
Evolution of the Electronic and Optical Properties of Meta-Stable Allotropic Forms of 2D Tellurium for Increasing Number of Layers


Abstract
:1. Introduction
2. Materials and Methods
3. Results
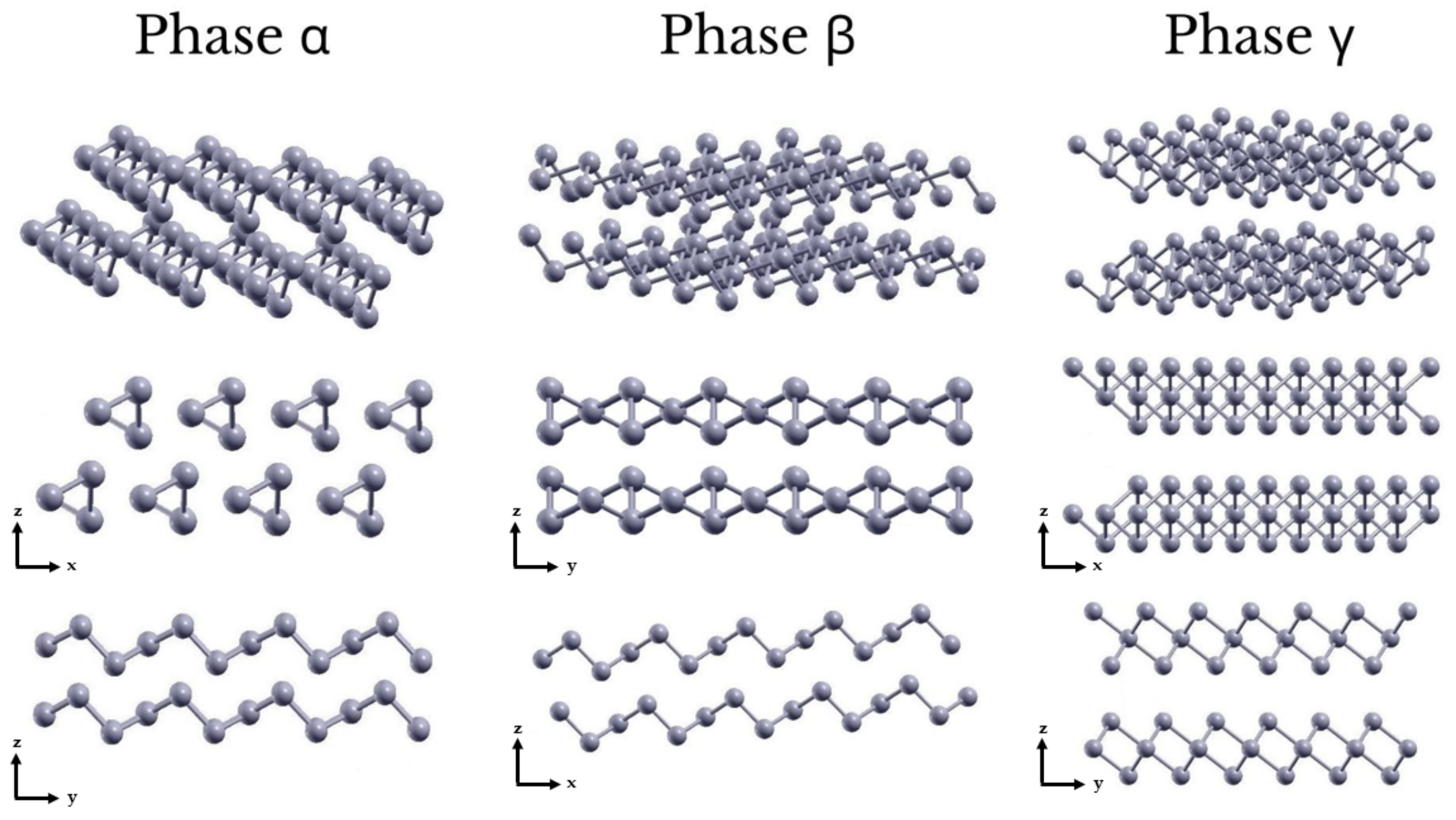
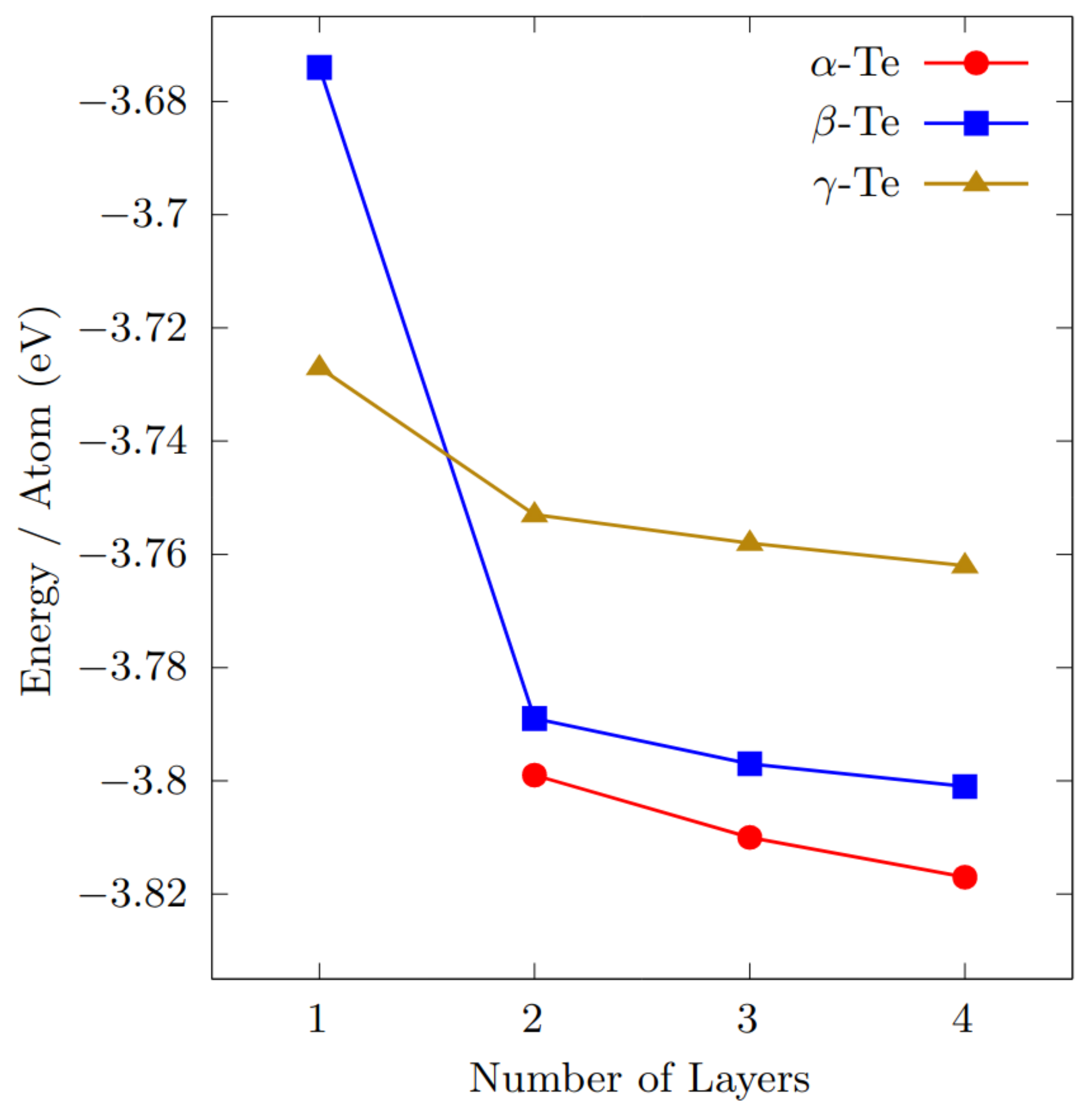
3.1. Geometry and Stability
