
Probing into the In-Situ Exsolution Mechanism of Metal Nanoparticles from Doped Ceria Host




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Computational Details
2. Results and Discussion
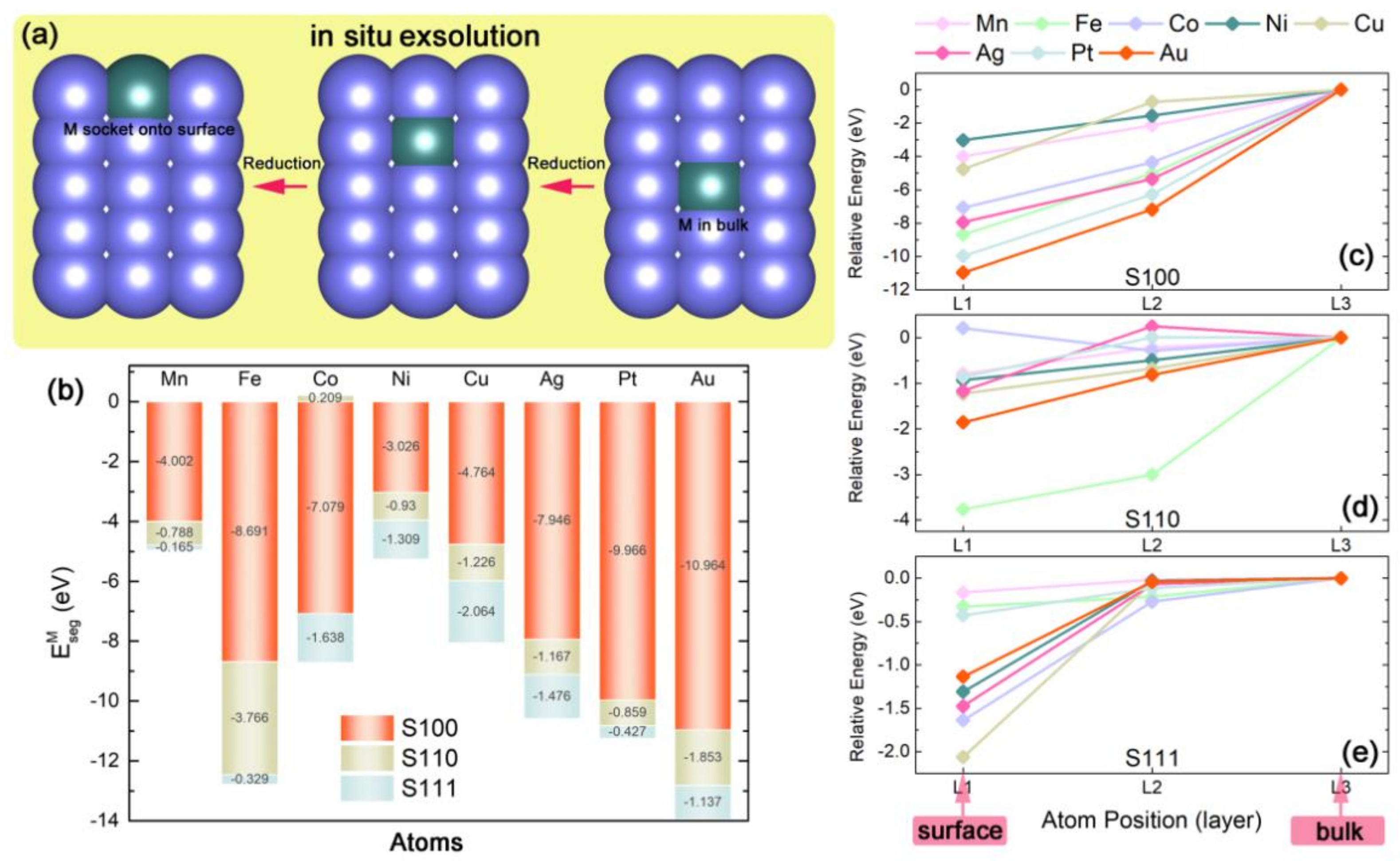
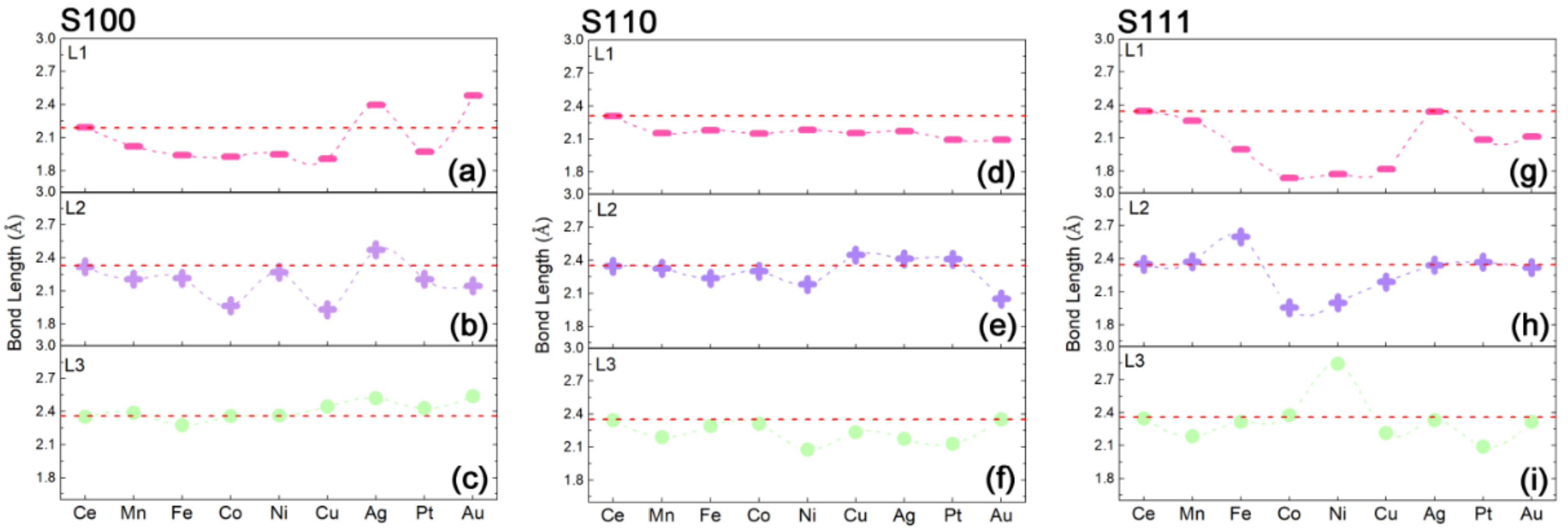
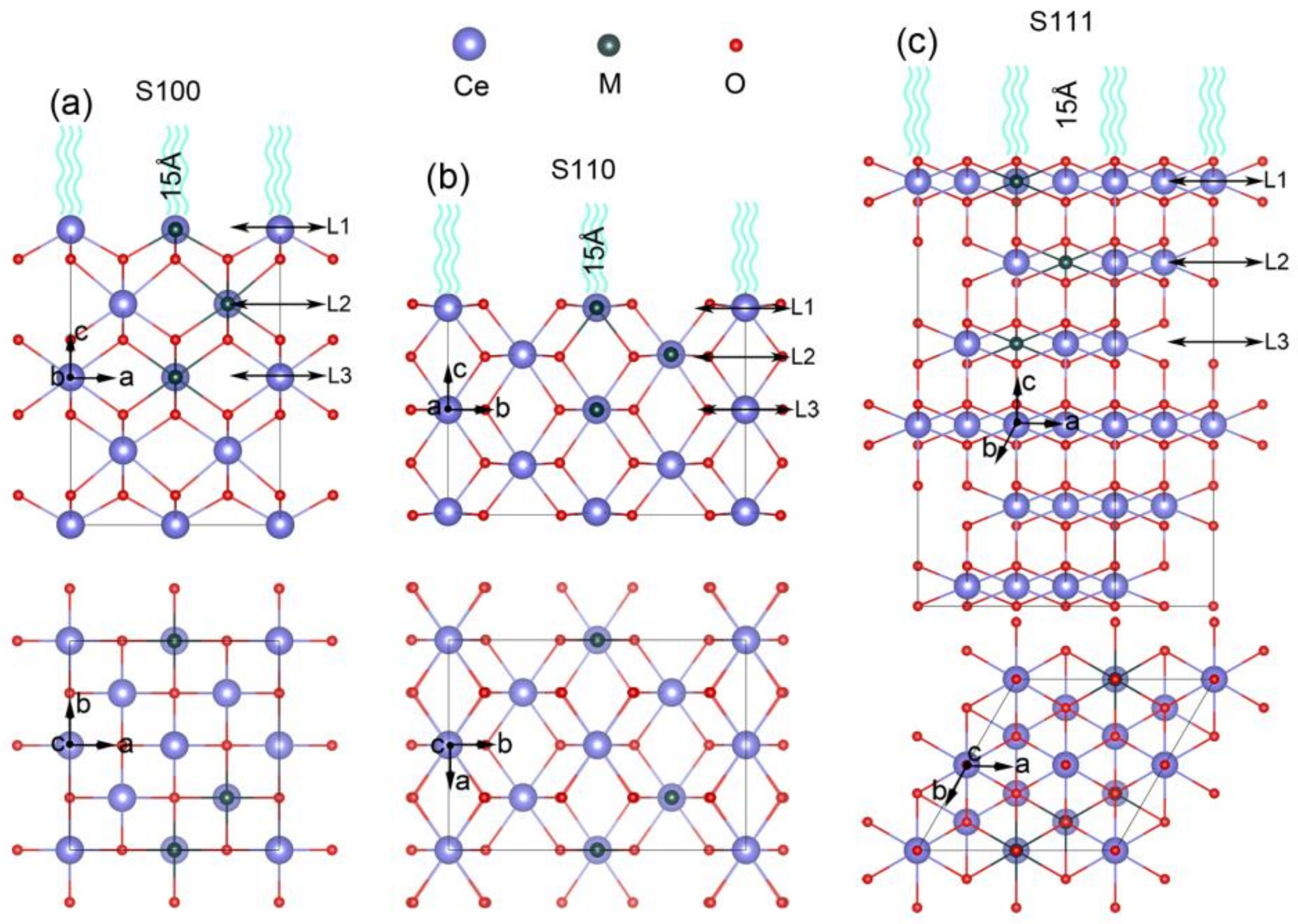
2.1. Construction of Metal Exsolved Surface Structures MxCe1−xO2
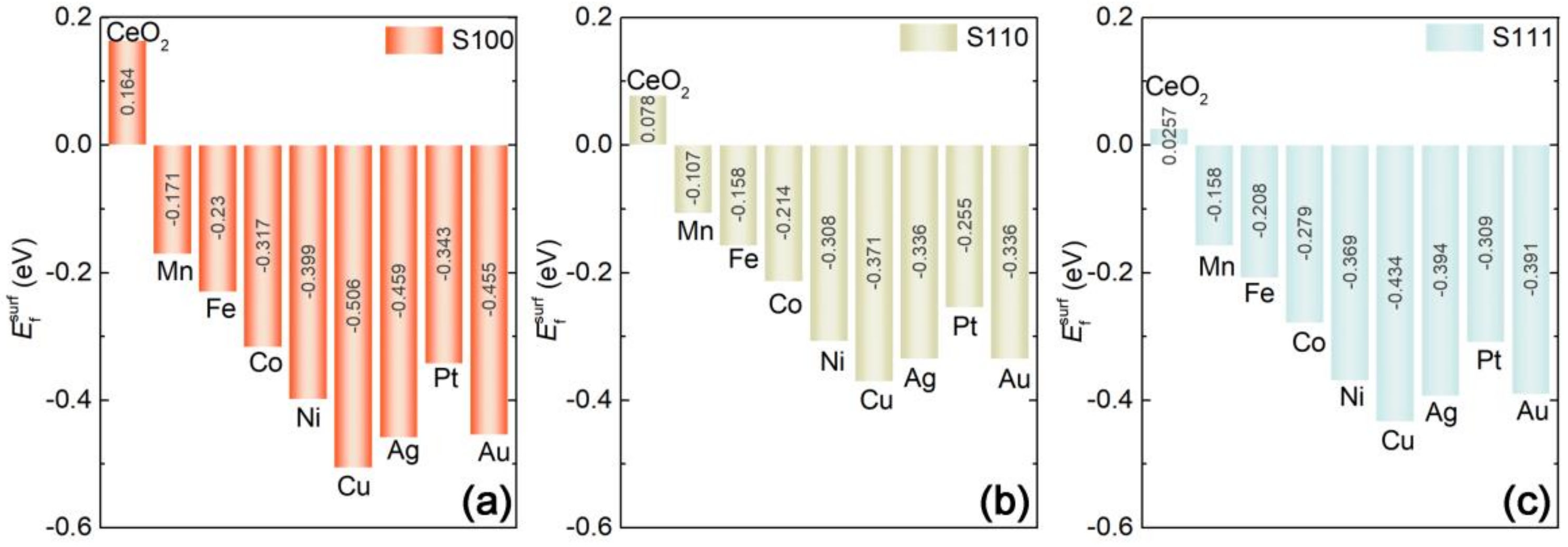
2.2. Capabilities of In-Situ Exsolution in MxCe1−xO2 Systems
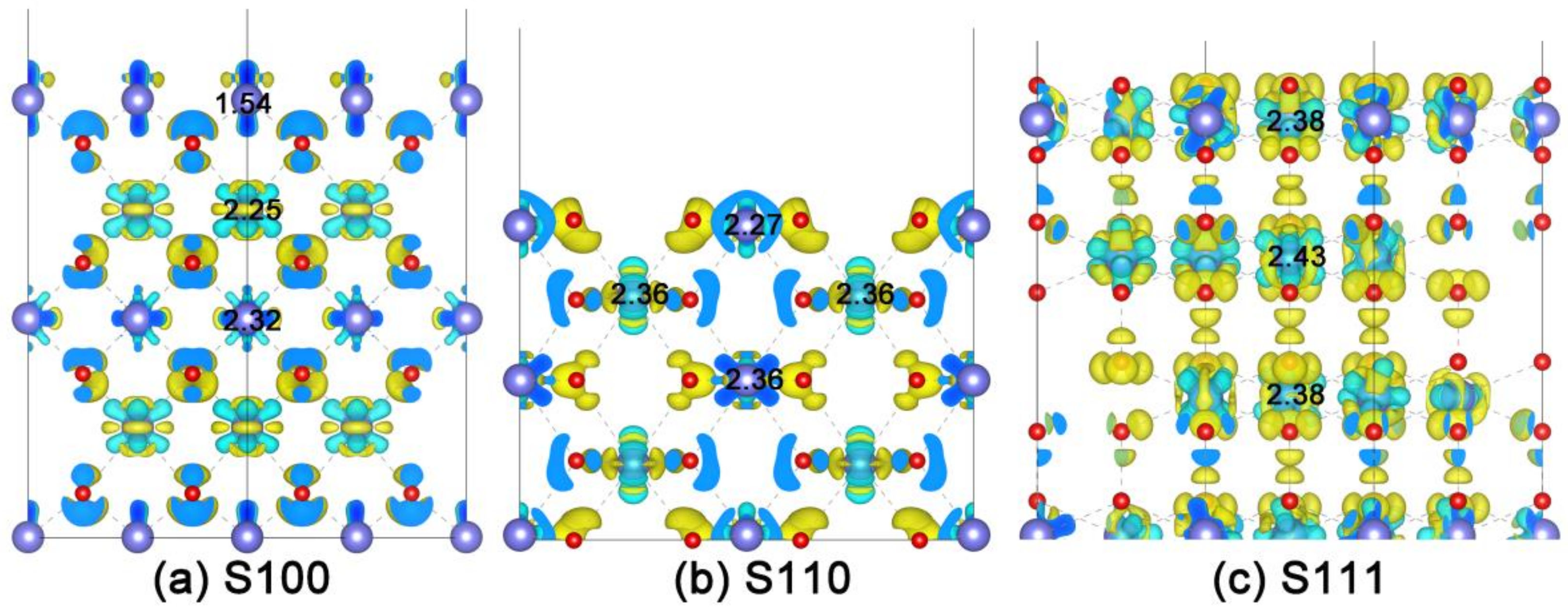
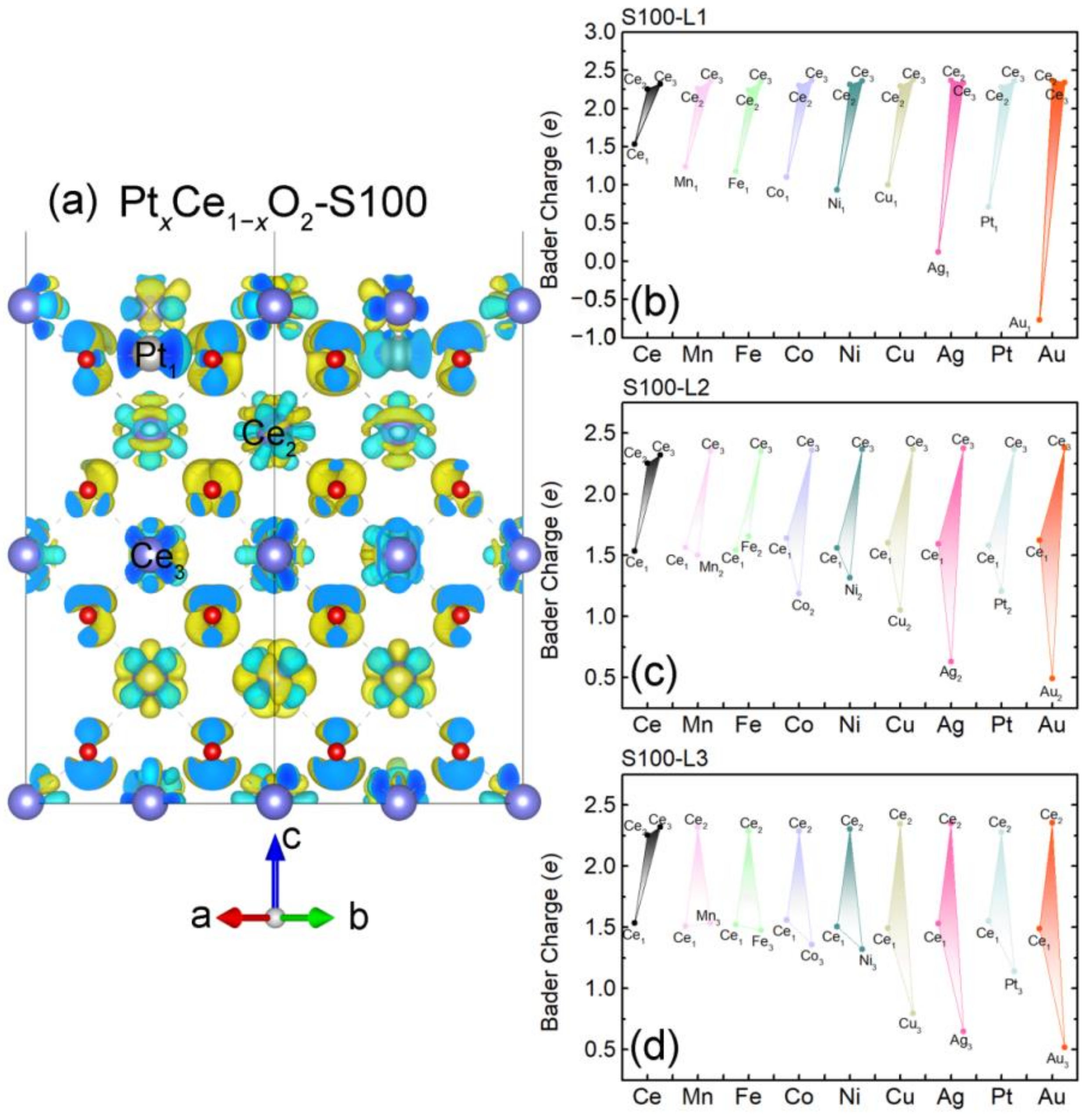
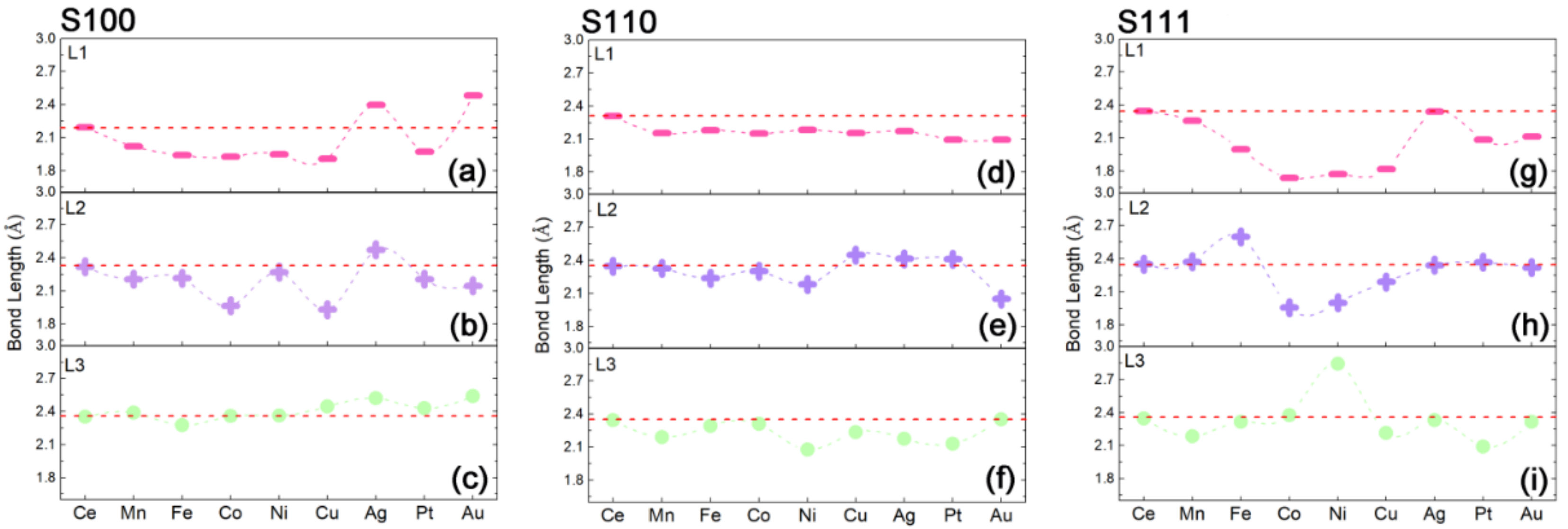
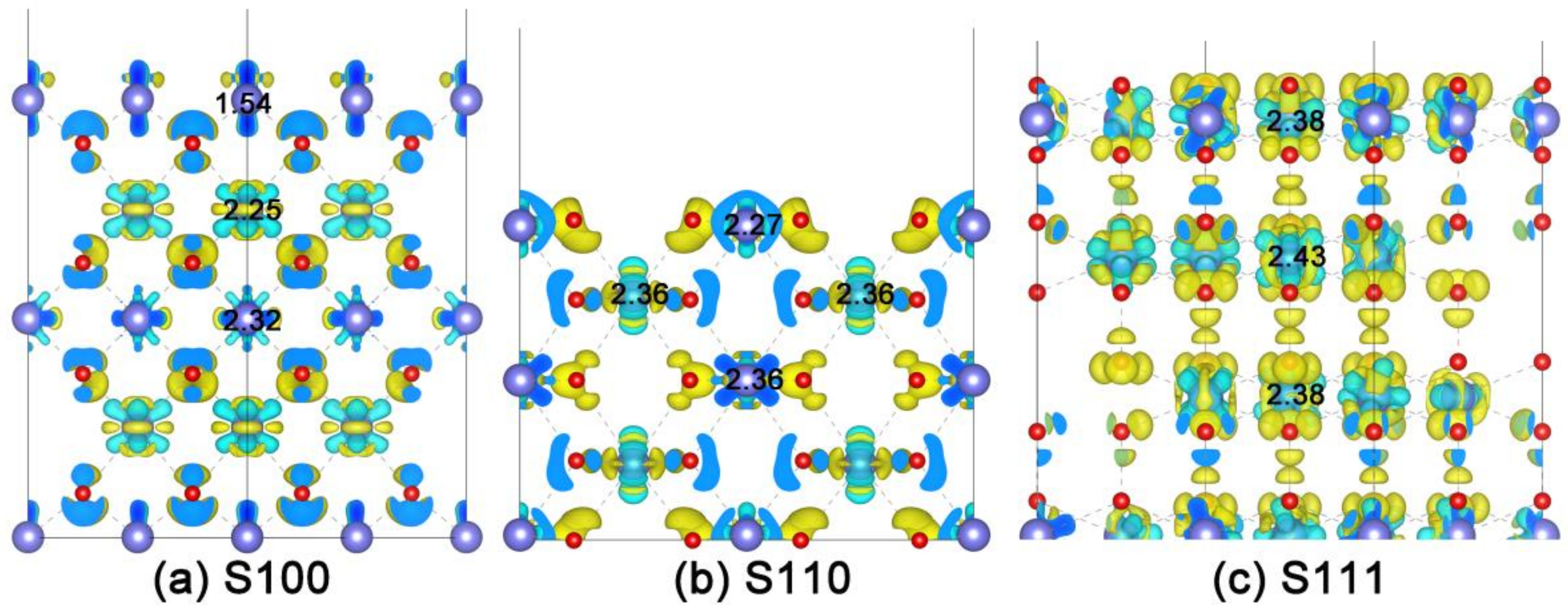
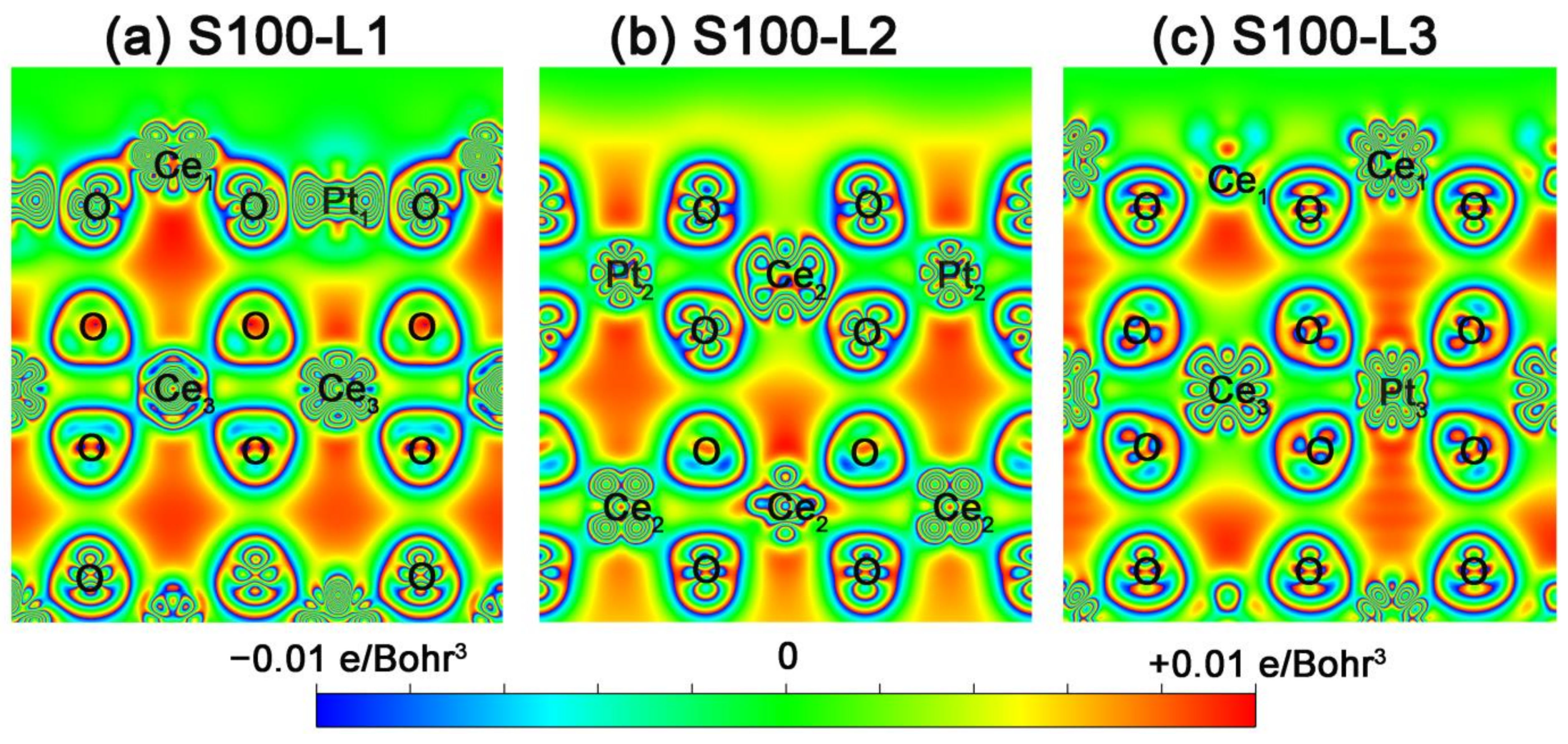
2.3. Intrinsic Mechanism of In-Situ Exsolution: Stair-Stepping Charge Difference
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishihata, Y.; Mizuki, J.I.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-Regeneration of a Pd-Perovskite Catalyst for Automotive Emissions Control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef]
- Arico, A.S.; Bruce, P.; Scrosati, B.; Tarascon, J.M.; Van Schalkwijk, W. Nanostructured Materials for Advanced Energy Con-version and Storage Devices. Nat. Mater. 2005, 4, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, M.; Ji, H.; Shen, X.; Yan, C.; Qian, T. Altering the Rate-Determining Step over Cobalt Single Clusters Leading to Highly Efficient Ammonia Synthesis. Natl. Sci. Rev. 2020, 8. [Google Scholar] [CrossRef]
- Ding, W.-L.; Cao, Y.-H.; Liu, H.; Wang, A.-X.; Zhang, C.-J.; Zheng, X.-R. In Situ Growth of NiSe@Co0.85Se Heterointerface Structure with Electronic Modulation on Nickel Foam for Overall Water Splitting. Rare Met. 2020, 40, 1373–1382. [Google Scholar] [CrossRef]
