
Effect of the Heating Rate to Prevent the Generation of Iron Oxides during the Hydrothermal Synthesis of LiFePO4

 ,
,  ,
,  ,
, 

Abstract
:
1. Introduction
2. Materials and Methods
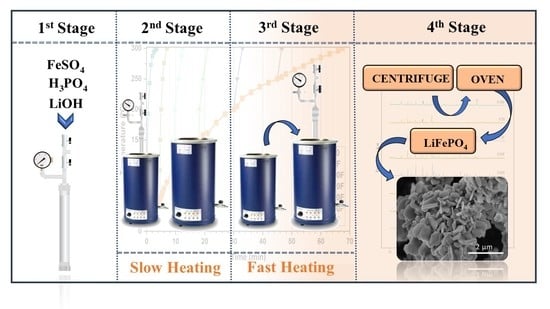
2.1. Equipment and Experimental Procedures
2.2. Reagents, Process Reactions and Products
2.3. Formation of the Base LiFePO4
2.4. Study of the Formation of the First Crystalline Nuclei of LiFePO4
2.5. Formation and Separation of LiFePO4 Microcrystals
2.6. Characterization
3. Results and Discussion
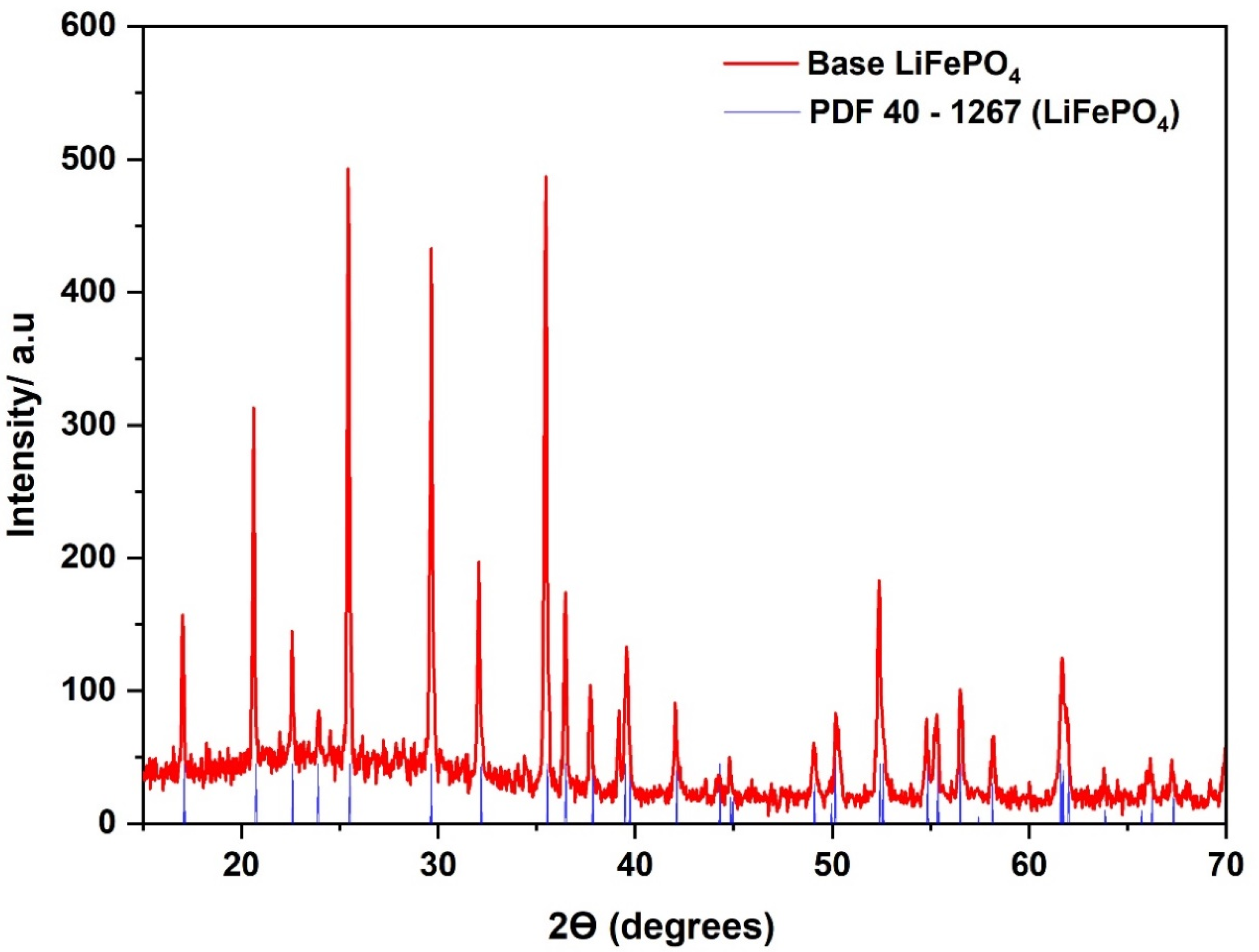
3.1. Analysis of the Synthesized Base LiFePO4
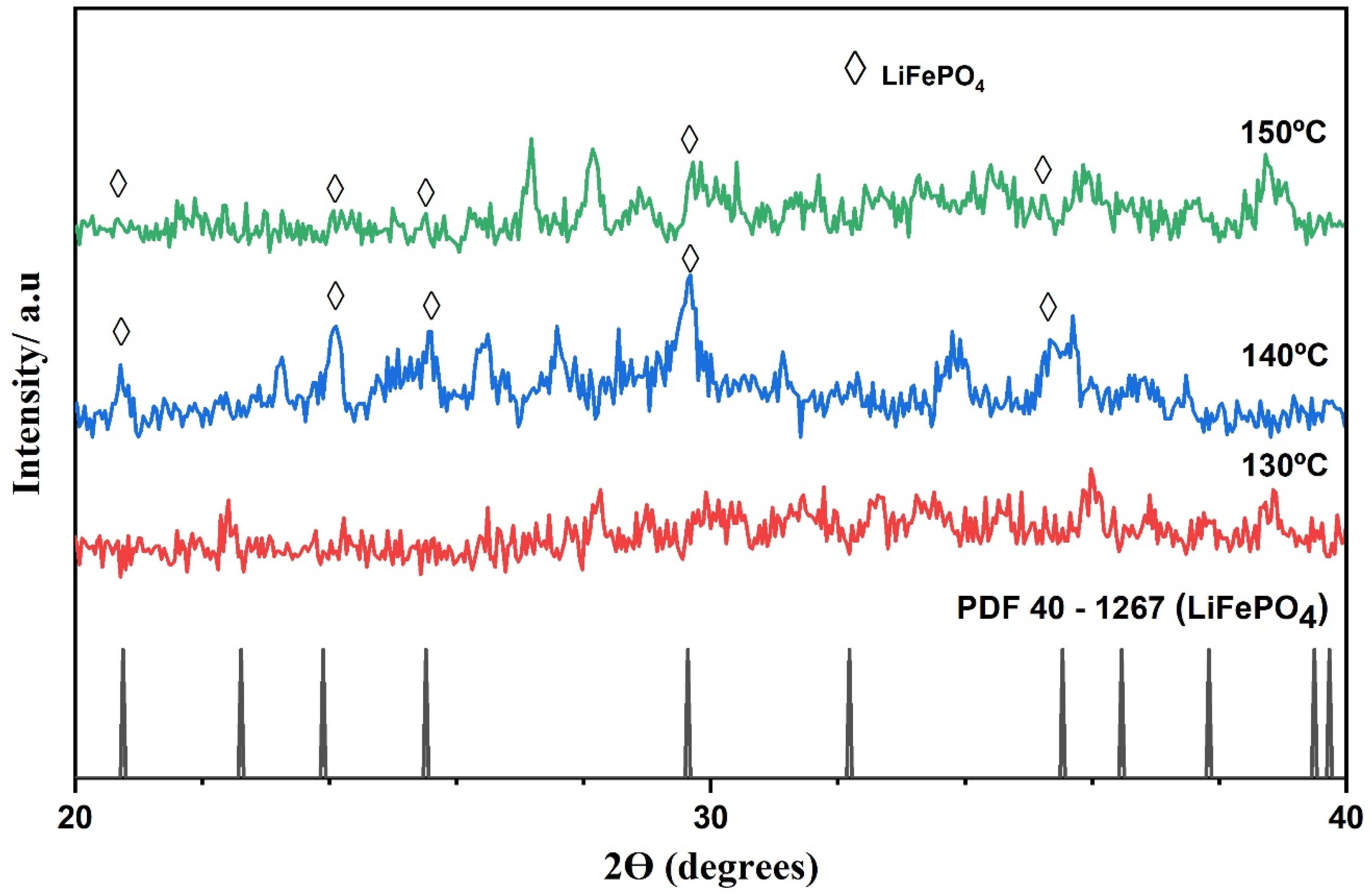
3.2. Study on the Formation of the First Crystalline Nuclei of LiFePO4
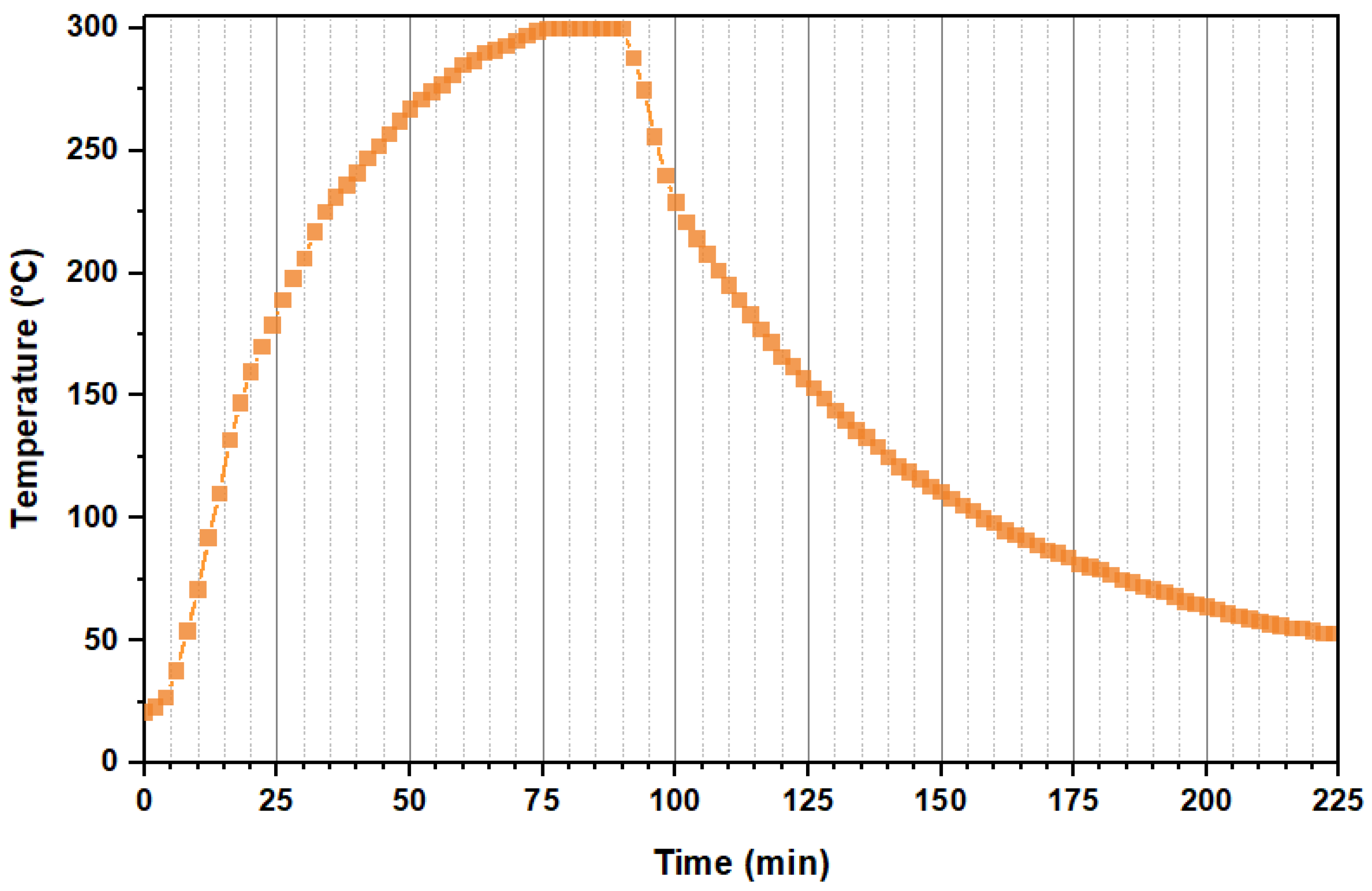
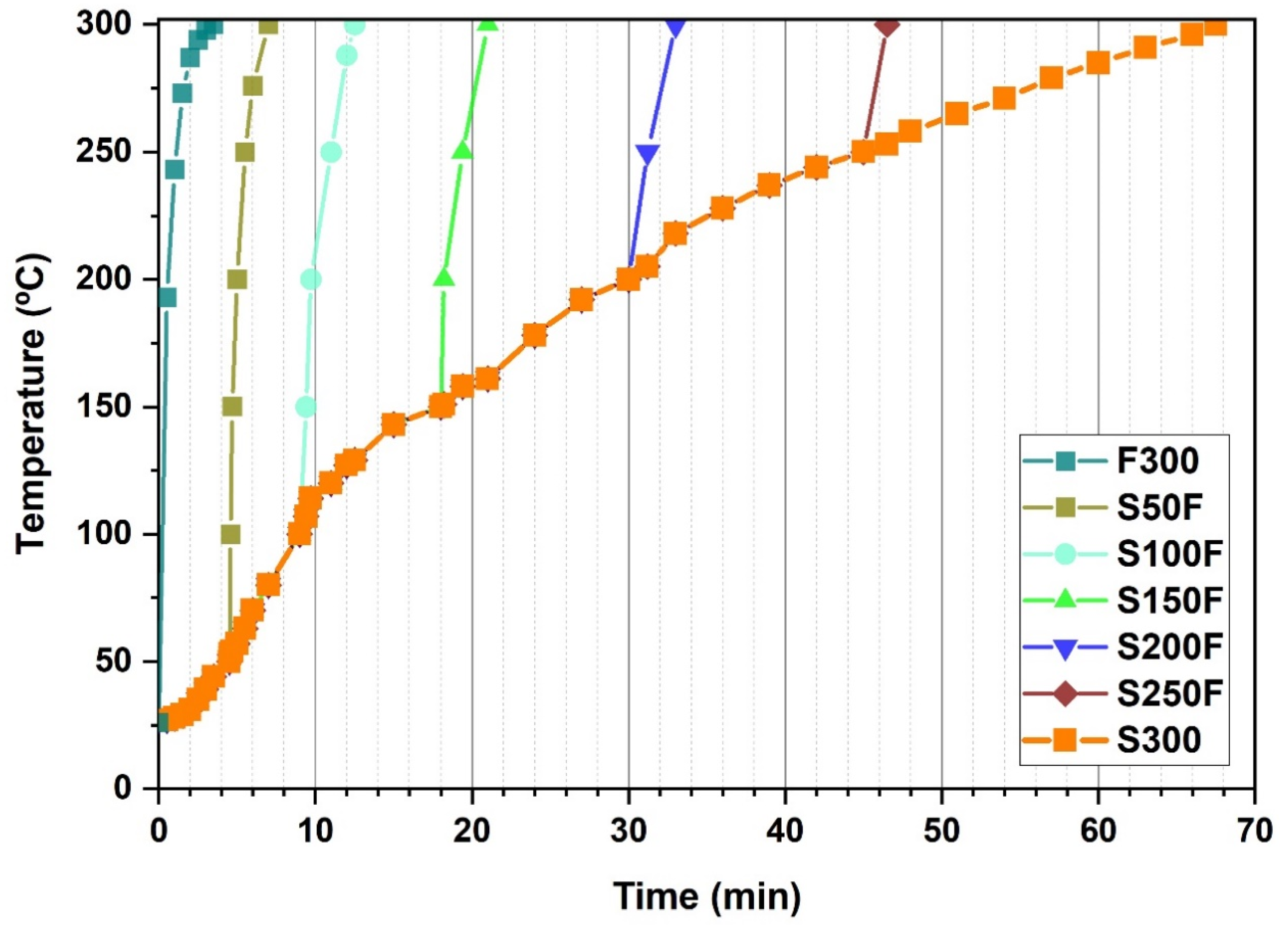
3.3. Study on the Combination of Slow-Fast Heating Rates during the HS of LiFePO4 Particles
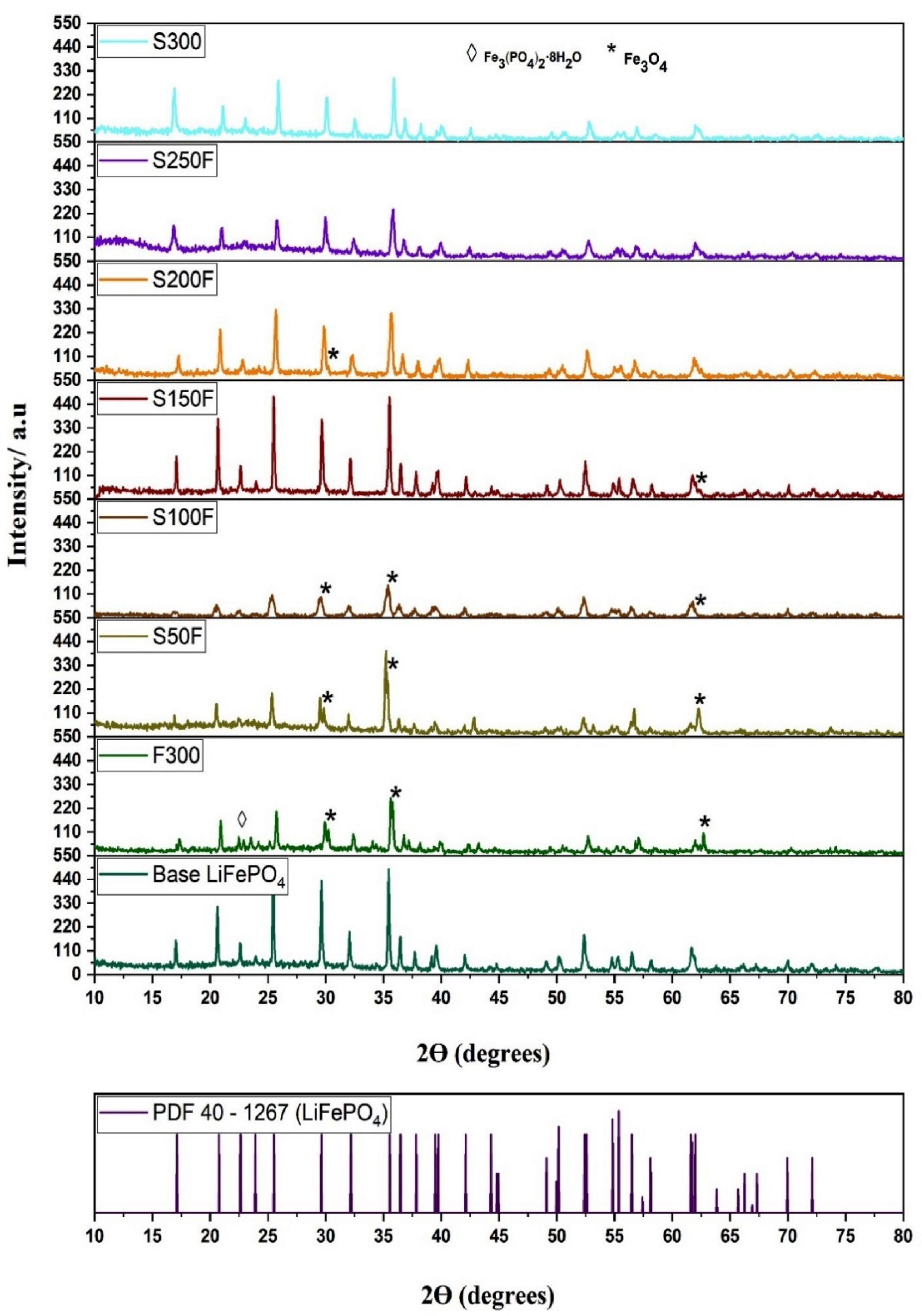
3.3.1. Crystallinity and Purity of the LiFePO4 Particles
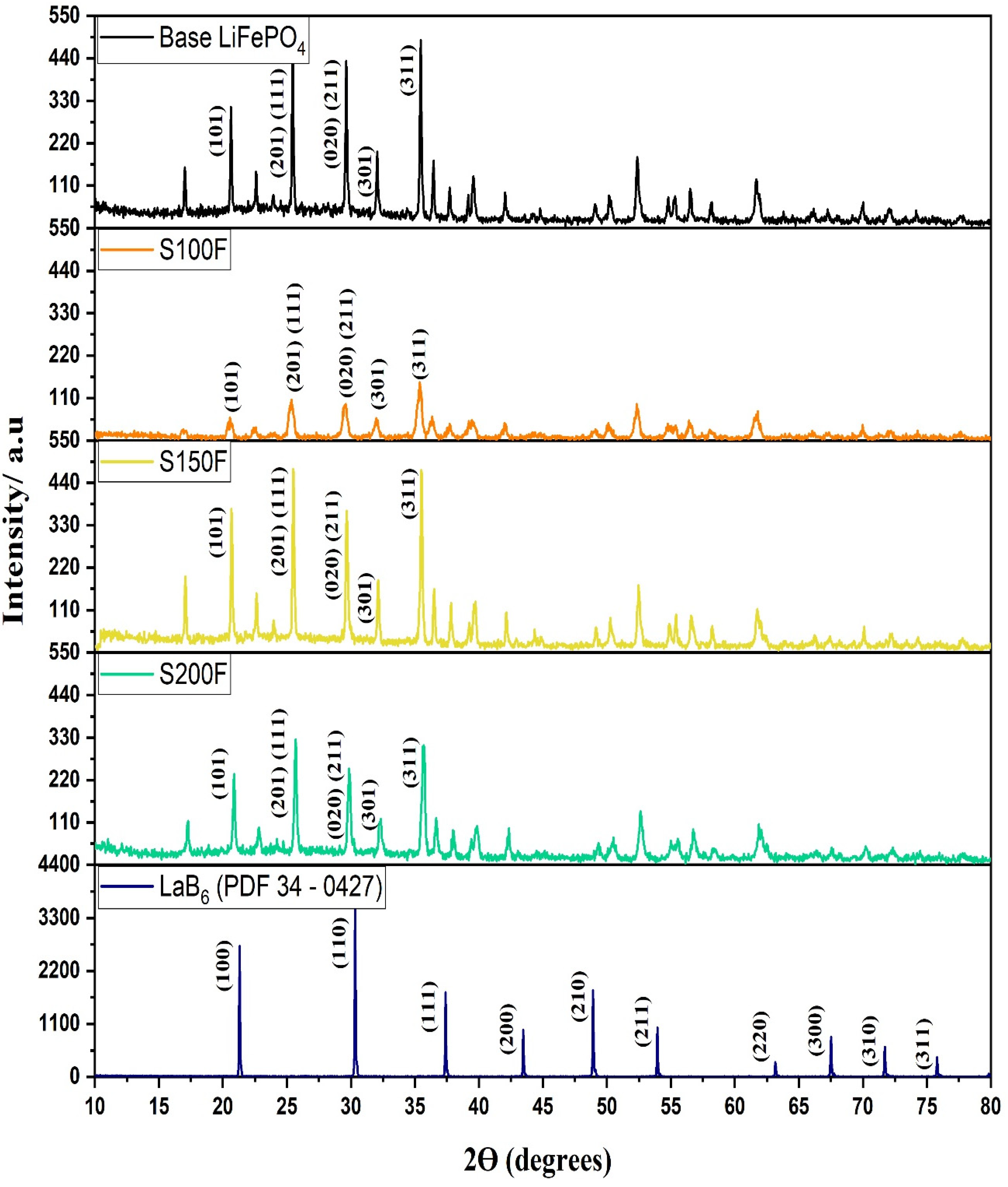
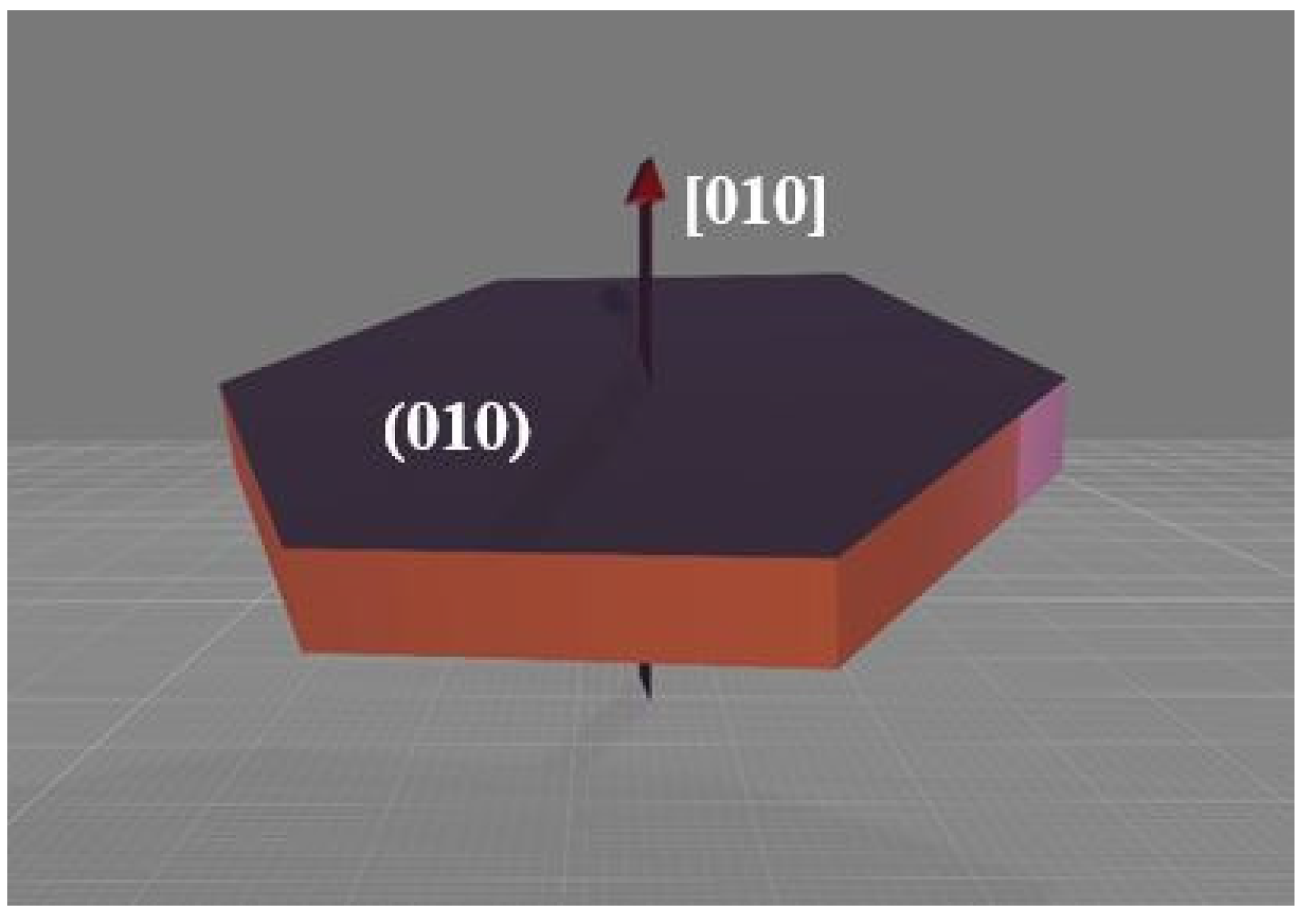
3.3.2. Growth Orientation and Morphology of the LiFePO4 Particles
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, F.; Zhang, L.; Li, Y.; An, Y.; Zhu, M.; Dai, B. Mechanism studies of LiFePO4 cathode material: Lithiation/delithiation process, electrochemical modification and synthetic reaction. RSC Adv. 2014, 4, 54576–54602. [Google Scholar] [CrossRef]
- Panda, P.K.; Grigoriev, A.; Mishra, Y.K.; Ahuja, R. Progress in supercapacitors: Roles of two dimensional nanotubular materials. Nanoscale Adv. 2020, 2, 70–108. [Google Scholar] [CrossRef] [Green Version]
- Benítez, A.; Morales, J.; Caballero, Á. Pistachio shell-derived carbon activated with phosphoric acid: A more efficient procedure to improve the performance of li–s batteries. Nanomaterials 2020, 10, 840. [Google Scholar] [CrossRef]
- Xin, S.; Yin, Y.X.; Guo, Y.G.; Wan, L.J. A high-energy room-temperature sodium-sulfur battery. Adv. Mater. 2014, 26, 1261–1265. [Google Scholar] [CrossRef]