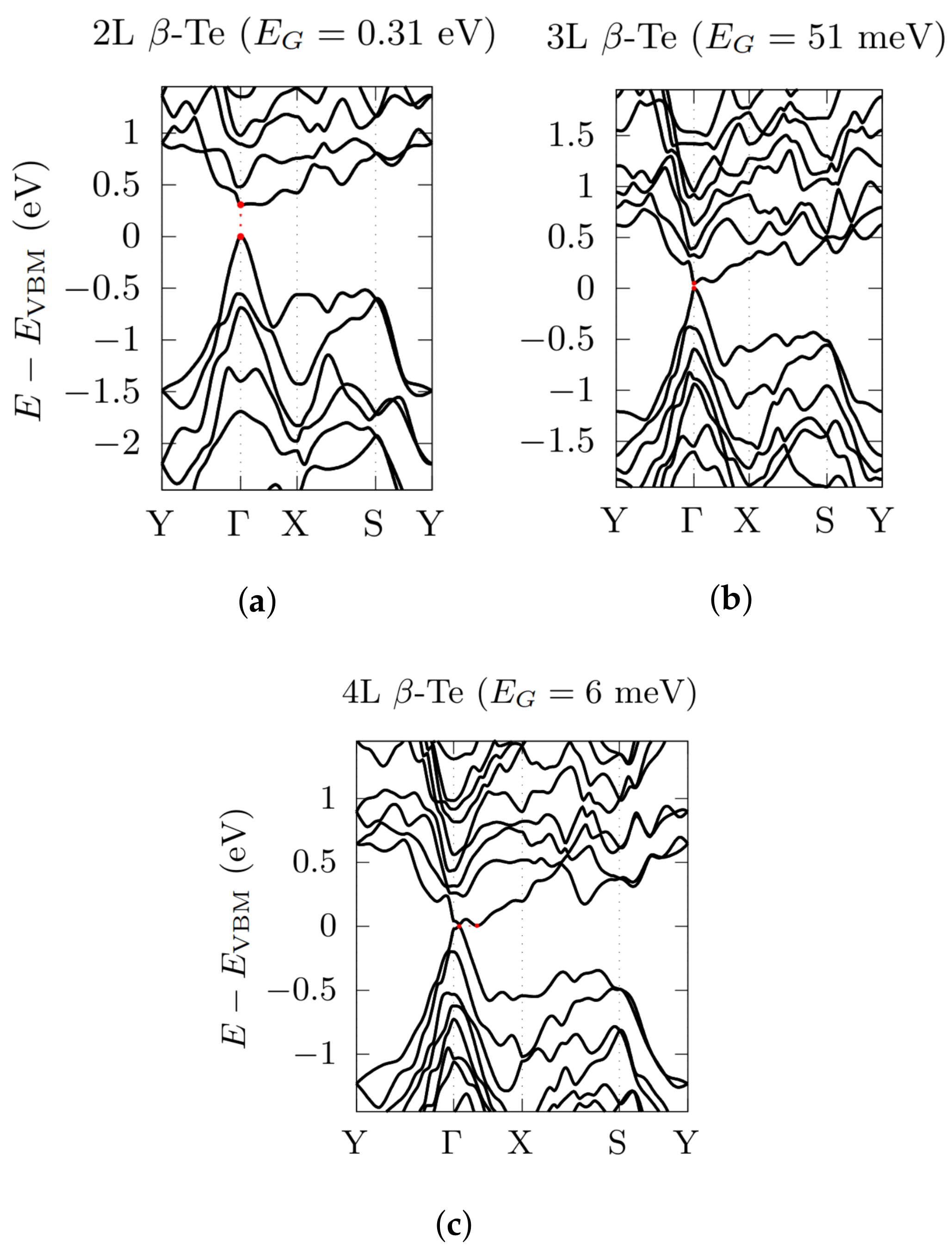
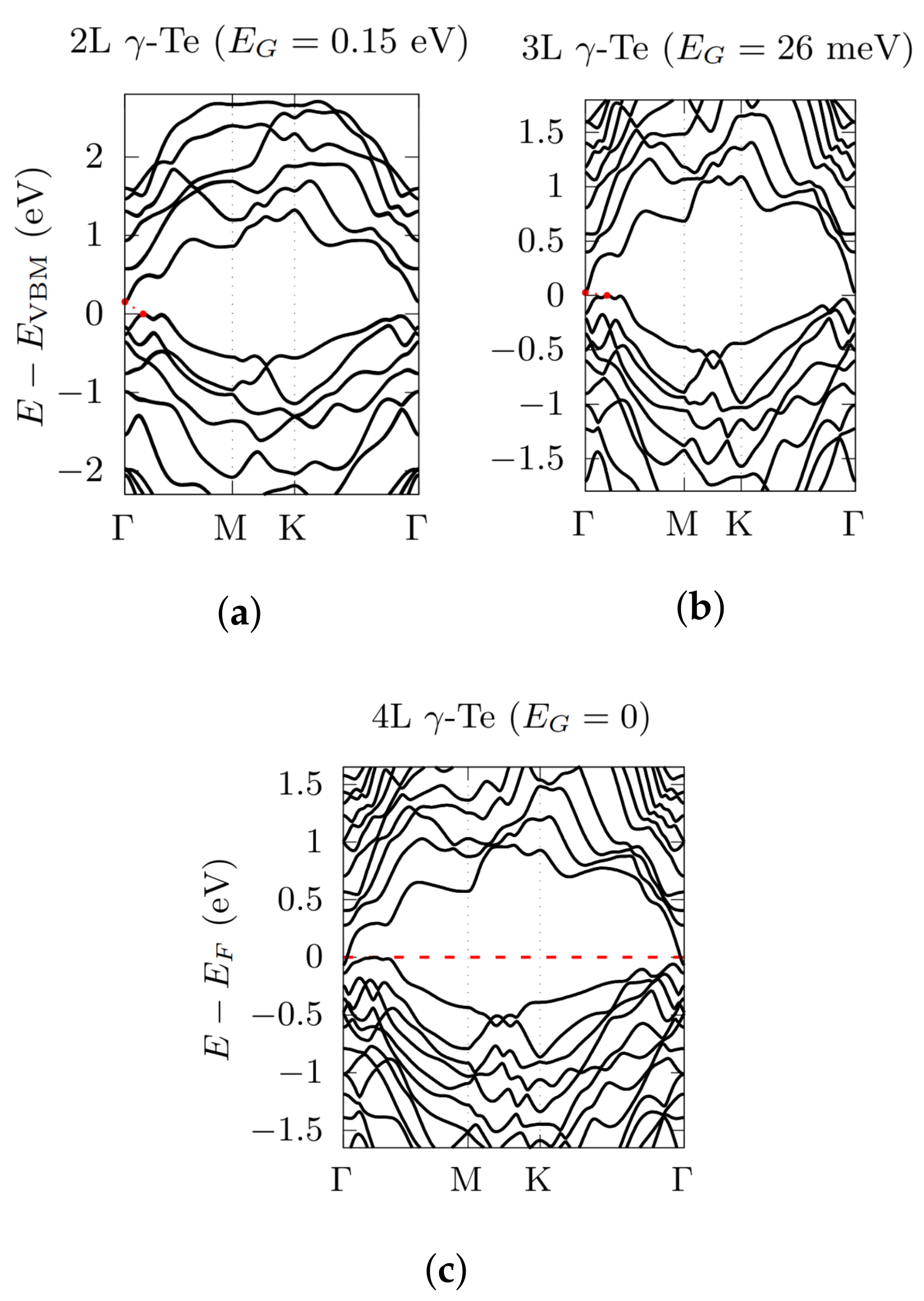
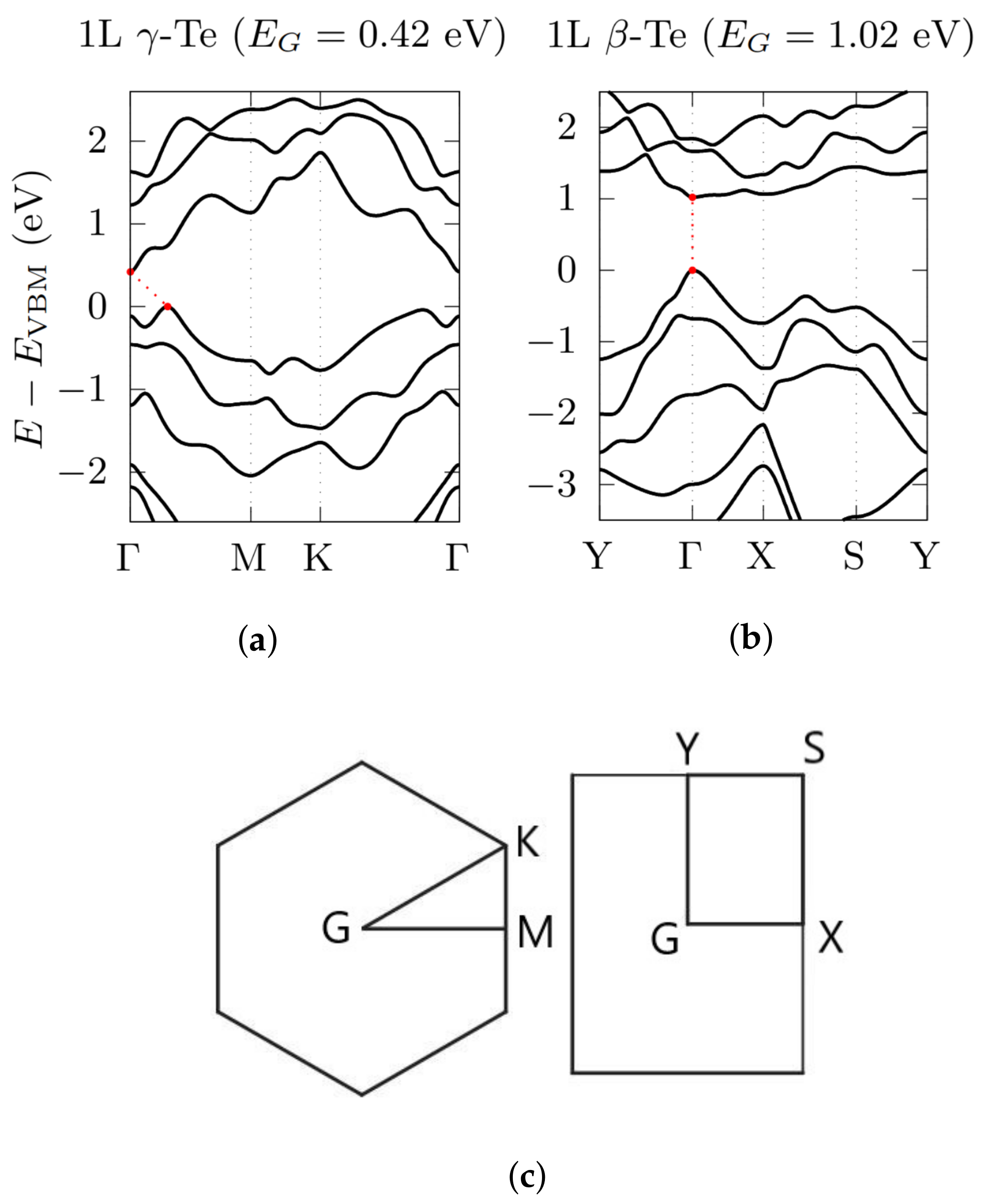
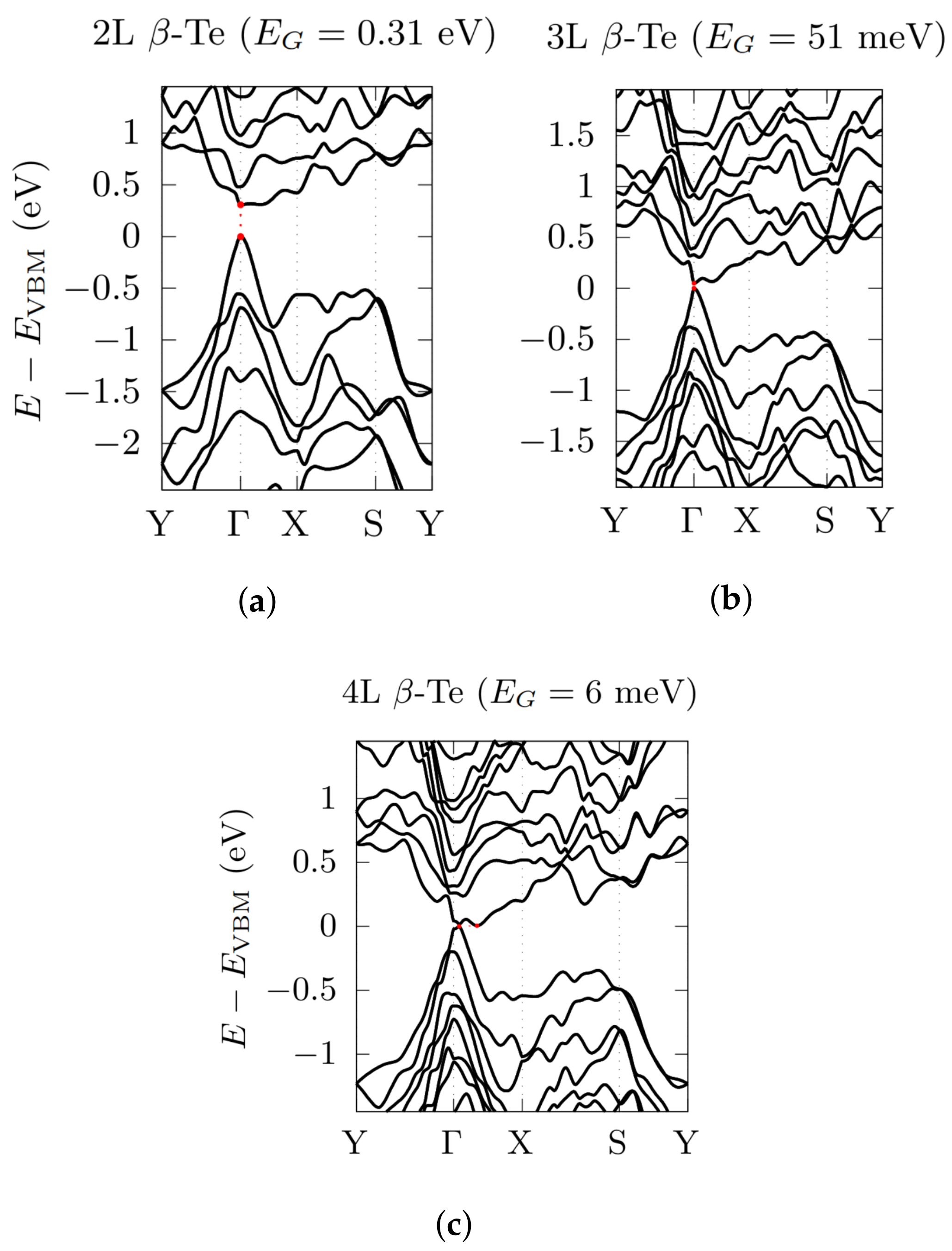
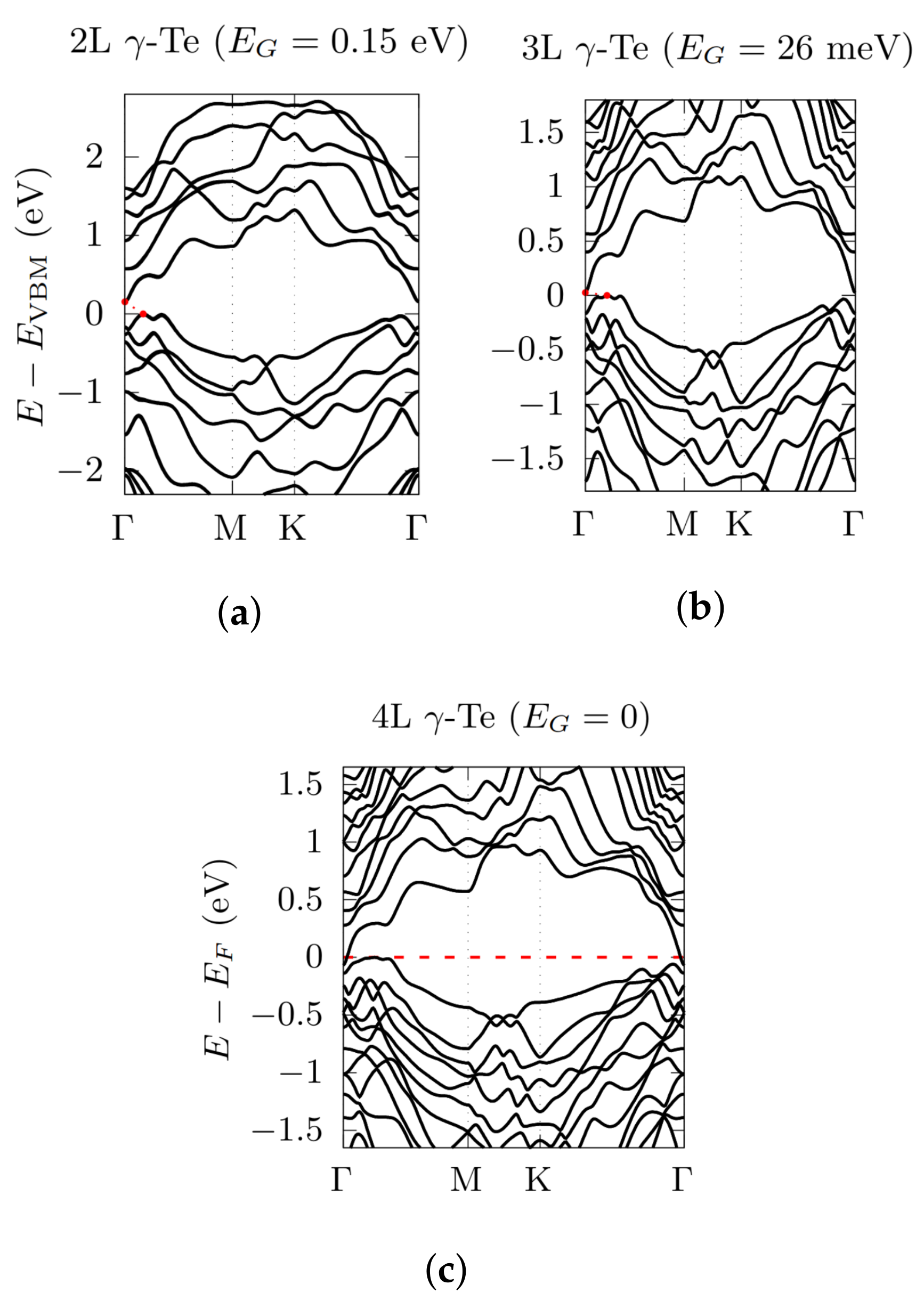
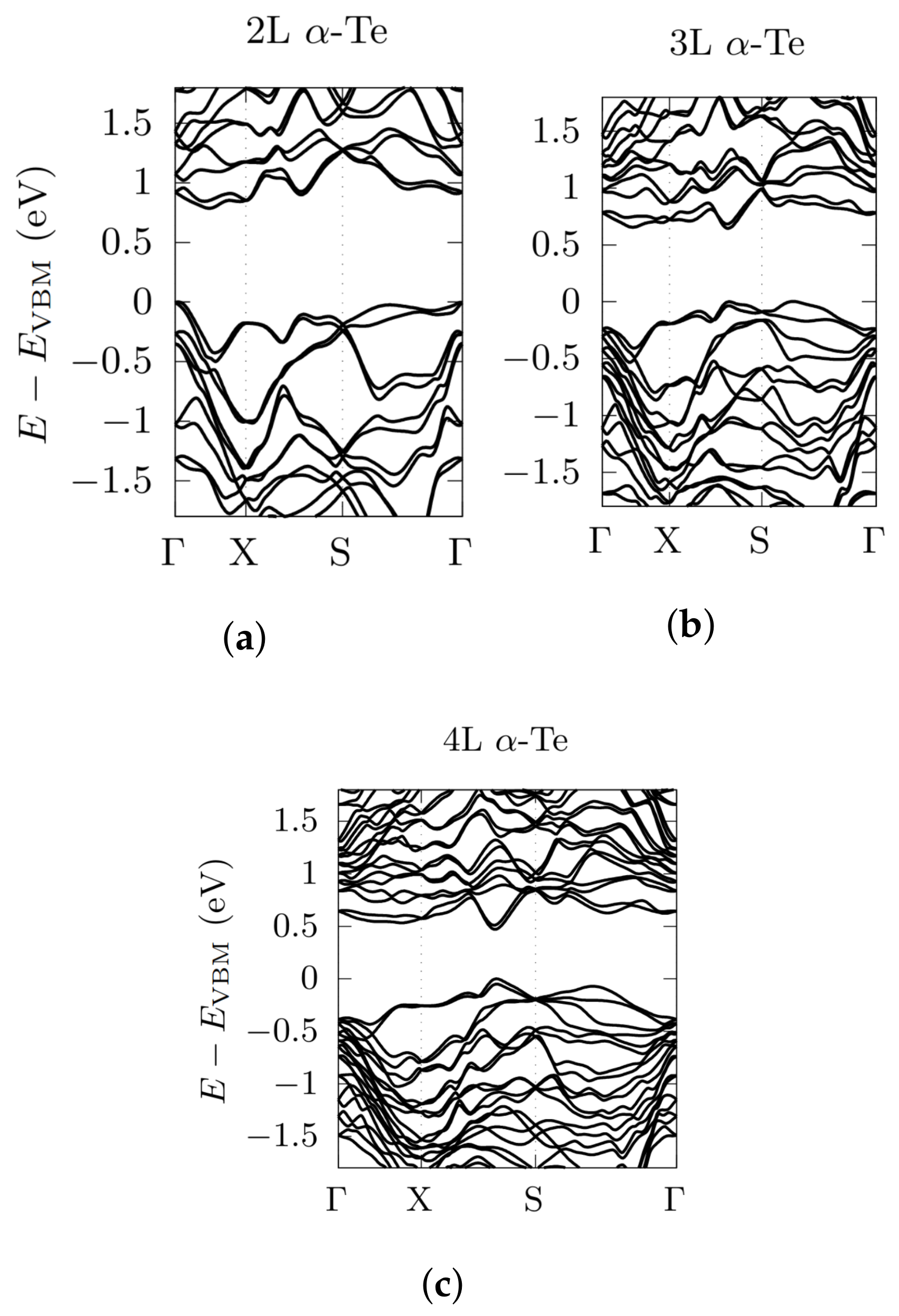
3.2. Electronic Bandstructures