- Xia, Z.; Liu, G.; Wen, J.; Mei, Z.; Balasubramanian, M.; Molokeev, M.S.; Peng, L.; Gu, L.; Miller, D.J.; Liu, Q.; et al. Tuning of Photoluminescence by Cation Nanosegregation in the (CaMg)x(NaSc)(1−x)Si2O6 Solid Solution. J. Am. Chem. Soc. 2016, 138, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Liu, J.; Qian, T.; Shen, X.; Zhou, J.; Yan, C. Single Lithiumion Channel Polymer Binder for Stabilizing Sulfur Cathodes. Natl. Sci. Rev. 2020, 7, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Lu, X.; Chu, G.; He, C.; Zhan, W.; Zhou, G. Surface Tuning of LaCoO3 Perovskite by Acid Etching to Enhance its Cat-alytic Performance. Rere Met. 2011, 40, 555–562. [Google Scholar] [CrossRef]
- Kim, K.J.; Han, H.; Defferriere, T.; Yoon, D.; Na, S.; Kim, S.J.; Dayaghi, A.M.; Son, J.; Oh, T.S.; Jang, H.M.; et al. Facet-Dependent in Situ Growth of Nanoparticles in Epitaxial Thin Films: The Role of Interfacial Energy. J. Am. Chem. Soc. 2019, 141, 7509–7517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, W.; Ran, R.; Chen, Y.; Shao, Z.; Liu, M. Promotion of Oxygen Reduction by Exsolved Silver Nanoparticles on a Perovskite Scaffold for Low-Temperature Solid Oxide Fuel Cells. Nano Lett. 2015, 16, 512–518. [Google Scholar] [CrossRef]
- Tsekouras, G.; Neagu, D.; Irvine, J.T.S. Step-Change in High Temperature Steam Electrolysis Performance of Perovskite Oxide Cathodes with Exsolution of B-Site Dopants. Energy Environ. Sci. 2013, 6, 256–266. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.; Zhang, L.; Meng, J.; Meng, F.; Liu, X.; Meng, J. Enhanced Electrochemical Property of La0.6Sr0.4Co0.8Fe0.2O3 as Cathode for Solid Oxide Fuel Cell by Efficient In Situ Polarization-Exsolution Treatment. Electrochim. Acta 2017, 258, 1096–1105. [Google Scholar] [CrossRef]