- Perry, M.L.; Weber, A.Z. Advanced Redox-Flow Batteries: A Perspective. J. Electrochem. Soc. 2016, 163, A5064–A5067. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, S.; Li, Y.; Sun, P.; Yang, C.; Wang, D.; Liu, L. Review: Phase transition mechanism and supercritical hydrothermal synthesis of nano lithium iron phosphate. Ceram. Int. 2020, 46, 27922–27939. [Google Scholar] [CrossRef]
- Satyavani, T.V.S.L.; Srinivas Kumar, A.; Subba Rao, P.S.V. Methods of synthesis and performance improvement of lithium iron phosphate for high rate Li-ion batteries: A review. Eng. Sci. Technol. Int. J. 2016, 19, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.Z.; Zhao, Y.M.; Chen, Y.H.; He, Z.F.; Kuang, Q. Optimized carbon-coated LiFePO4 cathode material for lithium-ion batteries. Mater. Chem. Phys. 2009, 115, 245–250. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, H.; Ji, S.; Fakir, R.; Linkov, V. Influence of carbon sources on electrochemical performances of LiFePO4/C composites. Solid State Ionics 2008, 179, 1810–1815. [Google Scholar] [CrossRef]
- Franger, S.; Le Cras, F.; Bourbon, C.; Rouault, H. Comparison between different LiFePO4 synthesis routes and their influence on its physico-chemical properties. J. Power Sources 2003, 119–121, 252–257. [Google Scholar] [CrossRef]
- Kosova, N.; Devyatkina, E. On mechanochemical preparation of materials with enhanced characteristics for lithium batteries. Solid State Ionics 2004, 172, 181–184. [Google Scholar] [CrossRef]
- Dominko, R.; Goupil, J.M.; Bele, M.; Gaberscek, M.; Remskar, M.; Hanzel, D.; Jamnik, J. Impact of LiFePO4/C composites porosity on their electrochemical performance. J. Electrochem. Soc. 2005, 152, A858. [Google Scholar] [CrossRef]
- Park, K.S.; Son, J.T.; Chung, H.T.; Kim, S.J.; Lee, C.H.; Kim, H.G. Synthesis of LiFePO4 by co-precipitation and microwave heating. Electrochem. Commun. 2003, 5, 839–842. [Google Scholar] [CrossRef]