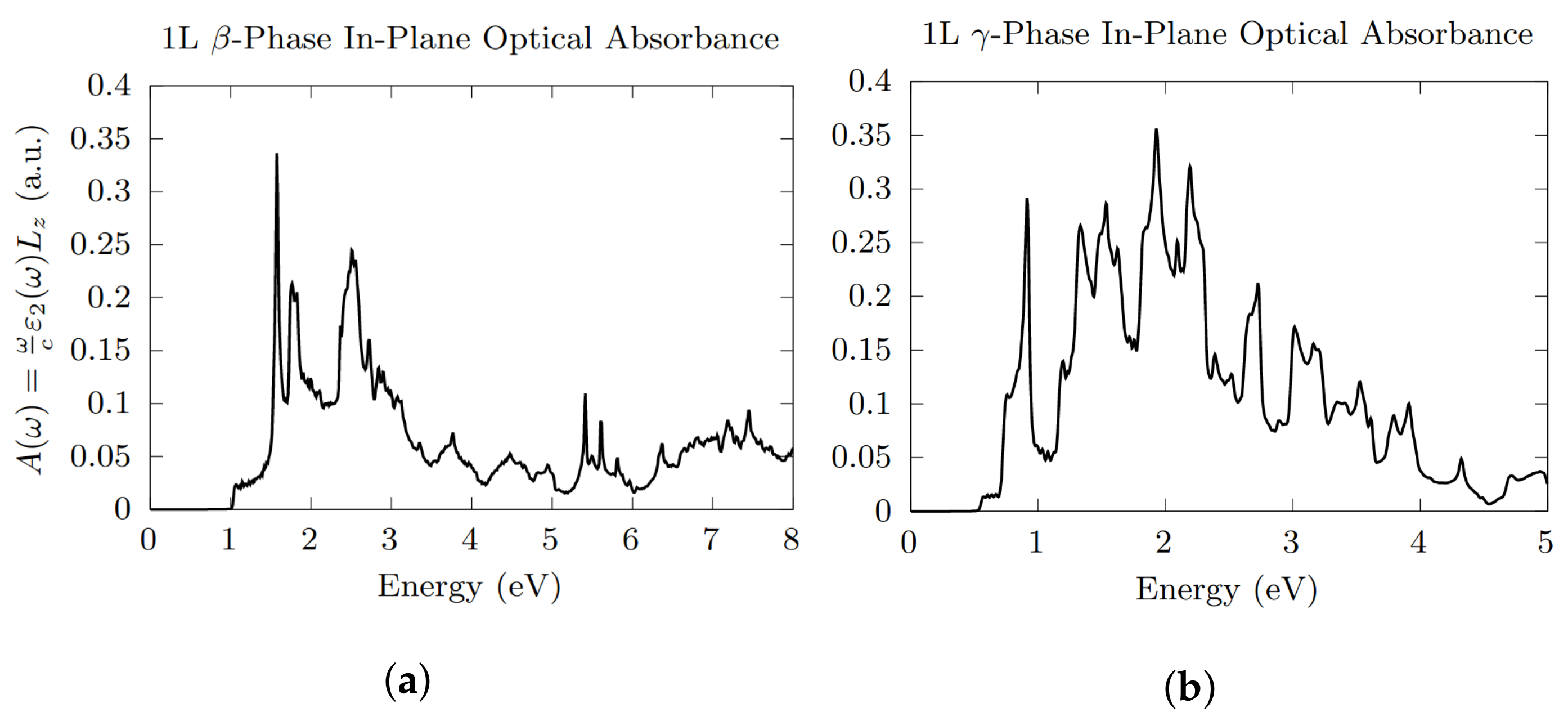
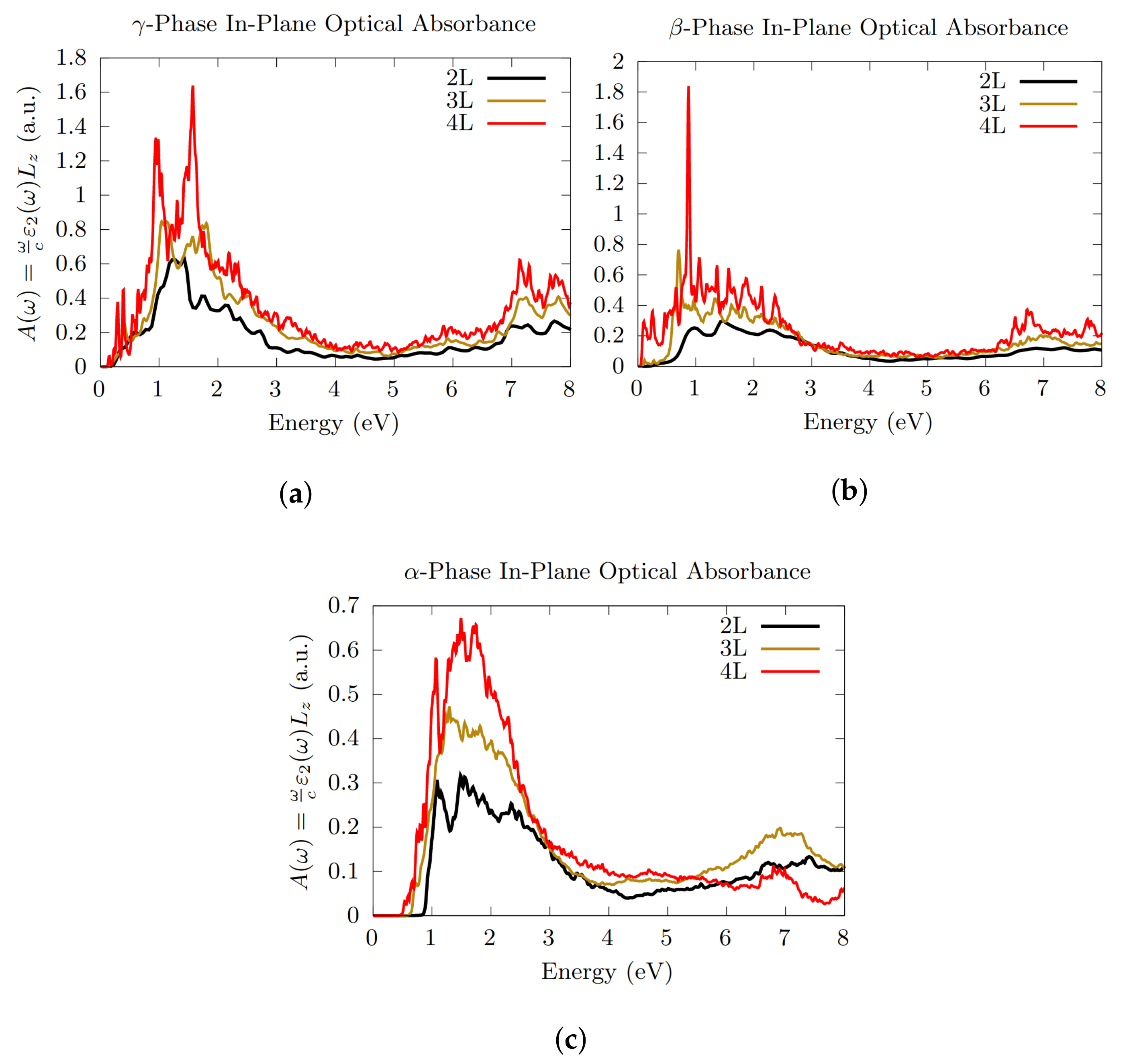
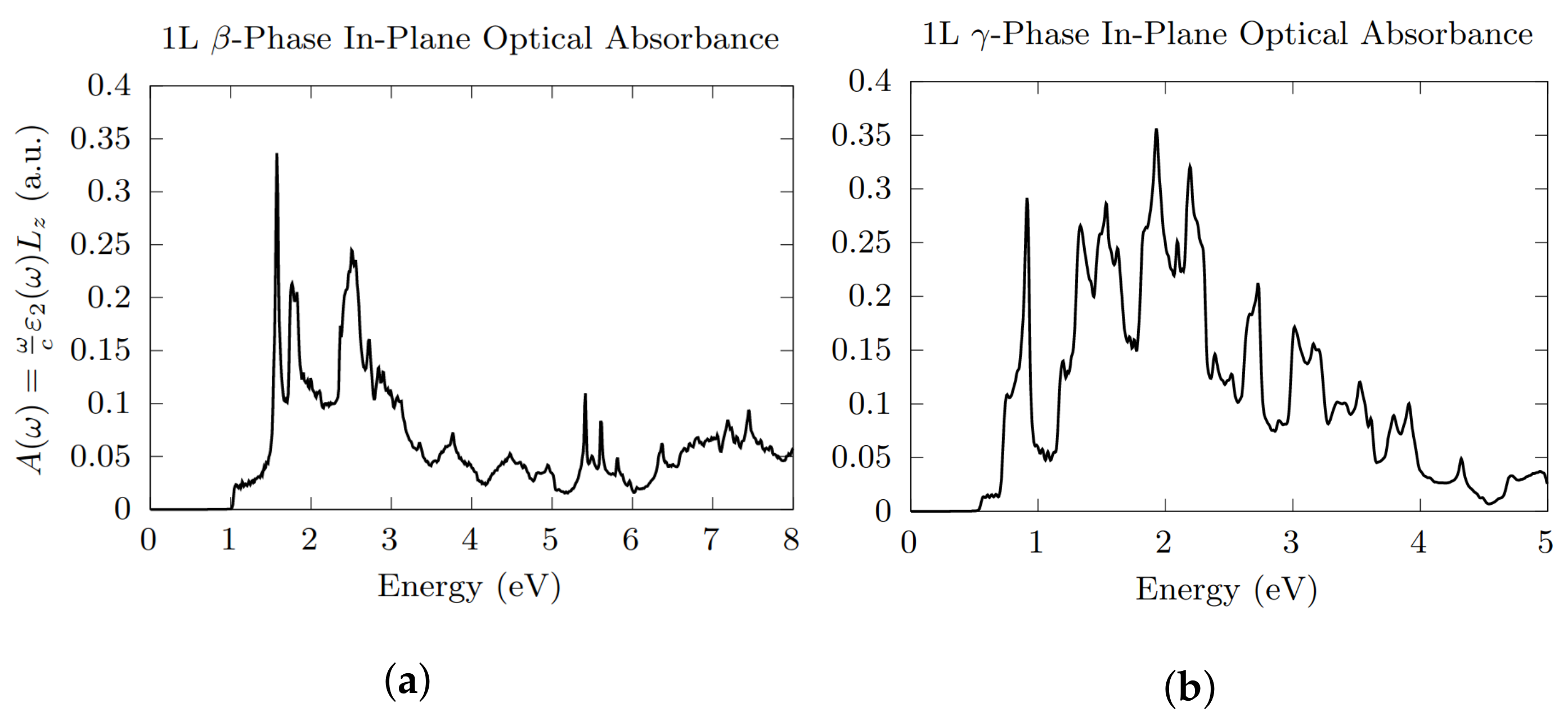
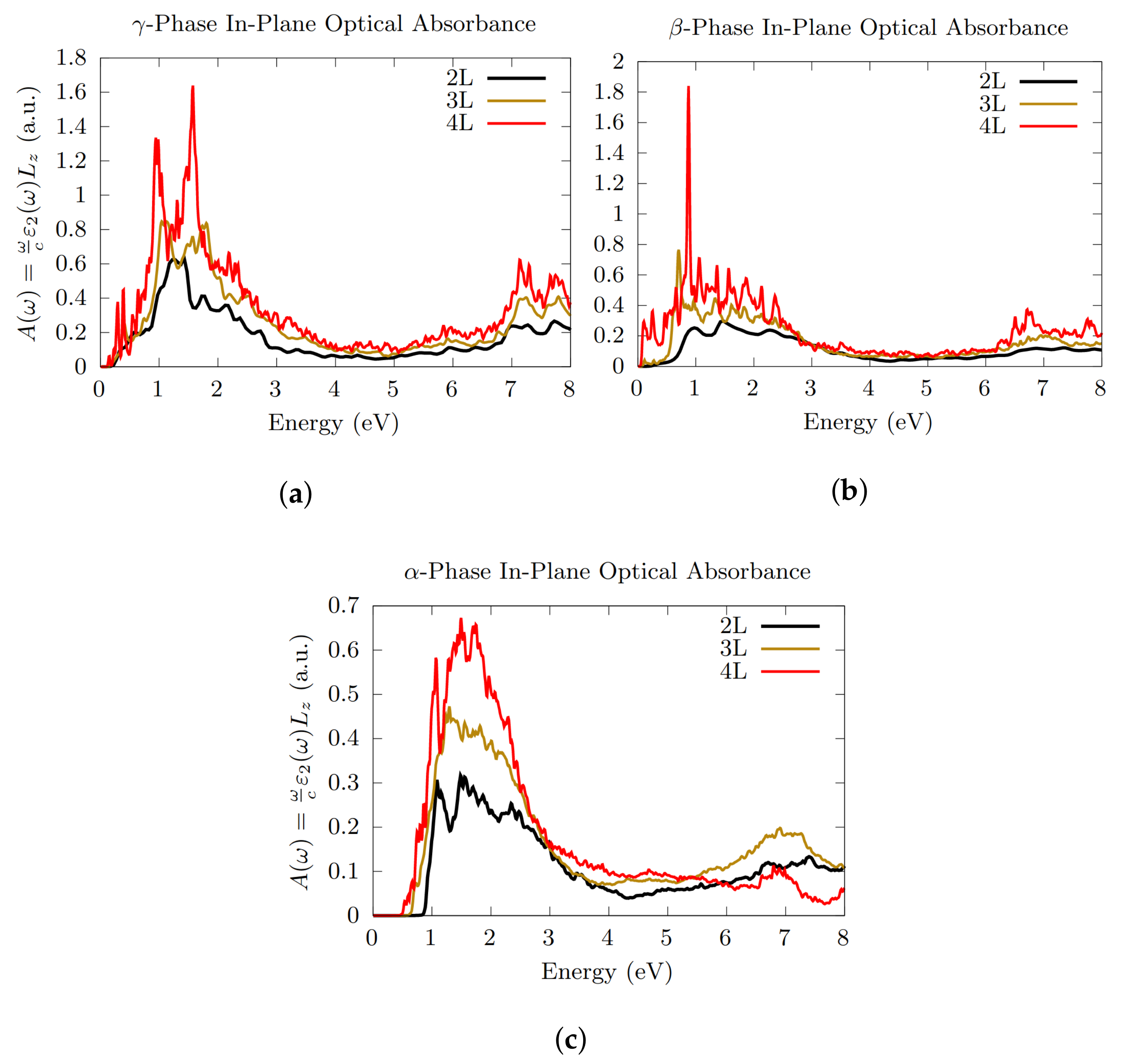
3.3. Optical Absorption
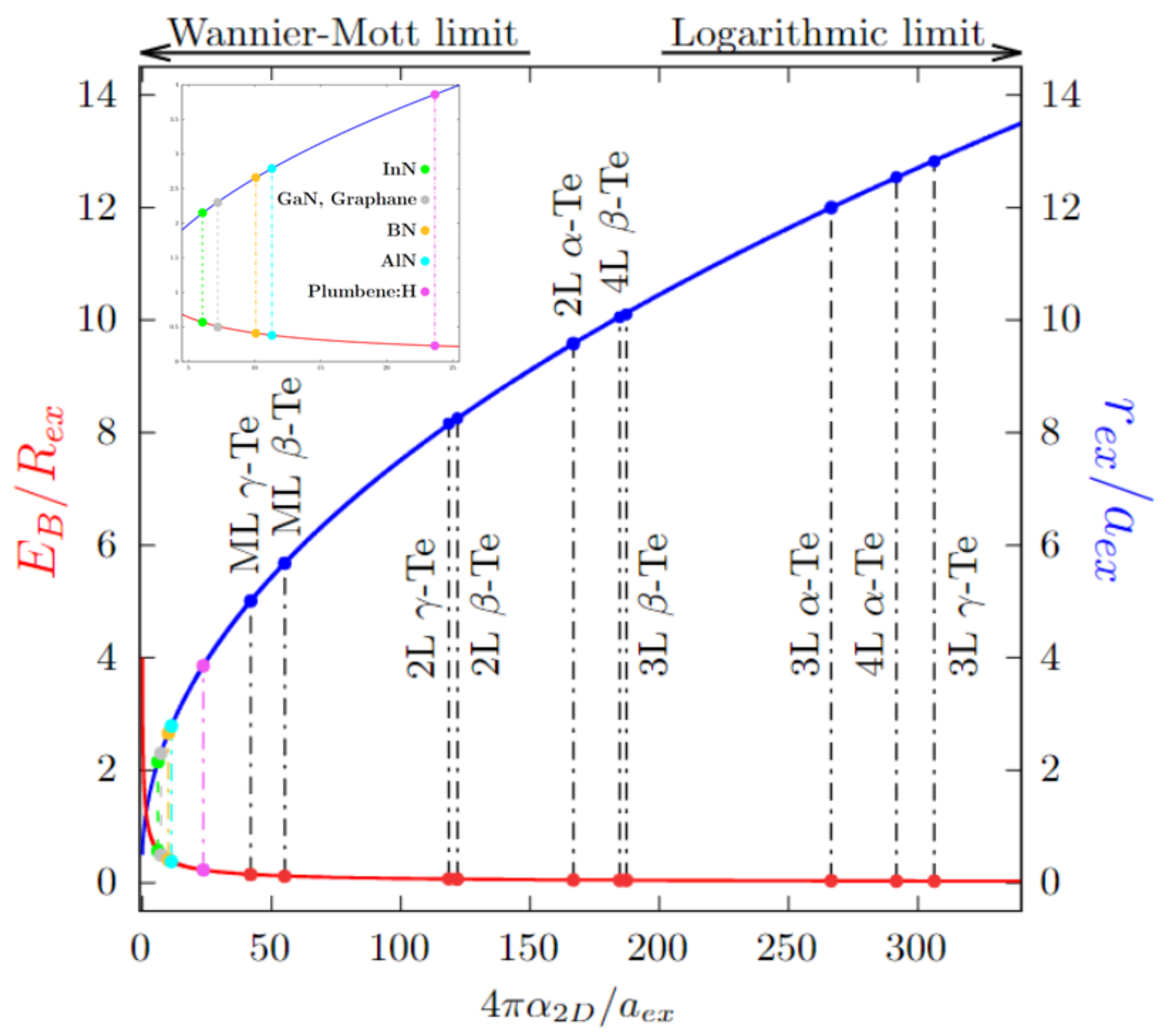
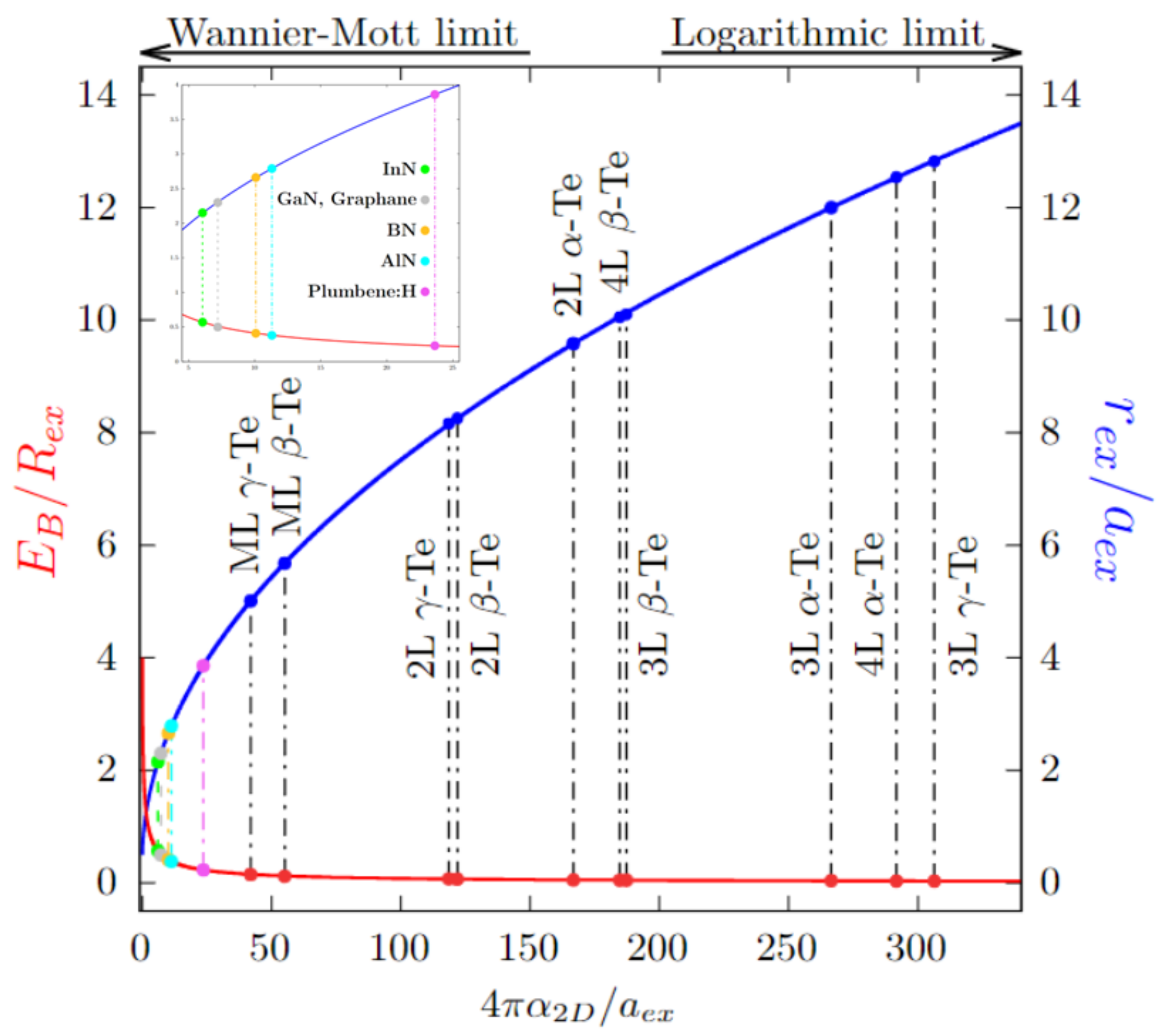
3.4. 2D Exciton Model