- Zhou, G.-F.; Ma, J.; Bai, S.; Wang, L.; Guo, Y. CO Catalytic Oxidation over Pd/CeO2 with Different Chemical States of Pd. Rare Met. 2019, 39, 800–805. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Y.; Chen, J.; Li, J.-H.; Zhu, Y.-T.; Zeng, Y.-M.; Amirkhiz, B.S.; Hua, B.; Luo, J.-L. New Opportunity for in Situ Exsolution of Metallic Nanoparticles on Perovskite Parent. Nano Lett. 2016, 16, 5303–5309. [Google Scholar] [CrossRef]
- Namba, K.; Ogura, S.; Ohno, S.; Di, W.; Kato, K.; Wilde, M.; Pletikosić, I.; Pervan, P.; Milun, M.; Fukutani, K. Acceleration of Hydrogen Absorption by Palladium through Surface Alloying with Gold. Proc. Natl. Acad. Sci. USA 2018, 115, 7896–7900. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tao, H.-B.; Liu, J.; Sun, Y.; Chen, J.; Hua, B.; Thundat, T.; Luo, J.-L. A Rational Design for Enhanced Oxygen Reduction: Strongly Coupled Silver Nanoparticles and Engineered Perovskite Nanofibers. Nano Energy 2017, 38, 392–400. [Google Scholar] [CrossRef]
- Xu, S.; Dong, D.; Wang, Y.; Doherty, W.; Xie, K.; Wu, Y. Perovskite Chromates Cathode with Resolved and Anchored Nickel Nano-Particles for Direct High-Temperature Steam Electrolysis. J. Power Sour. 2014, 246, 346–355. [Google Scholar] [CrossRef]
- Kwon, O.; Sengodan, S.; Kim, K.; Kim, G.; Jeong, H.Y.; Shin, J.; Ju, Y.-W.; Han, J.W.; Kim, G. Exsolution Trends and Co-Segregation Aspects of Self-Grown Catalyst Nanoparticles in Perovskites. Nat. Commun. 2017, 8, 15967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.P. Nanoscale and Nano-Structured Electrodes of Solid Oxide Fuel Cells by Infiltration: Advances and Challenges. Int. J. Hydrog. Energ. 2012, 37, 449–470. [Google Scholar] [CrossRef]
- Burnat, D.; Kontic, R.; Holzer, L.; Steiger, P.; Ferri, D.; Heel, A. Smart Material Concept: Reversible Microstructural Self-Regeneration for Catalytic Applications. J. Mater. Chem. A 2016, 4, 11939–11948. [Google Scholar] [CrossRef]