- Arnold, G.; Garche, J.; Hemmer, R.; Ströbele, S.; Vogler, C.; Wohlfahrt-Mehrens, M. Fine-particle lithium iron phosphate LiFePO4 synthesized by a new low-cost aqueous precipitation technique. J. Power Sources 2003, 119–121, 247–251. [Google Scholar] [CrossRef]
- Benedek, P.; Wenzler, N.; Yarema, M.; Wood, V.C. Low temperature hydrothermal synthesis of battery grade lithium iron phosphate. RSC Adv. 2017, 7, 17763–17767. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Teja, A.S. Characteristics of lithium iron phosphate (LiFePO4) particles synthesized in subcritical and supercritical water. J. Supercrit. Fluids 2005, 35, 83–90. [Google Scholar] [CrossRef]
- Xu, C.; Lee, J.; Teja, A.S. Continuous hydrothermal synthesis of lithium iron phosphate particles in subcritical and supercritical water. J. Supercrit. Fluids 2008, 44, 92–97. [Google Scholar] [CrossRef]
- Jin, E.M.; Jin, B.; Jun, D.K.; Park, K.H.; Gu, H.B.; Kim, K.W. A study on the electrochemical characteristics of LiFePO4 cathode for lithium polymer batteries by hydrothermal method. J. Power Sources 2008, 178, 801–806. [Google Scholar] [CrossRef]
- Gu, Y.J.; Li, C.J.; Cheng, L.; Lv, P.G.; Fu, F.J.; Liu, H.Q.; Ding, J.X.; Wang, Y.M.; Chen, Y.B.; Wang, H.F.; et al. Novel synthesis of plate-like LiFePO4 by hydrothermal method. J. New Mater. Electrochem. Syst. 2016, 19, 33–36. [Google Scholar] [CrossRef]
- He, L.H.; Zhao, Z.W.; Liu, X.H.; Chen, A.L.; Si, X.F. Thermodynamics analysis of LiFePO4 pecipitation from Li-Fe(II)-P-H2O system at 298 K. Trans. Nonferrous Met. Soc. China 2012, 22, 1766–1770. [Google Scholar] [CrossRef]
- Paolella, A.; Bertoni, G.; Hovington, P.; Feng, Z.; Flacau, R.; Prato, M.; Colombo, M.; Marras, S.; Manna, L.; Turner, S.; et al. Cation exchange mediated elimination of the Fe-antisites in the hydrothermal synthesis of LiFePO4. Nano Energy 2015, 16, 256–267. [Google Scholar] [CrossRef]