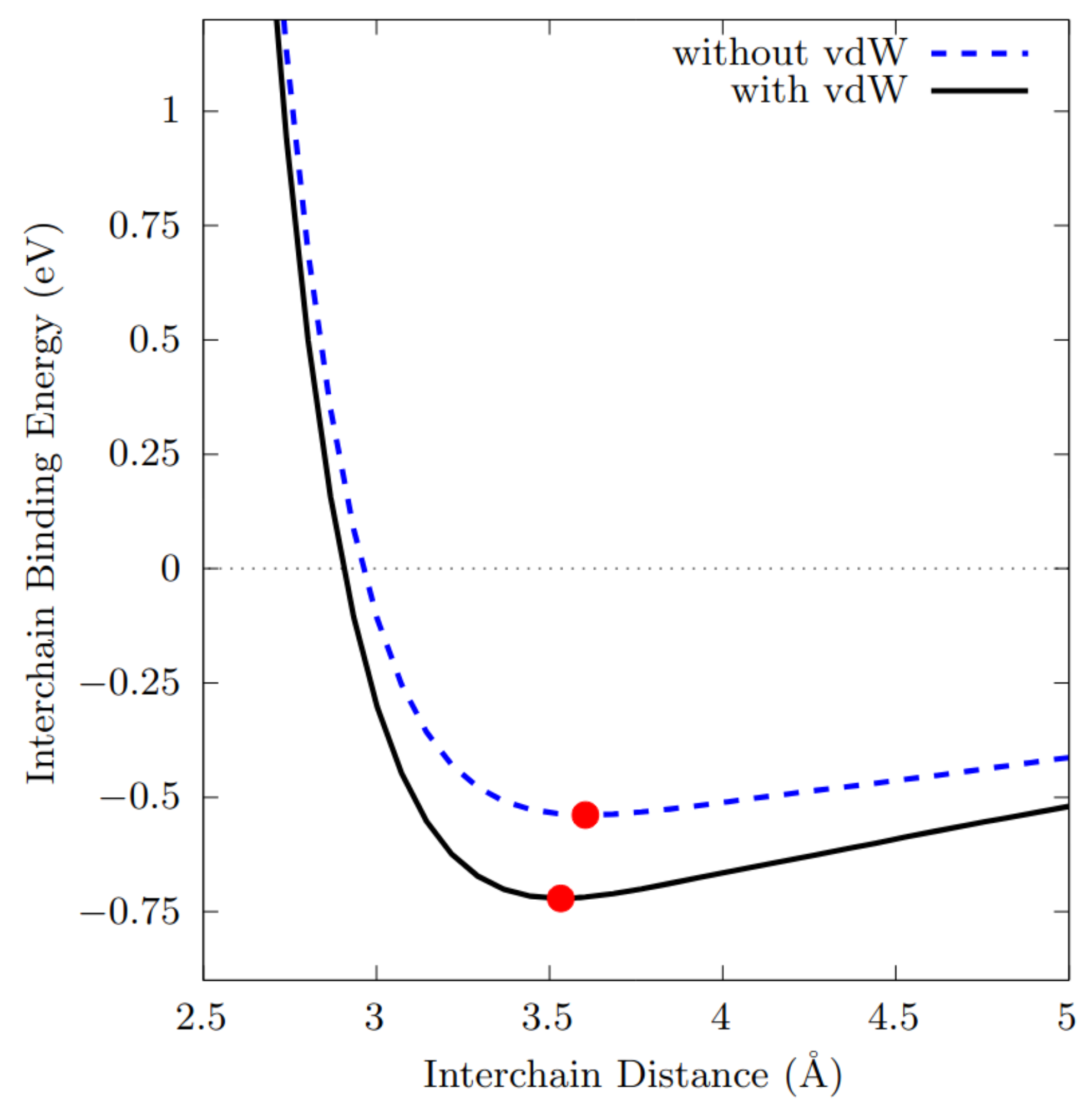
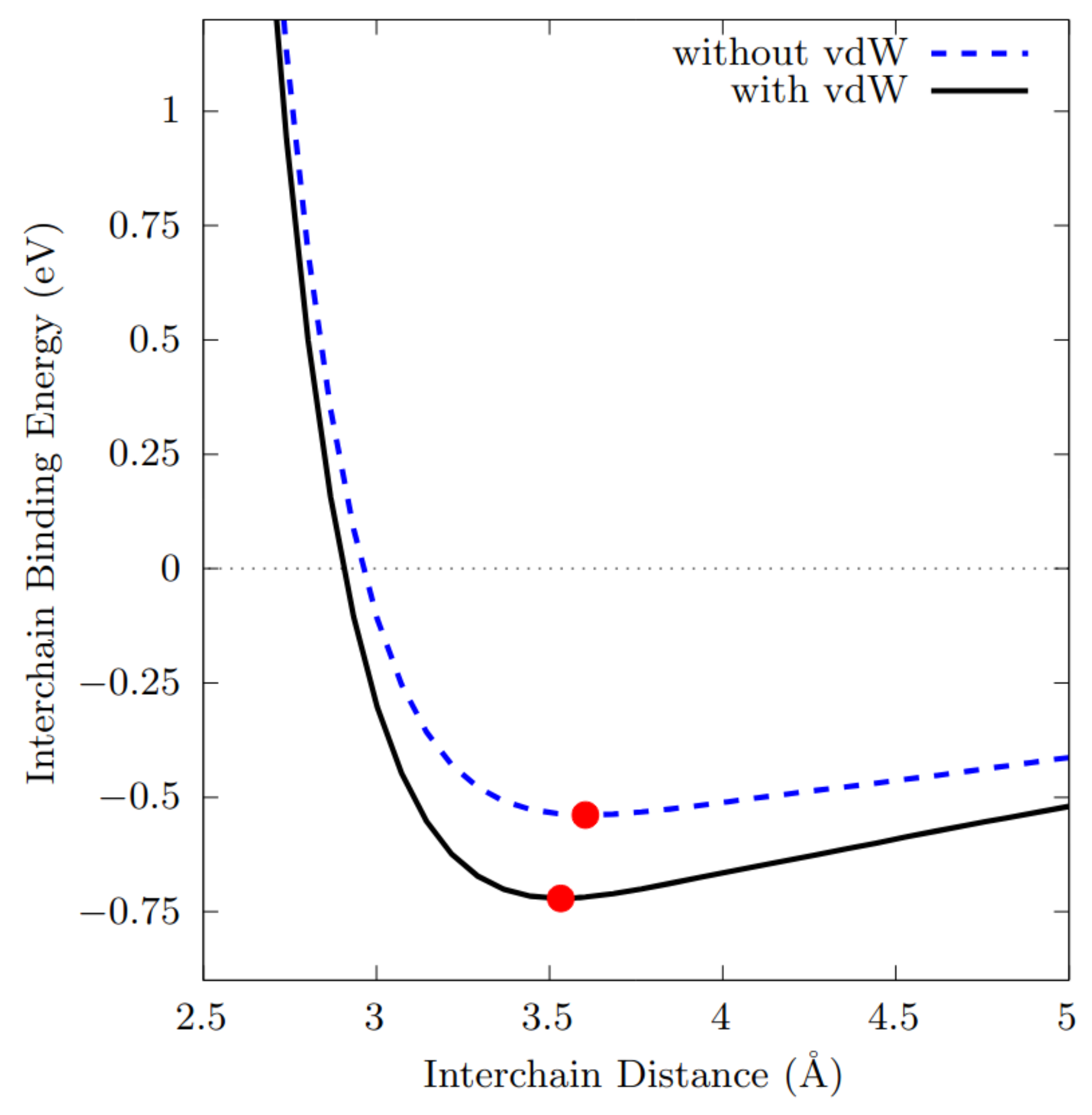
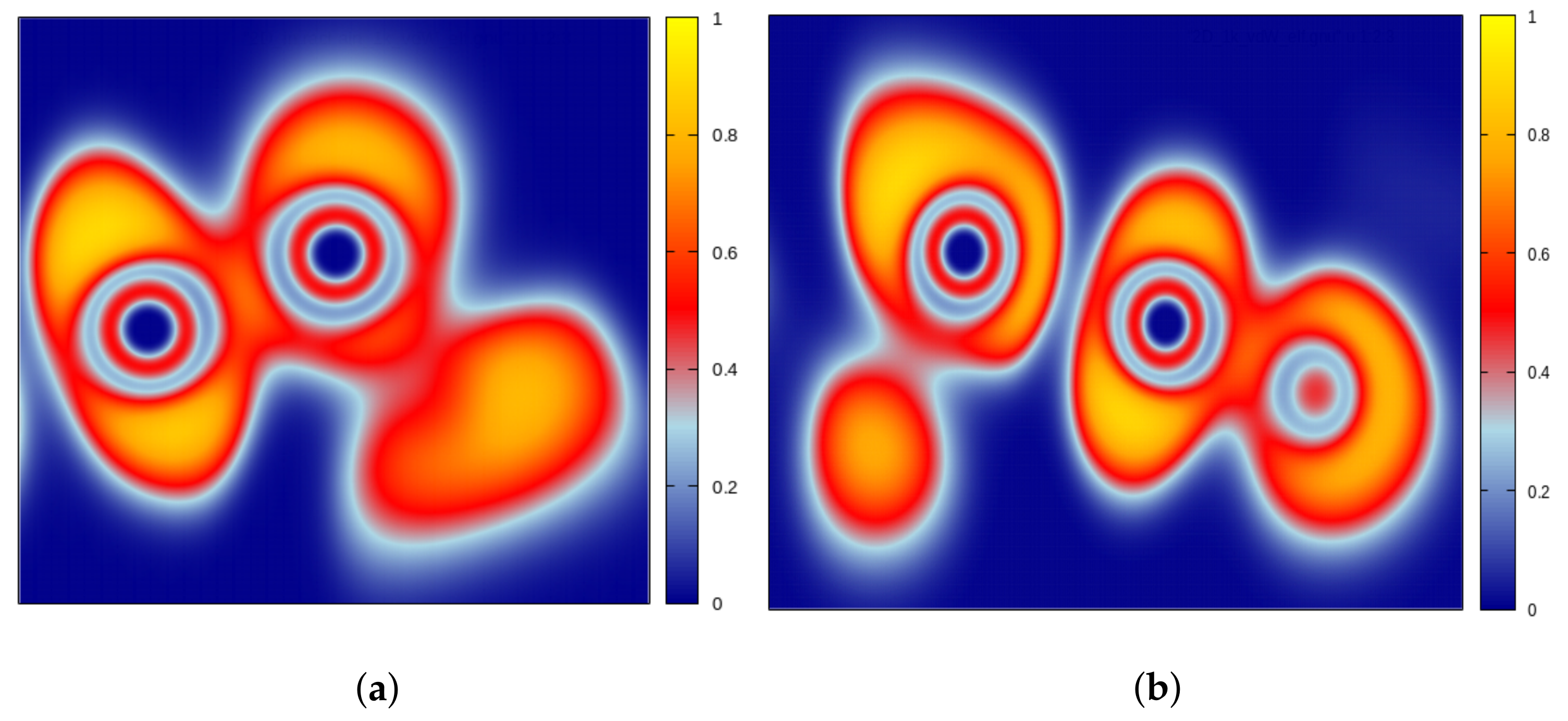
3.5. Tellurium Interchain Interaction
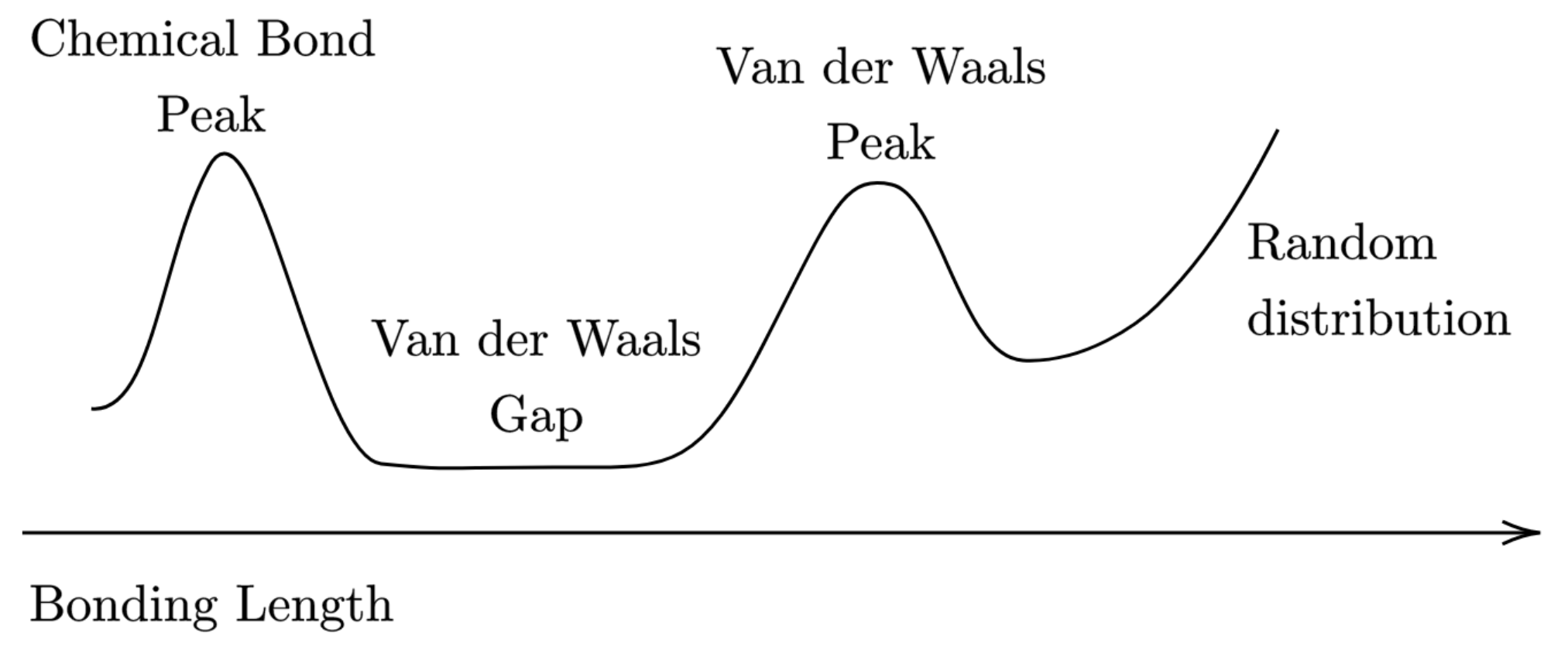
- interatomic distances between the vdW radii sum fall into the vdW peak, while longer distances should indicate non-interacting atoms;
- distances shorter than the vdW radii sum by more than about correspond most likely to a chemical bond, and those between to shorter fall within the so called “vdW gap” (see Figure 10), thus suggesting a special bonding situation that asks for a deeper analysis.