- Tanaka, H.; Uenishi, M.; Taniguchi, M.; Tan, I.; Narita, K.; Kimura, M.; Kaneko, K.; Nishihata, Y.; Mizuki, J. Intelligent Catalyst having the Self-Regenerative Function of Pd, Rh and Pt for Automotive Emissions Control. Catal. Today 2006, 117, 321–328. [Google Scholar] [CrossRef]
- Uenishi, M.; Tanaka, H.; Taniguchi, M.; Tan, I.; Nishihata, Y.; Mizuki, J.; Kobayashi, T. Time Evolution of Palladium Structure change with Redox Fluctuations in a LaFePdO3 Perovskite Automotive Catalyst by High-Speed Analysis with In Situ DXAFS. Catal. Commun. 2008, 9, 311–314. [Google Scholar] [CrossRef]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.; Suntivich, J.; Shao-Horn, Y. Toward the Rational Design of Non-Precious Transition Metal Oxides for Oxygen Electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef] [Green Version]
- Götsch, T.; Schlicker, L.; Bekheet, M.F.; Doran, A.; Grünbacher, M.; Praty, C.; Tada, M.; Matsui, H.; Ishiguro, N.; Gurlo, A.; et al. Structural Investigations of La0.6Sr0.4FeO3−δ under Reducing Conditions: Kinetic and Thermodynamic Limitations for Phase Transformations and Iron Exsolution Phenomena. RSC Adv. 2018, 8, 3120–3131. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; You, W.; Shen, Y.; Gu, X.; Ge, M.; Ahmadi, S.; Ahmad, S.; Kraatz, H.B. Facile Synthesis of Silver-rich Au/Ag Bimetallic Nanoparticles with Highly Active SERS Properties. New J. Chem. 2019, 43, 14772–14780. [Google Scholar] [CrossRef]
- Neagu, D.; Papaioannou, E.I.; Ramli, W.K.W.; Miller, D.N.; Murdoch, B.J.; Menard, H.; Umar, A.; Barlow, A.J.; Cumpson, P.J.; Irvine, J.T.S.; et al. Demonstration of Chemistry at a Point through Restructuring and Catalytic Activation at an-Chored Nanoparticles. Nat. Commun. 2017, 8, 1855. [Google Scholar] [CrossRef] [Green Version]
- Sengodan, S.; Yeo, H.J.; Shin, J.Y.; Kim, G. Assessment of Perovskite-Type La0.8Sr0.2ScxMn1−xO3-Delta Oxides as Anodes for in-Termediate-Temperature Solid Oxide Fuel Cells Using Hydrocarbon Fuels. J. Power Sour. 2011, 196, 3083–3088. [Google Scholar] [CrossRef]
