskaia, I.; Gromilov, S.; Atuchin, V. Optical properties and thermal stability of germanium oxide (GeO2) nanocrystals with α-quartz structure. Mater. Sci. Eng. B 2010, 174, 279–284. [Google Scholar] [CrossRef]
- Shi, L.; Fu, H.; Zhang, D.; Fang, J. Research progress of one-dimensional rare earth oxide nano-structure. J. Funct. Mater. 2006, 37, 1535–1538. [Google Scholar]
- Wu, G.; Zhang, L.; Cheng, B.; Xie, T.; Yuan, X. Synthesis of Eu2O3 nanotube arrays through a facile sol−gel template approach. J. Am. Chem. Soc. 2004, 126, 5976–5977. [Google Scholar] [CrossRef]
- Zhang, J.; Hong, G. Synthesis and photoluminescence of the Y2O3:Eu3+ phosphor nanowires in AAO template. J. Solid State Chem. 2004, 177, 1292–1296. [Google Scholar] [CrossRef]
- Rubio, E.J.; Atuchin, V.V.; Kruchinin, V.N.; Pokrovsky, L.D.; Prosvirin, I.P.; Ramana, C.V. Electronic Structure and Optical Quality of Nanocrystalline Y2O3 Film Surfaces and Interfaces on Silicon. J. Phys. Chem. C 2014, 118, 13644–13651. [Google Scholar] [CrossRef]
- Mudavakkat, V.; Atuchin, V.; Kruchinin, V.; Kayani, A.; Ramana, C. Structure, morphology and optical properties of nanocrystalline yttrium oxide (Y2O3) thin films. Opt. Mater. 2012, 34, 893–900. [Google Scholar] [CrossRef]
- Ramana, C.V.; Mudavakkat, V.H.; Bharathi, K.K.; Atuchin, V.V.; Pokrovsky, L.D.; Kruchinin, V.N. Enhanced optical constants of nanocrystalline yttrium oxide thin films. Appl. Phys. Lett. 2011, 98, 031905. [Google Scholar] [CrossRef]
- Gao, G.; Lai, A.; Zhou, X.; Liao, C.; Liu, J.; Xiao, Y. Precipitation crystallization process of yttrium carbonate and its influencing factors. Chin. J. Nonferrous Met. 2020, 30, 2457–2474. [Google Scholar]
- Li, N.; Yanagisawa, K. Controlling the morphology of yttrium oxide through different precursors synthesized by hydrothermal method. J. Solid State Chem. 2008, 181, 1738–1743. [Google Scholar] [CrossRef]
- Kaszewski, J.; Witkowski, B.S.; Wachnicki, Ł.; Przybylińska, H.; Kozankiewicz, B.; Mijowska, E.; Godlewski, M. Reduction of Tb4+ ions in luminescent Y2O3:Tb nanorods prepared by microwave hydrothermal method. J. Rare Earths 2016, 34, 774–781. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, X.; Yi, R.; Shi, R.; Gao, G.; Qiu, G. Selective and Controlled Synthesis of Single-Crystalline Yttrium Hydroxide/Oxide Nanosheets and Nanotubes. J. Phys. Chem. C 2008, 112, 17788–17795. [Google Scholar] [CrossRef]
- Chen, F.; Zhu, Y.-J.; Wang, K.-W.; Pan, Y.-B. A Simple Strategy for Preparation of a Series of One-dimensional Rare Earth Oxides Using Rare Earth Precursors as Templates. Curr. Nanosci. 2009, 5, 266–272. [Google Scholar] [CrossRef]
- Zhang, R.; Hao, X.; Li, J.; Zhao, Y.; Liu, H.; Wang, S.; Ma, X.; Xu, Z.; Siqin, B. Method for Preparing Needle-Shaped Yttrium Oxide by Controlling the Aging Time of Yttrium Carbonate Precipitation. Application CN201410185770.8, 5 May 2014. [Google Scholar]
- Jiang, L.; Zhang, R.; Du, F.; Cui, Z. Study on synthesis of needle-like nanometer calcium carbonate. J. Funct. Mater. 2002, 33, 545–547. [Google Scholar]
- Liu, X.; Yu, F.; Wang, H. New process for preparation of magnesium carbonate hydrate by carbonization at normal pressure. Inorg. Chem. Ind. 2015, 47, 31–34. [Google Scholar]
- Xiao, Y.; Feng, Z.; Huang, X.; Huang, L.; Long, Z.; Wang, Q.; Hou, Y. Synthesis of lanthanum oxide nanosheets by a green carbonation process. Chin. Sci. Bull. 2014, 59, 1864–1867. [Google Scholar] [CrossRef]
- Yan, Z.; Liu, H.; Tao, Z. Temperature effect on carbonic acid balance in water. Carsologica Sin. 2011, 30, 128–131. [Google Scholar]