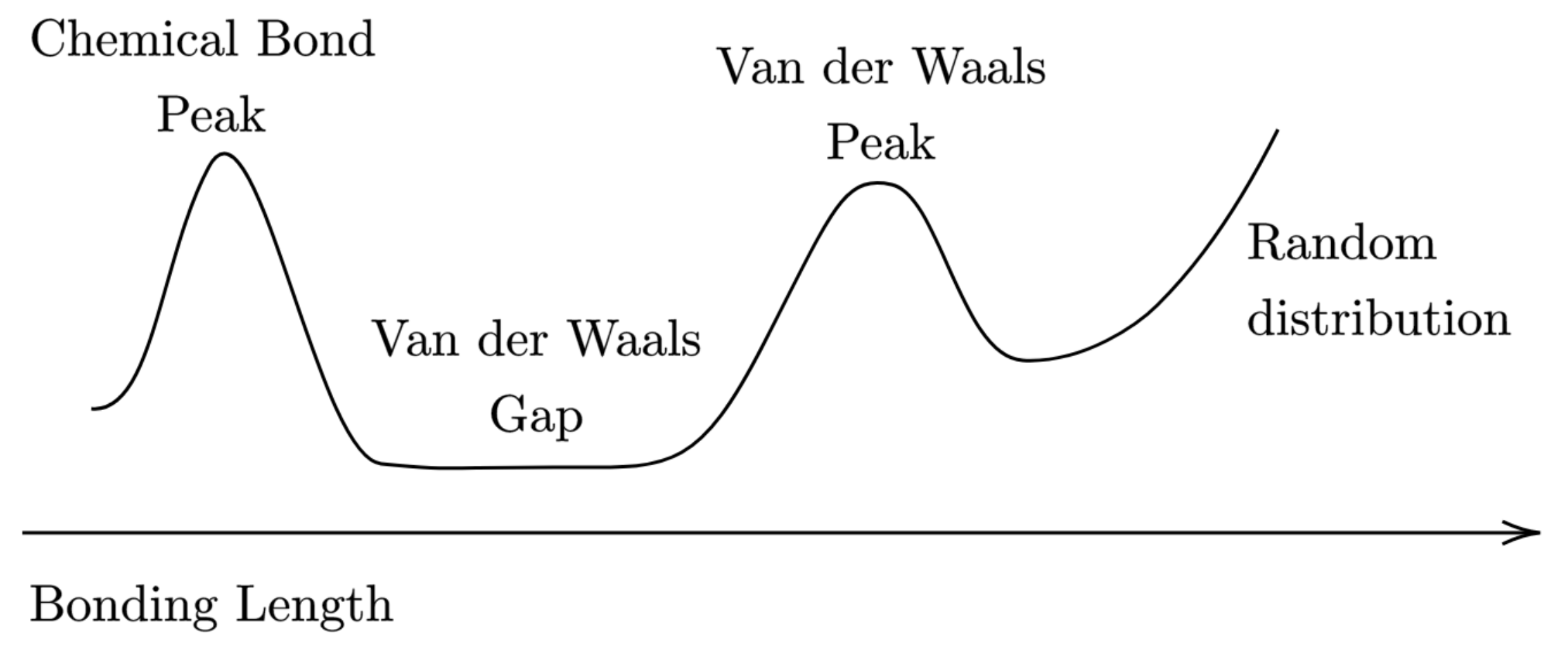
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, C.; Chakrabarti, A.; Ezawa, M. Aluminene as highly hole-doped graphene. New J. Phys. 2015, 17, 083014. [Google Scholar] [CrossRef]
- Tao, M.L.; Tu, Y.B.; Sun, K.; Wang, Y.L.; Xie, Z.B.; Liu, L.; Shi, M.X.; Wang, J.Z. Gallenene epitaxially grown on Si(1 1 1). 2D Mater. 2018, 5, 035009. [Google Scholar] [CrossRef]
- Singh, D.; Gupta, S.K.; Lukačević, I.; Sonvane, Y. Indiene 2D monolayer: A new nanoelectronic material. RSC Adv. 2016, 6, 8006–8014. [Google Scholar] [CrossRef]
- Vogt, P.; De Padova, P.; Quaresima, C.; Avila, J.; Frantzeskakis, E.; Asensio, M.C.; Resta, A.; Ealet, B.; Le Lay, G. Silicene: Compelling Experimental Evidence for Graphenelike Two-Dimensional Silicon. Phys. Rev. Lett. 2012, 108, 155501. [Google Scholar] [CrossRef]
- Bianco, E.; Butler, S.; Jiang, S.; Restrepo, O.D.; Windl, W.; Goldberger, J.E. Stability and Exfoliation of Germanane: A Germanium Graphane Analogue. ACS Nano 2013, 7, 4414–4421. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.F.; Chen, W.J.; Xu, Y.; Gao, C.L.; Guan, D.D.; Liu, C.H.; Qian, D.; Zhang, S.C.; Jia, J.F. Epitaxial growth of two-dimensional stanene. Nat. Mater. 2015, 14, 1020–1025. [Google Scholar] [CrossRef]
- Yuhara, J.; He, B.; Matsunami, N.; Nakatake, M.; Le Lay, G. Graphene’s Latest Cousin: Plumbene Epitaxial Growth on a “Nano WaterCube”. Adv. Mater. 2019, 31, 1901017. [Google Scholar] [CrossRef]
- Li, L.; Yu, Y.; Ye, G. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Yan, Z.; Li, Y.; Chen, Z.; Zeng, H. Atomically Thin Arsenene and Antimonene: Semimetal–Semiconductor and Indirect–Direct Band-Gap Transitions. Angew. Chem. Int. Ed. 2015, 54, 3112–3115. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Song, X.; Liu, J.; Yan, Z.; Huo, C.; Zhang, S.; Su, M.; Liao, L.; Wang, W.; Ni, Z.; et al. Two-dimensional antimonene single crystals grown by van der Waals epitaxy. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, F.; Li, G.; Dudy, L.; Bauernfeind, M.; Glass, S.; Hanke, W.; Thomale, R.; Schäfer, J.; Claessen, R. Bismuthene on a SiC substrate: A candidate for a high-temperature quantum spin Hall material. Science 2017, 357, 287–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Cai, X.; Yi, S.; Chen, J.; Dai, Y.; Niu, C.; Guo, Z.; Xie, M.; Liu, F.; Cho, J.H.; et al. Multivalency-Driven Formation of Te-Based Monolayer Materials: A Combined First-Principles and Experimental study. Phys. Rev. Lett. 2017, 119, 106101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Qiu, G.; Wang, R.; Huang, S.; Wang, Q.; Liu, Y.; Du, Y.; Goddard, W.A.; Kim, M.J.; Xu, X.; et al. Field-effect transistors made from solution-grown two-dimensional tellurene. Nat. Electron. 2018, 1, 228–236. [Google Scholar] [CrossRef]
- Du, Y.; Qiu, G.; Wang, Y.; Si, M.; Xu, X.; Wu, W.; Ye, P.D. One-Dimensional van der Waals Material Tellurium: Raman Spectroscopy under Strain and Magneto-Transport. Nano Lett. 2017, 17, 3965–3973. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Guan, J.; Lin, Z.; Liu, B.; Xing, S.; Wang, W.; Guo, J. Epitaxial Growth and Band Structure of Te Film on Graphene. Nano Lett. 2017, 17, 4619–4623. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Han, X.; Zhao, C.; Niu, C.; Jia, Y. Tellurene: An elemental 2D monolayer material beyond its bulk phases without van der Waals layered structures. J. Semicond. 2020, 41, 081002. [Google Scholar] [CrossRef]
- Grazianetti, C.; Martella, C.; Molle, A. 8—Two-dimensional Xenes and their device concepts for future micro- and nanoelectronics and energy applications. In Micro and Nano Technologies, Emerging 2D Materials and Devices for the Internet of Things; Elsevier: Amsterdam, The Netherlands, 2020; pp. 181–219. [Google Scholar] [CrossRef]
- Wu, W.; Qiu, G.; Wang, Y.; Wang, R.; Ye, P. Tellurene: Its physical properties, scalable nanomanufacturing, and device applications. Chem. Soc. Rev. 2018, 47, 7203–7212. [Google Scholar] [CrossRef]
- Pang, H.; Yan, J.; Yang, J.; Liu, S.; Pan, Y.; Zhang, X.; Shi, B.; Tang, H.; Yang, J.; Liu, Q.; et al. Bilayer tellurene–metal interfaces. J. Semicond. 2019, 40, 062003. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yang, A.; Lan, T.; Fan, C.; Pan, J.; Liu, Z.; Chu, J.; Yuan, H.; Wang, X.; Rong, M.; et al. Tellurene based chemical sensor. J. Mater. Chem. A 2019, 7, 26326–26333. [Google Scholar] [CrossRef]
- Cui, H.; Zheng, K.; Xie, Z.; Yu, J.; Zhu, X.; Ren, H.; Wang, Z.; Zhang, F.; Li, X.; Tao, L.Q.; et al. Tellurene Nanoflake-Based NO2 Sensors with Superior Sensitivity and a Sub-Parts-per-Billion Detection Limit. ACS Appl. Mater. Interfaces 2020, 12, 47704–47713. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Wang, D.W.; Yang, A.J.; Koratkar, N.; Chu, J.F.; Lv, P.L.; Rong, M.Z. Effects of adatom and gas molecule adsorption on the physical properties of tellurene: A first principles investigation. Phys. Chem. Chem. Phys. 2018, 20, 4058–4066. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Cheng, W.; Chai, G.; Zhang, H. Thermoelectric properties of two-dimensional selenene and tellurene from group-VI elements. Phys. Chem. Chem. Phys. 2018, 20, 24250–24256. [Google Scholar] [CrossRef] [PubMed]
- Deckoff-Jones, S.; Wang, Y.; Lin, H.; Wu, W.; Hu, J. Tellurene: A Multifunctional Material for Midinfrared Optoelectronics. ACS Photonics 2019, 6, 1632–1638. [Google Scholar] [CrossRef]
- Amani, M.; Tan, C.; Zhang, G.; Zhao, C.; Bullock, J.; Song, X.; Kim, H.; Shrestha, V.R.; Gao, Y.; Crozier, K.B.; et al. Solution-Synthesized High-Mobility Tellurium Nanoflakes for Short-Wave Infrared Photodetectors. ACS Nano 2018, 12, 7253–7263. [Google Scholar] [CrossRef]