- Hamada, I.; Uozumi, A.; Morikawa, Y.; Yanase, A.; Katayama-Yoshida, H. A Density Functional Theory Study of Self-Regenerating Catalysts LaFe(1−x)M(x)O(3−y) (M = Pd, Rh, Pt). J. Am. Chem. Soc. 2011, 133, 18506–18509. [Google Scholar] [CrossRef]
- Li, B.; Katz, M.B.; Zhang, Q.; Chen, L.; Graham, G.W.; Pan, X. Surface-Termination-Dependent Pd Bonding and Aggregation of Nanoparticles on LaFeO3 (001). J. Chem. Phys. 2013, 138, 144705. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.-S.; Miller, D.N.; Ménard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T.S. Nano-Socketed Nickel Particles with Enhanced Coking Resistance Grown In Situ by Redox Exsolution. Nat. Commun. 2015, 6, 8120. [Google Scholar] [CrossRef] [Green Version]
- Neagu, D.; Tsekouras, G.; Miller, D.N.; Ménard, H.; Irvine, J. In Situ Growth of Nanoparticles through Control of Non-Stoichiometry. Nat. Chem. 2013, 5, 916–923. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Lu, Z.; You, T.L.; Wang, J.; Xie, L.; He, J.; Ciucci, F. Energetics of Nanoparticle Exsolution from Perovskite Oxides. J. Phys. Chem. Lett. 2018, 9, 3772–3778. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.J. Materials for Solid Oxide Fuel Cells. Chem. Mater. 2009, 22, 660–674. [Google Scholar] [CrossRef]
- Liu, L.; Liu, R.; Xu, T.; Zhang, Q.; Tan, Y.; Zhang, Q.; Ding, J.; Tang, Y. Enhanced Catalytic Oxidation of Chlorobenzene over MnO2 Grafted In Situ by Rare Earth Oxide: Surface Doping Induces Lattice Oxygen Activation. Inorg. Chem. 2020, 59, 14407–14414. [Google Scholar] [CrossRef] [PubMed]
- Bera, P.; Patil, K.; Jayaram, V.; Subbanna, G.; Hegde, M. Ionic Dispersion of Pt and Pd on CeO2 by Combustion Method: Effect of Metal–Ceria Interaction on Catalytic Activities for NO Reduction and CO and Hydrocarbon Oxidation. J. Catal. 2000, 196, 293–301. [Google Scholar] [CrossRef]