- Vediappan, K.; Guerfi, A.; Gariépy, V.; Demopoulos, G.P.; Hovington, P.; Trottier, J.; Mauger, A.; Julien, C.M.; Zaghib, K. Stirring effect in hydrothermal synthesis of nano C-LiFePO4. J. Power Sources 2014, 266, 99–106. [Google Scholar] [CrossRef]
- Wu, G.; Liu, N.; Gao, X.; Tian, X.; Zhu, Y.; Zhou, Y.; Zhu, Q. A hydrothermally synthesized LiFePO4/C composite with superior low-temperature performance and cycle life. Appl. Surf. Sci. 2018, 435, 1329–1336. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, J.; Zhang, J.; Yu, F.; Wang, J.; Nie, N.; Li, W. LiFePO4 nanoparticles growth with preferential (010) face modulated by Tween-80. RSC Adv. 2015, 5, 9745–9751. [Google Scholar] [CrossRef]
- Liang, Y.P.; Li, C.C.; Chen, W.J.; Lee, J.T. Hydrothermal synthesis of lithium iron phosphate using pyrrole as an efficient reducing agent. Electrochim. Acta 2013, 87, 763–769. [Google Scholar] [CrossRef]
- Gunawan, I.; Wagiyo, H. Synthesis of LiFePO4 in the Presence of Organic Reductant by Hydrothermal Method and its Characterization. Macromol. Symp. 2015, 353, 225–230. [Google Scholar] [CrossRef]
- Golestani, E.; Javanbakht, M.; Ghafarian-Zahmatkesh, H.; Beydaghi, H.; Ghaemi, M. Tartaric acid assisted carbonization of LiFePO4 synthesized through in situ hydrothermal process in aqueous glycerol solution. Electrochim. Acta 2018, 259, 903–915. [Google Scholar] [CrossRef]
- Qin, X.; Wang, J.; Xie, J.; Li, F.; Wen, L.; Wang, X. Hydrothermally synthesized LiFePO4 crystals with enhanced electrochemical properties: Simultaneous suppression of crystal growth along [10] and antisite defect formation. Phys. Chem. Chem. Phys. 2012, 14, 2669–2677. [Google Scholar] [CrossRef]
- Johnson, I.D.; Lübke, M.; Wu, O.Y.; Makwana, N.M.; Smales, G.J.; Islam, H.U.; Dedigama, R.Y.; Gruar, R.I.; Tighe, C.J.; Scanlon, D.O.; et al. Pilot-scale continuous synthesis of a vanadium-doped LiFePO4/C nanocomposite high-rate cathodes for lithium-ion batteries. J. Power Sources 2016, 302, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Paolella, A.; Turner, S.; Bertoni, G.; Hovington, P.; Flacau, R.; Boyer, C.; Feng, Z.; Colombo, M.; Marras, S.; Prato, M.; et al. Accelerated Removal of Fe-Antisite Defects while Nanosizing Hydrothermal LiFePO4 with Ca2+. Nano Lett. 2016, 16, 2692–2697. [Google Scholar] [CrossRef] [Green Version]