- Wang, Q.; Safdar, M.; Xu, K.; Mirza, M.; Wang, Z.; He, J. Van der Waals Epitaxy and Photoresponse of Hexagonal Tellurium Nanoplates on Flexible Mica Sheets. ACS Nano 2014, 8, 7497–7505. [Google Scholar] [CrossRef]
- Wu, L.; Huang, W.; Wang, Y.; Zhao, J.; Ma, D.; Xiang, Y.; Li, J.; Ponraj, J.S.; Dhanabalan, S.C.; Zhang, H. 2D Tellurium Based High-Performance All-Optical Nonlinear Photonic Devices. Adv. Funct. Mater. 2019, 29, 1806346. [Google Scholar] [CrossRef]
- Qiao, J.; Pan, Y.; Yang, F.; Wang, C.; Chai, Y.; Ji, W. Few-layer Tellurium: One-dimensional-like layered elementary semiconductor with striking physical properties. Sci. Bull. 2018, 63, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Dai, Y.; Ma, Y.; Dai, X.; Ho, W.; Xie, M. Ultrathin β,-tellurium layers grown on highly oriented pyrolytic graphite by molecular-beam epitaxy. Nanoscale 2017, 9, 15945–15948. [Google Scholar] [CrossRef] [Green Version]
- Khatun, S.; Banerjee, A.; Pal, A.J. Nonlayered tellurene as an elemental 2D topological insulator: Experimental evidence from scanning tunneling spectroscopy. Nanoscale 2019, 11, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, C.; Chen, M.; Hua, C.; Zou, J.; Wu, C.; Jiang, J.; Yang, S.A.; Lu, Y.; Ji, W. Two-dimensional ferroelectricity and switchable spin-textures in ultra-thin elemental Te multilayers. Mater. Horiz. 2018, 5, 521–528. [Google Scholar] [CrossRef]
- Xiang, Y.; Gao, S.; Xu, R.G.; Wu, W.; Leng, Y. Phase transition in two-dimensional tellurene under mechanical strain modulation. Nano Energy 2019, 58, 202–210. [Google Scholar] [CrossRef]
- Keldysh, L.V. Coulomb interaction in thin semiconductor and semimetal films. Sov. J. Exp. Theor. Phys. Lett. 1979, 29, 658. [Google Scholar]
- Rytova, N.S. Screened potential of a point charge in a thin. arXiv 2018, arXiv:1806.00976. [Google Scholar]
- Pulci, O.; Gori, P.; Marsili, M.; Garbuio, V.; Sole, R.D.; Bechstedt, F. Strong excitons in novel two-dimensional crystals: Silicane and germanane. EPL Europhys. Lett. 2012, 98, 37004. [Google Scholar] [CrossRef] [Green Version]
- Pulci, O.; Marsili, M.; Garbuio, V.; Gori, P.; Kupchak, I.; Bechstedt, F. Excitons in two-dimensional sheets with honeycomb symmetry. Phys. Status Solidi 2015, 252, 72–77. [Google Scholar] [CrossRef]
- Prete, M.S.; Grassano, D.; Pulci, O.; Kupchak, I.; Olevano, V.; Bechstedt, F. Giant excitonic absorption and emission in two-dimensional group-III nitrides. arXiv 2019, arXiv:1903.12031. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Zeng, B.; Zhang, X.; Li, D.; He, J.; Long, M. Study on the strain-induced mechanical property modulations in monolayer Tellurene. J. Appl. Phys. 2019, 125, 064304. [Google Scholar] [CrossRef]
- Wu, B.; Liu, X.; Yin, J.; Lee, H. Bulkβ-Te to few layeredβ-tellurenes: Indirect to direct band-Gap transitions showing semiconducting property. Mater. Res. Express 2017, 4, 095902. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Gao, S.; Yang, L.; Lu, J. Dependence of excited-state properties of tellurium on dimensionality: From bulk to two dimensions to one dimensions. Phys. Rev. B 2018, 98, 085135. [Google Scholar] [CrossRef] [Green Version]
- Shinada, M.; Sugano, S. Interband Optical Transitions in Extremely Anisotropic Semiconductors. I. Bound and Unbound Exciton Absorption. J. Phys. Soc. Jpn. 1966, 21, 1936–1946. [Google Scholar] [CrossRef]
- Flügge, S. Rechenmethoden der Quantentheorie: Elementare Quantenmechanik. Dargestellt in Aufgaben und Lösungen; Springer: Berlin/Heidelberg, Germany, 2013; Volume 6. [Google Scholar]
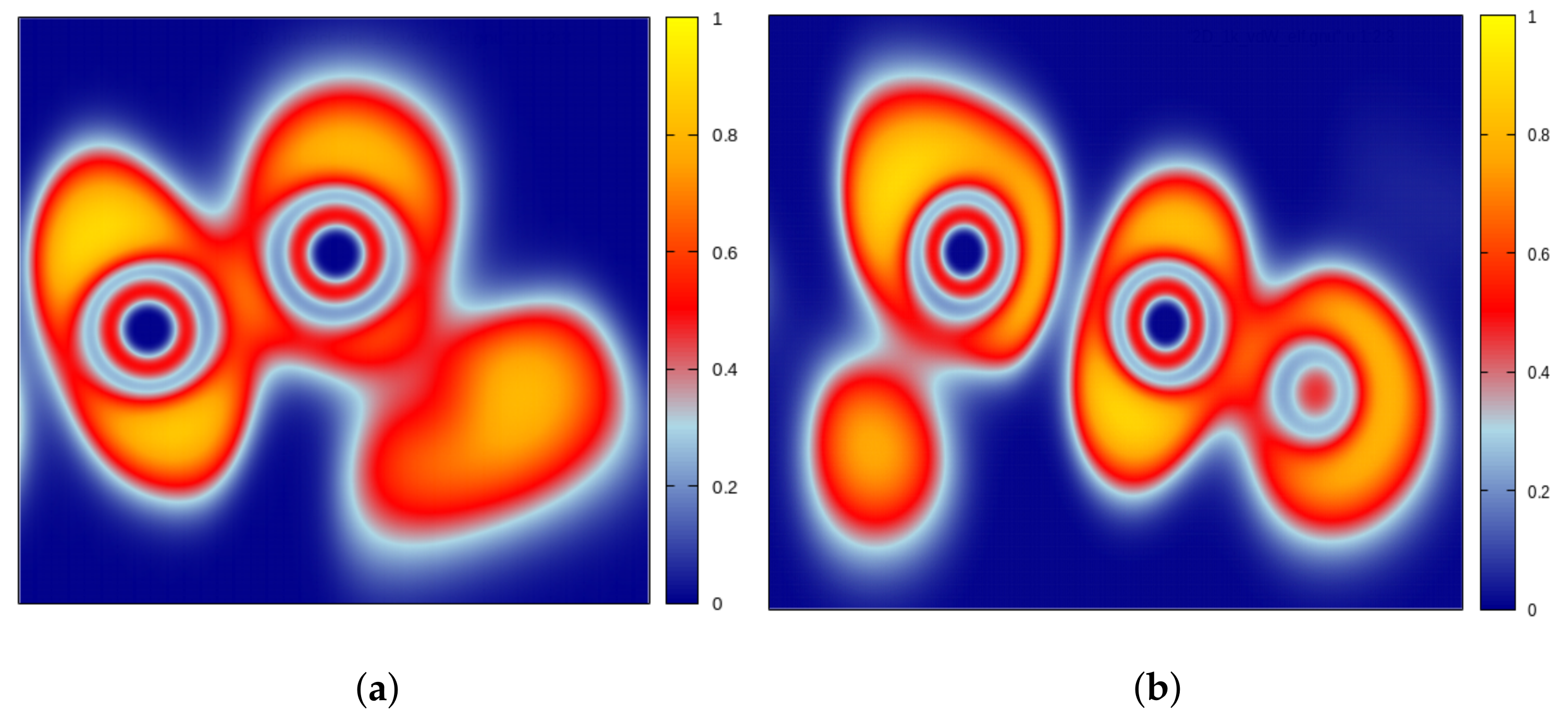
- In the case of all α-Te, the states involved in the first optical transition did not correspond to the VBM and CBM of the respective bands. Since we are interested in calculating the electron and hole effective masses—using a quadratic fit nearby the VBM and CBM—this situation could in principle represent a non-negligible problem. Luckily enough, by plotting the interband difference, the energy showed a perfectly parabolic behavior, with a clearly pronounced minimum. Thus, to reduce the numerical error, the electron-hole effective mass, directly corresponding to the exciton reduced mass, has been extracted using the quadratic fit of this difference.
- Gao, Q.; Li, X.; Fang, L.; Wang, T.; Wei, S.; Xia, C.; Jia, Y. Exciton states and oscillator strength in few-layer α,-tellurene. Appl. Phys. Lett. 2019, 114, 092101. [Google Scholar] [CrossRef]
- Reitz, J.R. Electronic Band Structure of Selenium and Tellurium. Phys. Rev. 1957, 105, 1233–1240. [Google Scholar] [CrossRef]
- Yi, S.; Zhu, Z.; Cai, X.; Jia, Y.; Cho, J.H. The nature of bonding in bulk tellurium composed of one-dimensional helical chains. Inorg. Chem. 2018, 57, 5083–5088. [Google Scholar] [CrossRef] [Green Version]
- Epstein, A.S.; Fritzsche, H.; Lark-Horovitz, K. Electrical Properties of Tellurium at the Melting Point and in the Liquid State. Phys. Rev. 1957, 107, 412–419. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounet, N.; Gibertini, M.; Schwaller, P.; Campi, D.; Merkys, A.; Marrazzo, A.; Sohier, T.; Castelli, I.E.; Cepellotti, A.; Pizzi, G.; et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018, 13, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The intrachain bond lengths extracted from the single chain are 2.75Å and 2.79Å when the relaxation is performed with and without the inclusions of the vdW corrections, independently on the correction adopted.