- Gui, L.; Pan, G.; Ma, X.; You, M.; He, B.; Yang, Z.; Sun, J.; Zhou, W.; Xu, J.; Zhao, L. In-Situ Exsolution of CoNi Alloy Nanoparticles on LiFe0.8Co0.1Ni0.1O2 Parent: New opportunity for Boosting Oxygen Evolution and Reduction Reaction. Appl. Surf. Sci. 2020, 543, 148817. [Google Scholar] [CrossRef]
- Boulfrad, S.; Cassidy, M.; Irvine, J.T. NbTi0.5Ni0.5O4 as Anode Compound Material for SOFCs. Solid State Ionics 2011, 197, 37–41. [Google Scholar] [CrossRef]
- Sengodan, S.; Ju, Y.W.; Kwon, O.; Jun, A.; Jeong, H.Y.; Ishihara, T.; Shin, J.; Kim, G. Self-Decorated MnO Nanoparticles on Double Perovskite Solid Oxide Fuel Cell Anode by in Situ Exsolution. ACS Sustain. Chem. Eng. 2017, 5, 9207–9213. [Google Scholar] [CrossRef]
- Pilger, F.; Testino, A.; Carino, A.; Proff, C.; Kampolis, A.; Cervellino, A.; Ludwig, C. Size Control of Pt Clusters on CeO2 Nanoparticles via an Incorporation–Segregation Mechanism and Study of Segregation Kinetics. ACS Catal. 2016, 6, 3688–3699. [Google Scholar] [CrossRef]
- Wachsman, E.D.; Lee, K.T. Lowering the Temperature of Solid Oxide Fuel Cells. Science 2011, 334, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, J.; Yao, F.; Zhang, W.; Liu, X.; Meng, J.; Zhang, H. Insight into the Mechanism of the Ionic Conductivity for Ln-Doped Ceria (Ln = La, Pr, Nd, Pm, Sm, Gd, Tb, Dy, Ho, Er, and Tm) through First-Principles Calculation. Inorg. Chem. 2018, 57, 12690–12696. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cao, F.-X.; Gao, W.; Dong, Q.-C.; Qu, Y.-Q. Uniform Small Metal Nanoparticles Anchored on CeO2 Nanorods Driven by Electroless Chemical Deposition. Rare Met. 2019, 39, 806–814. [Google Scholar] [CrossRef]