- Yen, H.; Rohan, R.; Chiou, C.Y.; Hsieh, C.J.; Bolloju, S.; Li, C.C.; Yang, Y.F.; Ong, C.W.; Lee, J.T. Hierarchy concomitant in situ stable iron(II)−carbon source manipulation using ferrocenecarboxylic acid for hydrothermal synthesis of LiFePO4 as high-capacity battery cathode. Electrochim. Acta 2017, 253, 227–238. [Google Scholar] [CrossRef]
- Kim, J.K. Supercritical synthesis in combination with a spray process for 3D porous microsphere lithium iron phosphate. CrystEngComm 2014, 16, 2818–2822. [Google Scholar] [CrossRef]
- Hong, S.A.; Kim, S.J.; Chung, K.Y.; Chun, M.S.; Lee, B.G.; Kim, J. Continuous synthesis of lithium iron phosphate (LiFePO4) nanoparticles in supercritical water: Effect of mixing tee. J. Supercrit. Fluids 2013, 73, 70–79. [Google Scholar] [CrossRef]
- Ruiz-Jorge, F.; Benítez, A.; Fernández-García, S.; Sánchez-Oneto, J.; Portela, J.R. Effect of Fast Heating and Cooling in the Hydrothermal Synthesis on LiFePO4 Microparticles. Ind. Eng. Chem. Res. 2020, 59, 9318–9327. [Google Scholar] [CrossRef]
- Vasquez-Elizondo, L.J.; Rendón-Ángeles, J.C.; Matamoros-Veloza, Z.; López-Cuevas, J.; Yanagisawa, K. Urea decomposition enhancing the hydrothermal synthesis of lithium iron phosphate powders: Effect of the lithium precursor. Adv. Powder Technol. 2017, 28, 1593–1602. [Google Scholar] [CrossRef]
- Chen, J.; Wang, S.; Whittingham, M.S. Hydrothermal synthesis of cathode materials. J. Power Sources 2007, 174, 442–448. [Google Scholar] [CrossRef]
- Sánchez-Oneto, J.; Portela, J.R.; Nebot, E.; de la Ossa, E.M. Hydrothermal oxidation: Application to the treatment of different cutting fluid wastes. J. Hazard. Mater. 2007, 144, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Fiore, J.; Li, D.; Kinsinger, N.M.; Wang, Q.; DiMasi, E.; Guo, J.; Kisailus, D. Solvothermal synthesis, development, and performance of LiFePO4 nanostructures. Cryst. Growth Des. 2013, 13, 4659–4666. [Google Scholar] [CrossRef]