- Liu, Z.; Liu, J.Z.; Cheng, Y.; Li, Z.; Wang, L.; Zheng, Q. Interlayer binding energy of graphite: A mesoscopic determination from deformation. Phys. Rev. B 2012, 85, 205418. [Google Scholar] [CrossRef] [Green Version]
- Gould, T.; Liu, Z.; Liu, J.Z.; Dobson, J.F.; Zheng, Q.; Lebègue, S. Binding and interlayer force in the near-contact region of two graphite slabs: Experiment and theory. J. Chem. Phys. 2013, 139, 224704. [Google Scholar] [CrossRef] [Green Version]
- Mostaani, E.; Drummond, N.; Fal’ko, V. Quantum Monte Carlo Calculation of the Binding Energy of Bilayer Graphene. Phys. Rev. Lett. 2015, 115, 115501. [Google Scholar] [CrossRef]
- Dappe, Y.J.; Basanta, M.A.; Flores, F.; Ortega, J. Weak chemical interaction and van der Waals forces between graphene layers: A combined density functional and intermolecular perturbation theory approach. Phys. Rev. B 2006, 74, 205434. [Google Scholar] [CrossRef]
- Podeszwa, R. Interactions of graphene sheets deduced from properties of polycyclic aromatic hydrocarbons. J. Chem. Phys. 2010, 132, 044704. [Google Scholar] [CrossRef]
- Chakarova-Käck, S.D.; Schröder, E.; Lundqvist, B.I.; Langreth, D.C. Application of van der Waals Density Functional to an Extended System: Adsorption of Benzene and Naphthalene on Graphite. Phys. Rev. Lett. 2006, 96, 146107. [Google Scholar] [CrossRef] [Green Version]
- Lebedeva, I.V.; Knizhnik, A.A.; Popov, A.M.; Lozovik, Y.E.; Potapkin, B.V. Interlayer interaction and relative vibrations of bilayer graphene. Phys. Chem. Chem. Phys. 2011, 13, 5687–5695. [Google Scholar] [CrossRef]
- Gould, T.; Lebègue, S.; Dobson, J.F. Dispersion corrections in graphenic systems: A simple and effective model of binding. J. Phys. Condens. Matter 2013, 25, 445010. [Google Scholar] [CrossRef]
- Koumpouras, K.; Larsson, J.A. Distinguishing between chemical bonding and physical binding using electron localization function (ELF). J. Phys. Condens. Matter 2020, 32, 315502. [Google Scholar] [CrossRef] [PubMed]
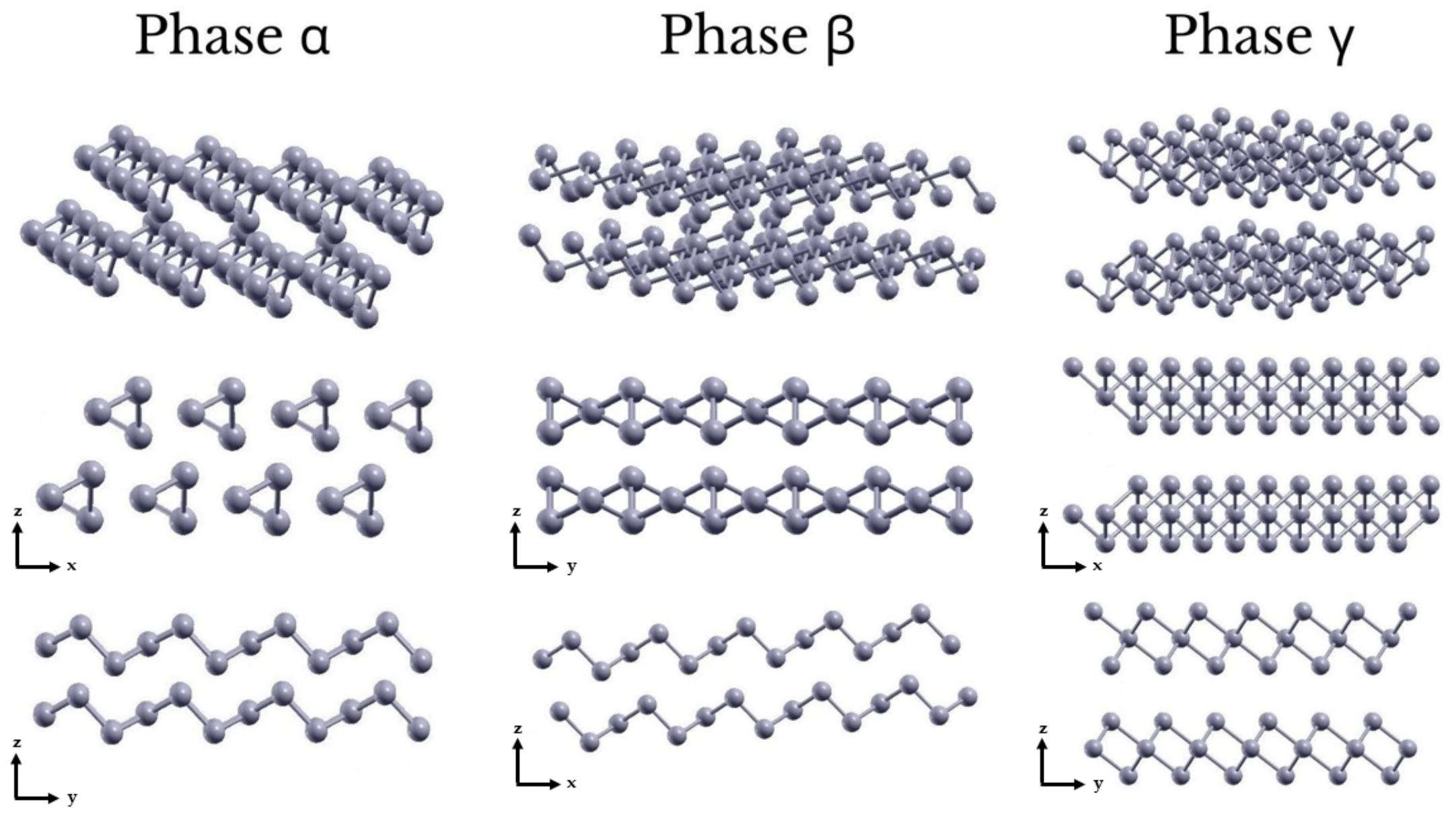
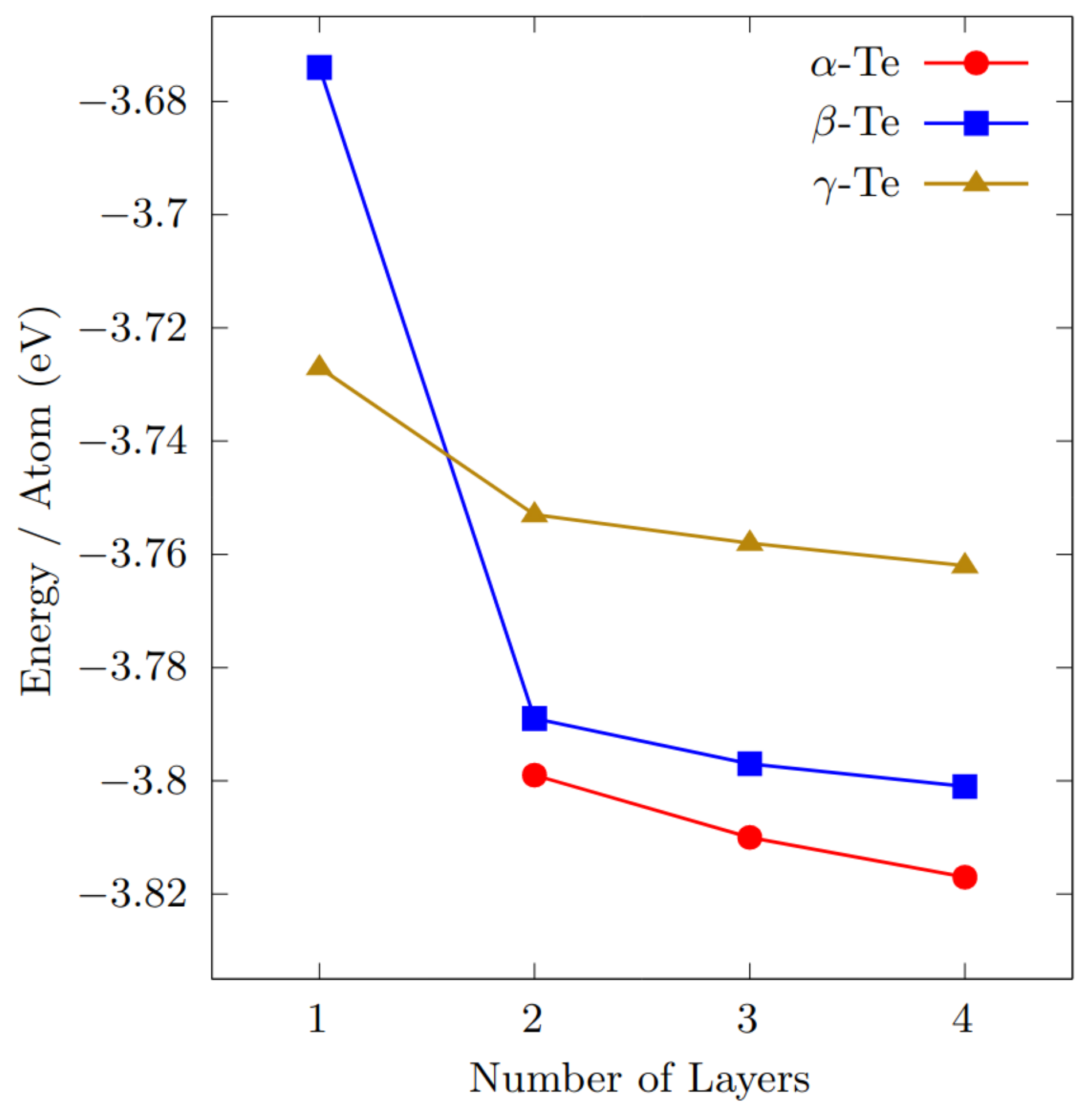
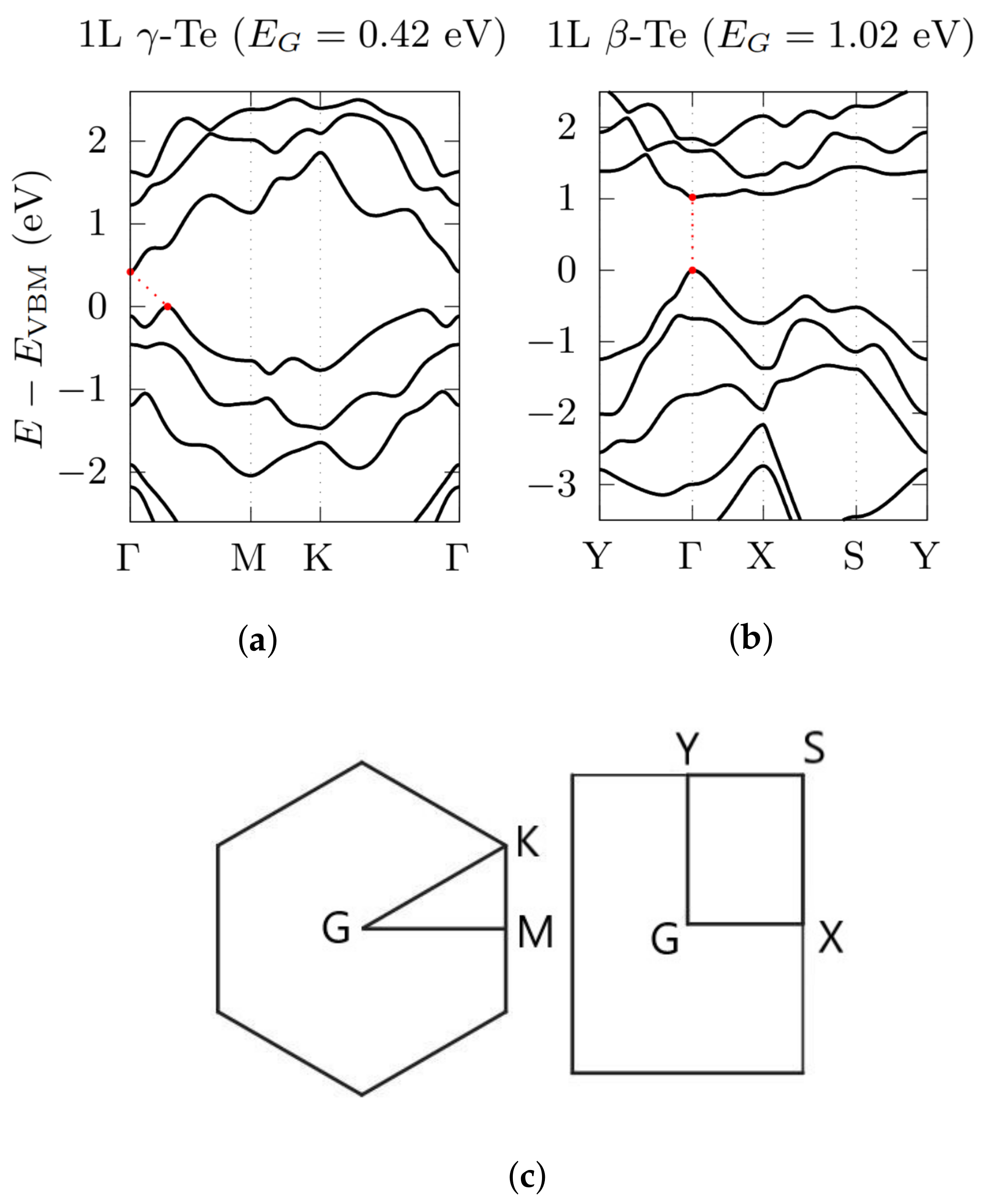


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1L | 2L | 3L | 4L | |||||
|---|---|---|---|---|---|---|---|---|
| a, b (Å) | (Å) | a, b (Å) | (Å) | a, b (Å) | (Å) | a, b (Å) | (Å) | |
| -phase | ||||||||
| This work | - | - | ||||||
| [30] | ||||||||
| -phase | ||||||||
| This work | ||||||||
| [14,45] | ||||||||
| -phase | ||||||||
| This work | ||||||||
| [14] | ||||||||
| 1L | 2L | 3L | 4L | |
|---|---|---|---|---|
| (eV) | (eV) | (eV) | (eV) | |
| -phase | − | |||
| -phase | ||||
| -phase | 0 |
| (eV) | (eV) | |||
|---|---|---|---|---|
| 2L -Te | 15 | |||
| 3L -Te | 19 | |||
| 4L -Te | 25 | |||
| 1L -Te | 9 | [46] | ||
| 2L -Te | 26 | |||
| 3L -Te | 57 | |||
| 4L -Te | 267 | |||
| 1L -Te | 27 | [50] | ||
| 2L -Te | 63 | [50] | ||
| 3L -Te | 52 | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grillo, S.; Pulci, O.; Marri, I. Evolution of the Electronic and Optical Properties of Meta-Stable Allotropic Forms of 2D Tellurium for Increasing Number of Layers. Nanomaterials 2022, 12, 2503. https://doi.org/10.3390/nano12142503
Grillo S, Pulci O, Marri I. Evolution of the Electronic and Optical Properties of Meta-Stable Allotropic Forms of 2D Tellurium for Increasing Number of Layers. Nanomaterials. 2022; 12(14):2503. https://doi.org/10.3390/nano12142503
Chicago/Turabian StyleGrillo, Simone, Olivia Pulci, and Ivan Marri. 2022. "Evolution of the Electronic and Optical Properties of Meta-Stable Allotropic Forms of 2D Tellurium for Increasing Number of Layers" Nanomaterials 12, no. 14: 2503. https://doi.org/10.3390/nano12142503
APA StyleGrillo, S., Pulci, O., & Marri, I. (2022). Evolution of the Electronic and Optical Properties of Meta-Stable Allotropic Forms of 2D Tellurium for Increasing Number of Layers. Nanomaterials, 12(14), 2503. https://doi.org/10.3390/nano12142503