- Tan, J.; Lee, D.; Ahn, J.; Kim, B.; Kim, J.; Moon, J. Thermally Driven In-Situ Exsolution of Ni Nanoparticles from (Ni, Gd)CeO2 for High-Performance Solid Oxide Fuel Cells. J. Mater. Chem. A. 2018, 6, 18133–18142. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [Green Version]
- Petit, L.; Svane, A.; Szotek, Z.; Temmerman, W.M. First-Principles Study of Rare-Earth Oxides. Phys. Rev. B 2005, 72, 205118. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Ponits for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pourovskii, L.; Amadon, B.; Biermann, S.; Georges, A. Self-Consistency over the Charge Density in Dynamical Mean-Field Theory: A Linear Muffintin Implementation and Some Physical Implications. Phys. Rev. B 2007, 76. [Google Scholar] [CrossRef] [Green Version]
- Sanville, E.; Kenny, S.; Smith, R.; Henkelman, G. Improved Grid-Based Algorithm for Bader Charge Allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Gerward, L.; Olsen, J.S.; Petit, L.; Vaitheeswaran, G.; Kanchana, V.; Svane, A. Bulk Modulus of CeO2 and PrO2-An Experimental and Theoretical Study. J. Alloys Compd. 2005, 400, 56–61. [Google Scholar] [CrossRef]
- Anglès, X.A.; Roldan, A.; De Leeuw, N.H. Gadolinium-Vacancy Clusters in the (111) Surface of Gadolinium-Doped Ceria: A Density Functional Theory Study. Chem. Mater. 2015, 27, 7910–7917. [Google Scholar] [CrossRef]
- Ma, H.; Chen, X.-Q.; Li, R.; Wang, S.; Dong, J.; Ke, W. First-Principles Modeling of Anisotropic Anodic Dissolution of Metals and Alloys in Corrosive Environments. Acta Mater. 2017, 130, 137–146. [Google Scholar] [CrossRef]
- Andersson, D.A.; Simak, S.I.; Skorodumova, N.V.; Abrikosov, I.A.; Johansson, B. Optimization of Ionic Conductivity in Doped Ceria. Proc. Natl. Acad. Sci. USA 2006, 103, 3518–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padi, S.P.; Shelly, L.; Komarala, E.P.; Schweke, D.; Hayun, S.; Rosen, B.A. Coke-Free Methane Dry Reforming over Nano-Sized NiO-CeO2 Solid Solution after Exsolution. Catal. Commun. 2020, 138, 105951. [Google Scholar] [CrossRef]