- Gerhild, T.L. Schikorr Reaction: Iron(II) Hydroxide, Iron(II,III) Oxide; Betascript Publishing: Beau Bassin, Mauritius, 2012. [Google Scholar]
- Min, C.; Ou, X.; Shi, Z.; Liang, G.; Wang, L. Effects of heating rate on morphology and performance of lithium iron phosphate synthesized by hydrothermal route in organic-free solution. Ionics 2017, 24, 1285–1292. [Google Scholar] [CrossRef]
- Ellis, B.; Kan, W.H.; Makahnouk, W.R.M.; Nazar, L.F. Synthesis of nanocrystals and morphology control of hydrothermally prepared LiFePO4. J. Mater. Chem. 2007, 17, 3248–3254. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Islam, M.S. Surface structures and crystal morphologies of LiFePO4: Relevance to electrochemical behaviour. J. Mater. Chem. 2008, 18, 1209–1215. [Google Scholar] [CrossRef]
- Amaro-Gahete, J.; Benítez, A.; Otero, R.; Esquivel, D.; Jiménez-Sanchidrián, C.; Morales, J.; Caballero, Á.; Romero-Salguero, F.J. A comparative study of particle size distribution of graphene nanosheets synthesized by an ultrasound-assisted method. Nanomaterials 2019, 9, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, D.; Van der Ven, A.; Ceder, G. Li Conductivity in LixMPO4(M = Mn, Fe, Co, Ni) Olivine Materials. Electrochem. Solid-State Lett. 2004, 7, A30. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.J.; Slater, P.R. Atomic-scale investigation of defects, dopants, and lithium transport in the LiFePO4 olivine-type battery material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Dokko, K.; Koizumi, S.; Nakano, H.; Kanamura, K. Particle morphology, crystal orientation, and electrochemical reactivity of LiFePO4 synthesized by the hydrothermal method at 443 K. J. Mater. Chem. 2007, 17, 4803. [Google Scholar] [CrossRef]