- Yang, T.; Yan, X. Study on “Four Films Model” of Carbonization Process on Calcium Carbonate Nano-particles. China Powder Sci. Technol. 2012, 18, 66–68. [Google Scholar]
- Liu, B.; Qiao, M.; Hu, Q.; Zhai, X. Kinetics of the carbonation of dolomite slurry. J. Hebei Univ. Sci. Technol. 2005, 26, 118–123. [Google Scholar]
- Feng, Z.; Huang, X.; Wang, M.; Sun, X.; Xu, Y.; Xue, Q.; Chen, S. Green synthesis of ceria powders with special physical properties by carbon dioxide carbonization. J. Rare Earths 2018, 36, 1084–1089. [Google Scholar] [CrossRef]
- Lai, A.; He, Q.; Rao, M.; Gao, G.; Xiao, Y. Synthesis of highly uniform ceria nanosheets by carbon dioxide carbonization and their growth mechanism. J. Solid State Chem. 2020, 290, 121593. [Google Scholar] [CrossRef]
- Yildirim, M.; Akarsu, H. Kinetics of Calcium Carbonate (CaCO3) Precipitation from a Icel-Yavca Dolomite Leach Solution by a Gas (Carbon Dioxide)/Liquid Reaction. Helv. Chim. Acta 2009, 92, 502–513. [Google Scholar] [CrossRef]
- Liu, H.; Lan, P.; Lu, S.; Wu, S. The crystallization kinetic model of nano-CaCO3 in CO2-ammonia-phosphogypsum three-phase reaction system. J. Cryst. Growth 2018, 492, 114–121. [Google Scholar] [CrossRef]
- Qian, Y. Study on the Precipitation and Crystallization Process of Ammonium Bicarbonate in Low Concentration Rare Earth Solution; Jiangxi University of Science and Technology: Ganzhou, China, 2013; pp. 29–31. [Google Scholar]
- Yin, J.-Q.; Zou, Z.-Q.; Tian, J. Preparation of crystalline rare earth carbonates with large particle size from the lixivium of weathered crust elution-deposited rare earth ores. Int. J. Miner. Met. Mater. 2020, 27, 1482–1488. [Google Scholar] [CrossRef]
- Miyawaki, R.; Matsubara, S. A Refinement of the Crystal Structure of Synthetic Tengerite-(Y). Bulletin of the National Science Museum. Ser. C Geol. Paleontol. 2004, 30, 1–4. [Google Scholar]
- Zhu, W.; Qiu, D.; Pei, H.; Zhou, X.; Zhu, W.; Li, J.; Liu, Y.; Li, D.; Li, Y. Precipitation Crystallization Process of Yttrium Carbonate and Size Controlling Synthesis of Yttrium Oxide Particles. J. Chin. Soc. Rare Earths 2016, 34, 180–188. [Google Scholar]
- Xue, X.; Huang, F.; Lin, Z. Oriented attachment crystal growth kinetics and its roles in tailoring optical properties of quantum dots. Sci. Sin. Chim. 2012, 42, 1553–1574. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, X.; Yang, Y.; Zeng, Z.; Chen, S.; Lin, Y.; Xin, H.; Dai, Z.; Song, C.; Zhu, X.; et al. Chemical reaction directed oriented attachment: From precursor particles to new substances. arXiv 2015, arXiv:1512.00666. [Google Scholar]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, M.; Lai, A.; Zan, M.; Gao, M.; Xiao, Y. Synthesis of Yttrium Oxide Nanoneedles with Carbon Dioxide Carbonization. Nanomaterials 2022, 12, 3440. https://doi.org/10.3390/nano12193440
Rao M, Lai A, Zan M, Gao M, Xiao Y. Synthesis of Yttrium Oxide Nanoneedles with Carbon Dioxide Carbonization. Nanomaterials. 2022; 12(19):3440. https://doi.org/10.3390/nano12193440
Chicago/Turabian StyleRao, Minglu, Anbang Lai, Miaomiao Zan, Menglan Gao, and Yanfei Xiao. 2022. "Synthesis of Yttrium Oxide Nanoneedles with Carbon Dioxide Carbonization" Nanomaterials 12, no. 19: 3440. https://doi.org/10.3390/nano12193440
APA StyleRao, M., Lai, A., Zan, M., Gao, M., & Xiao, Y. (2022). Synthesis of Yttrium Oxide Nanoneedles with Carbon Dioxide Carbonization. Nanomaterials, 12(19), 3440. https://doi.org/10.3390/nano12193440