- Akbay, T.; Staykov, A.; Druce, J.; Tellez, H.; Ishihara, T.; Kilner, J.A. The Interaction of Molecular Oxygen on LaO Terminated Surfaces of La2NiO4. J. Mater. Chem. A 2016, 4, 13113–13124. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, W.; Zhu, Z.; Peng, R.; Wu, X.; Xia, C.; Lu, Y. First-Principles Study of O2 Reduction on BaZr1−xCoxO3 Cathodes in Protonic-Solid Oxide Fuel Cells. J. Mater. Chem. A 2014, 2, 16707–16714. [Google Scholar] [CrossRef]
- Li, X.; Benedek, N.A. Enhancement of Ionic Transport in Complex Oxides through Soft Lattice Modes and Epitaxial Strain. Chem. Mater. 2015, 27, 2647–2652. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Analysis Algorithm without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Ji, W.; Guo, Q.; Cheng, Y.; Liu, X.; Lu, H.; Dai, H. Probing into the In-Situ Exsolution Mechanism of Metal Nanoparticles from Doped Ceria Host. Nanomaterials 2021, 11, 2114. https://doi.org/10.3390/nano11082114
Zhang L, Ji W, Guo Q, Cheng Y, Liu X, Lu H, Dai H. Probing into the In-Situ Exsolution Mechanism of Metal Nanoparticles from Doped Ceria Host. Nanomaterials. 2021; 11(8):2114. https://doi.org/10.3390/nano11082114
Chicago/Turabian StyleZhang, Lifang, Weiwei Ji, Qiyang Guo, Yu Cheng, Xiaojuan Liu, Hongbin Lu, and Hong Dai. 2021. "Probing into the In-Situ Exsolution Mechanism of Metal Nanoparticles from Doped Ceria Host" Nanomaterials 11, no. 8: 2114. https://doi.org/10.3390/nano11082114
APA StyleZhang, L., Ji, W., Guo, Q., Cheng, Y., Liu, X., Lu, H., & Dai, H. (2021). Probing into the In-Situ Exsolution Mechanism of Metal Nanoparticles from Doped Ceria Host. Nanomaterials, 11(8), 2114. https://doi.org/10.3390/nano11082114