- Ni, J.; Morishita, M.; Kawabe, Y.; Watada, M.; Takeichi, N.; Sakai, T. Hydrothermal preparation of LiFePO4 nanocrystals mediated by organic acid. J. Power Sources 2010, 195, 2877–2882. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
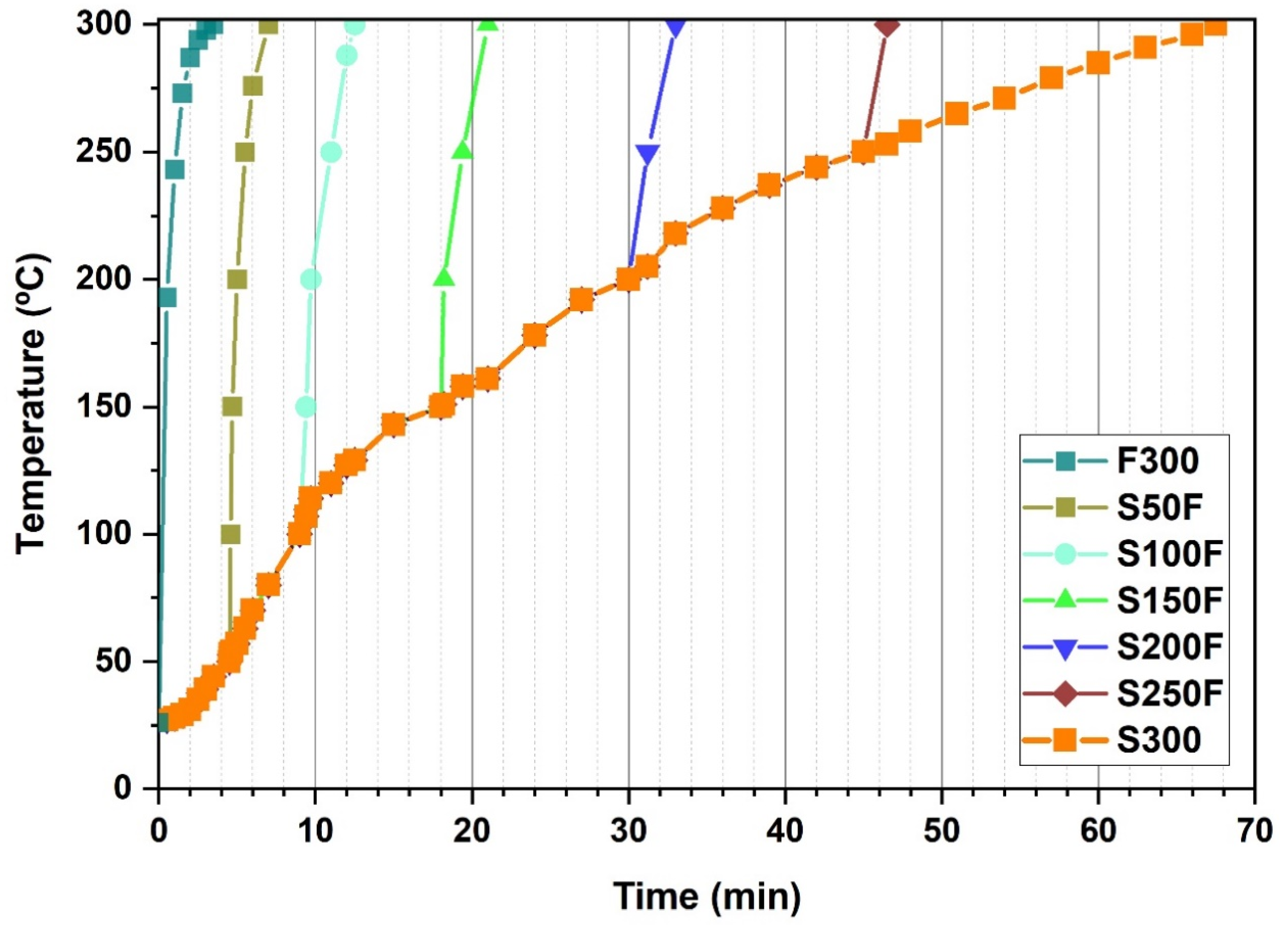
| HS Experiments | Slow Heating To (°C) | Fast Heating To (°C) | Reaction Time (min) | Total Time Length of the Synthesis Process * (min) |
|---|---|---|---|---|
| F300 | - | 300 | 5 | 0 + 3.5 + 5 = 8.5 |
| S50F | 50 | 300 | 5 | 4.5 + 3 + 5 = 12.5 |
| S100F | 100 | 300 | 5 | 9 + 3 + 5 = 17 |
| S150F | 150 | 300 | 5 | 18 + 3 + 5 = 26 |
| S200F | 200 | 300 | 5 | 30 + 2.5 + 5 = 37.5 |
| S250F | 250 | 300 | 5 | 45 + 2 + 5 = 52 |
| S300 | 300 | - | 5 | 67.5 + 0 + 5 = 72.5 |
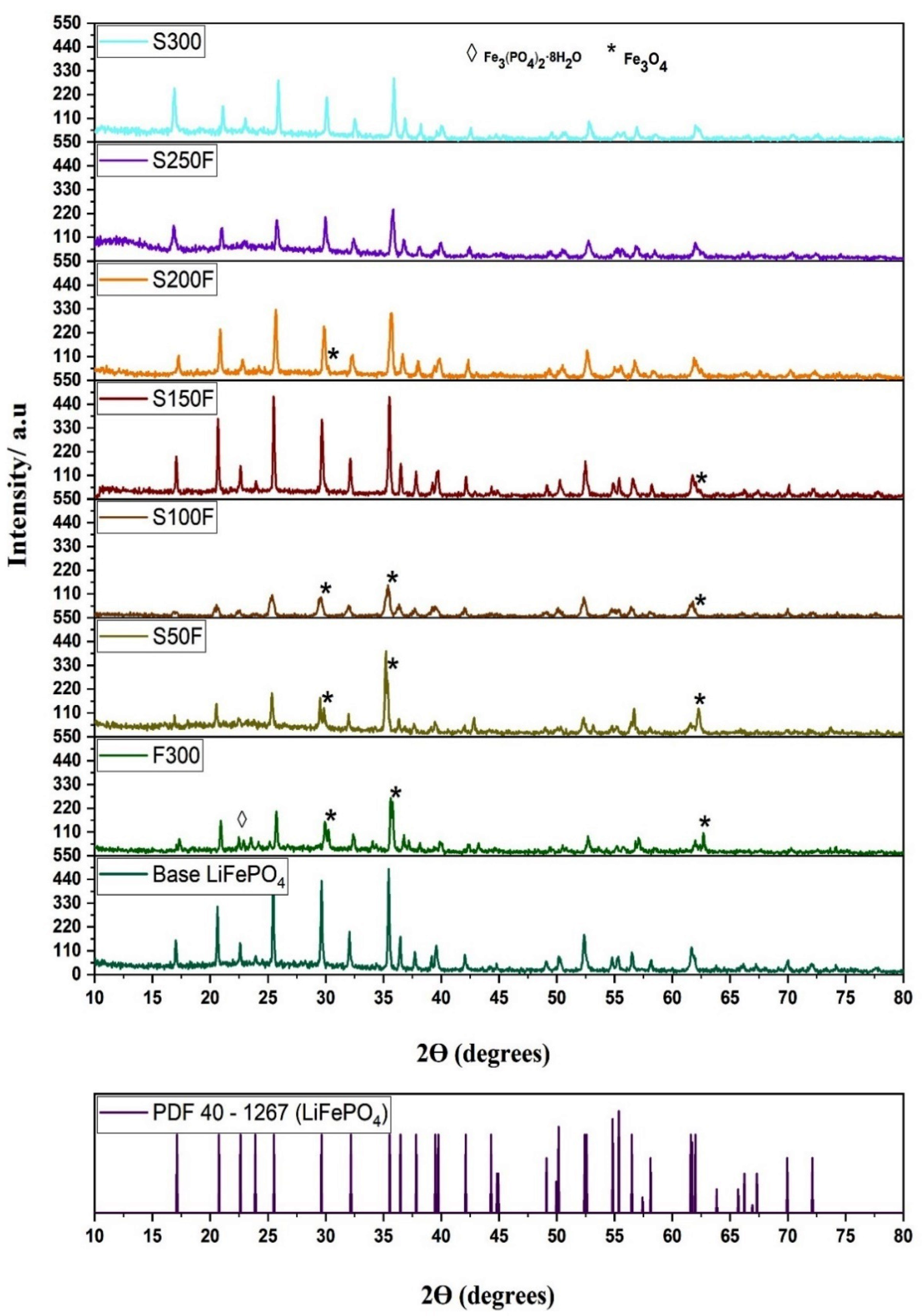
| HS Experiment | Crystalline Phases |
|---|---|
| F300 | LiFePO4 + Fe3O4 + Fe3(PO4)2·8H2O |
| S50F | LiFePO4 + Fe3O4 |
| S100F | LiFePO4 + Fe3O4 |
| S150F | LiFePO4 + Fe3O4 |
| S200F | LiFePO4 + Fe3O4 |
| S250F | LiFePO4 |
| S300 | LiFePO4 |
| HS Experiments | Ti * | Ts * | Tf * |
|---|---|---|---|
| S150F | 12.5 | 5.5 | 3 |
| S200F | 12.5 | 17.5 | 2.5 |
| S250F | 12.5 | 32.5 | 2 |
| S300 | 12.5 | 55 | - |
| Samples | Planes | Peak º (2ϴ) | FWHM | Crystallite Size * (nm) | Average Crystallite Size * (nm) | LiFePO4 (%) | Fe3O4 (%) |
|---|---|---|---|---|---|---|---|
| Base LiFePO4 | (101) | 20.643 | 0.110 | 109 | 82.5 | 99.74 | 0.26 |
| (301) | 32.506 | 0.136 | 73.5 | ||||
| (311) | 35.443 | 0.144 | 65.0 | ||||
| S100F | (101) | 20.949 | 0.195 | 45.4 | 34.26 | 82.11 | 17.89 |
| (301) | 29.715 | 0.320 | 26.5 | ||||
| (311) | 35.539 | 0.278 | 30.9 | ||||
| S150F | (101) | 20.685 | 0.120 | 88.2 | 66.63 | 96.96 | 3.04 |
| (301) | 32.133 | 0.162 | 57.5 | ||||
| (311) | 35.515 | 0.168 | 54.2 | ||||
| S200F | (101) | 20.860 | 0.173 | 52.0 | 39.23 | 97.19 | 2.81 |
| (301) | 32.292 | 0.257 | 33.2 | ||||
| (311) | 35.681 | 0.265 | 32.5 |
| Intensity Ratios | F300 | S50F | S100F | S150F | S200F | S250F | S300 |
|---|---|---|---|---|---|---|---|
| I (200)/(311) | 0.31 | 0.38 | 0.22 | 0.42 | 0.37 | 0.69 | 0.84 |
| I (101)/(311) | 0.64 | 0.59 | 0.47 | 0.79 | 0.76 | 0.64 | 0.56 |
| I (111)/(311) | 0.82 | 0.78 | 0.81 | 1.01 | 1.06 | 0.64 | 0.56 |
| I (020)/(311) | 0.63 | 0.70 | 0.71 | 0.78 | 0.77 | 0.85 | 0.70 |
| I (301)/(311) | 0.40 | 0.41 | 0.44 | 0.40 | 0.38 | 0.43 | 0.36 |
| Samples | Purity and Crystallinity | Morphology and Size | Synthesis Process Time * (min) |
|---|---|---|---|
| F300 |
| Widely variable morphology | 8.5 |
| S50F |
| Widely variable morphology | 12 |
| S100F |
| Widely variable morphology | 17.5 |
| S150F |
|
| 26 |
| S200F |
|
| 38 |
| S250F |
|
| 52 |
| S300 |
|
| 67.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Jorge, F.; Benítez, A.; García-Jarana, M.B.; Sánchez-Oneto, J.; Portela, J.R.; Martínez de la Ossa, E.J. Effect of the Heating Rate to Prevent the Generation of Iron Oxides during the Hydrothermal Synthesis of LiFePO4. Nanomaterials 2021, 11, 2412. https://doi.org/10.3390/nano11092412
Ruiz-Jorge F, Benítez A, García-Jarana MB, Sánchez-Oneto J, Portela JR, Martínez de la Ossa EJ. Effect of the Heating Rate to Prevent the Generation of Iron Oxides during the Hydrothermal Synthesis of LiFePO4. Nanomaterials. 2021; 11(9):2412. https://doi.org/10.3390/nano11092412
Chicago/Turabian StyleRuiz-Jorge, Francisco, Almudena Benítez, M. Belén García-Jarana, Jezabel Sánchez-Oneto, Juan R. Portela, and Enrique J. Martínez de la Ossa. 2021. "Effect of the Heating Rate to Prevent the Generation of Iron Oxides during the Hydrothermal Synthesis of LiFePO4" Nanomaterials 11, no. 9: 2412. https://doi.org/10.3390/nano11092412
APA StyleRuiz-Jorge, F., Benítez, A., García-Jarana, M. B., Sánchez-Oneto, J., Portela, J. R., & Martínez de la Ossa, E. J. (2021). Effect of the Heating Rate to Prevent the Generation of Iron Oxides during the Hydrothermal Synthesis of LiFePO4. Nanomaterials, 11(9), 2412. https://doi.org/10.3390/nano11092412