
Factors Affecting Hydrogen Adsorption in Metal–Organic Frameworks: A Short Review



Abstract
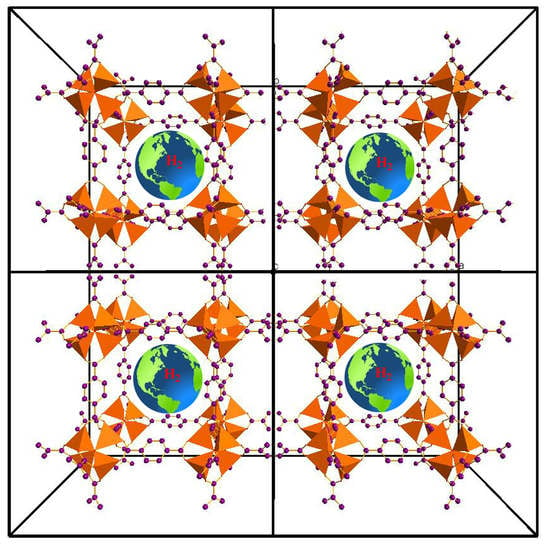
:
1. Introduction
2. Basic Knowledge of Interaction between Solid Porous MOFs and Gaseous Hydrogen
2.1. Hydrogen Sorption
2.2. Surface Area
2.3. Surface Excess and the Total Adsorbed Amount of Hydrogen
2.4. Isosteric Enthalpy of Hydrogen Adsorption
3. Structure and Topology of MOFs Studied for Hydrogen Adsorption
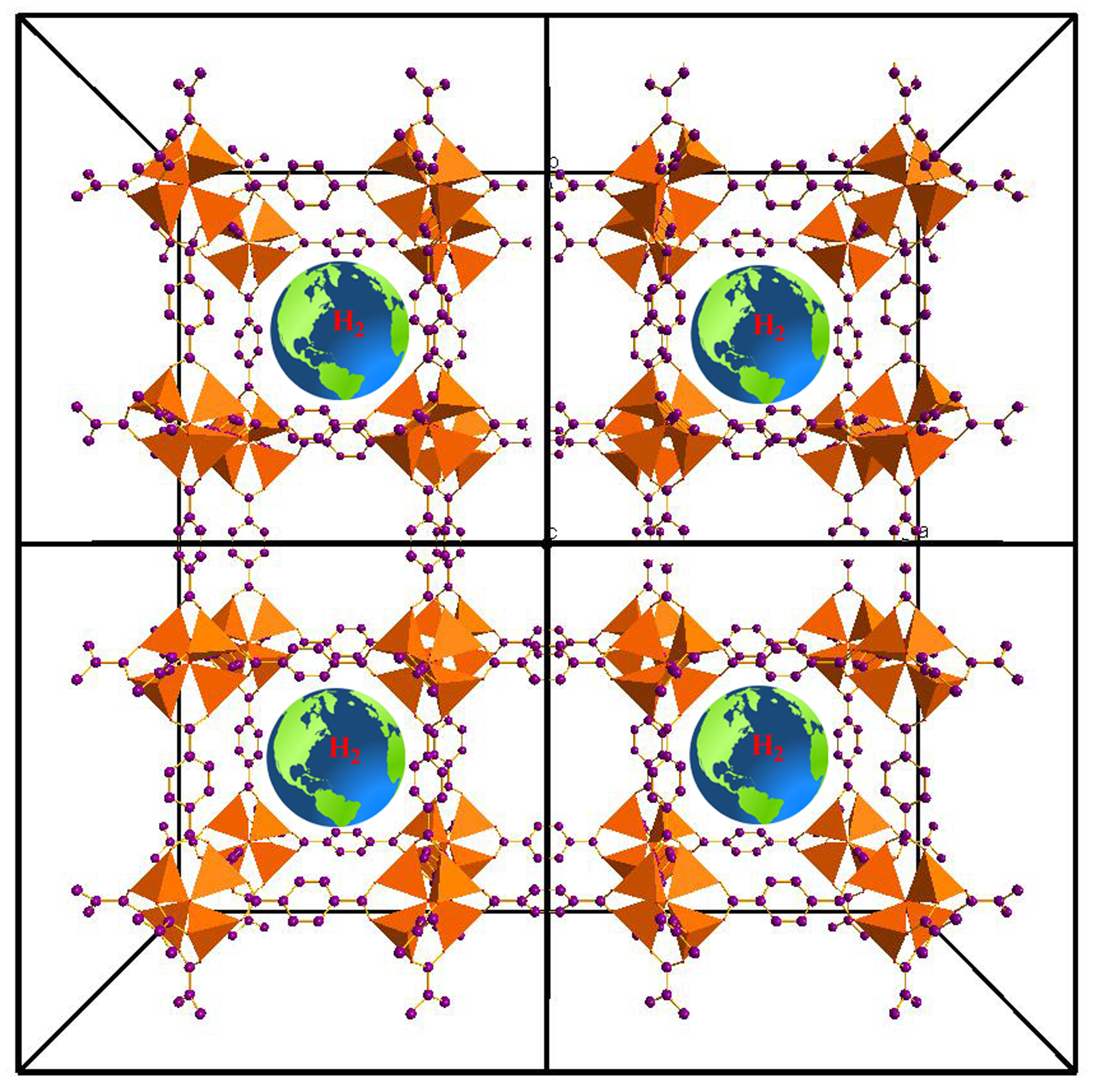
3.1. MOF-5 and Isorecticular Compounds
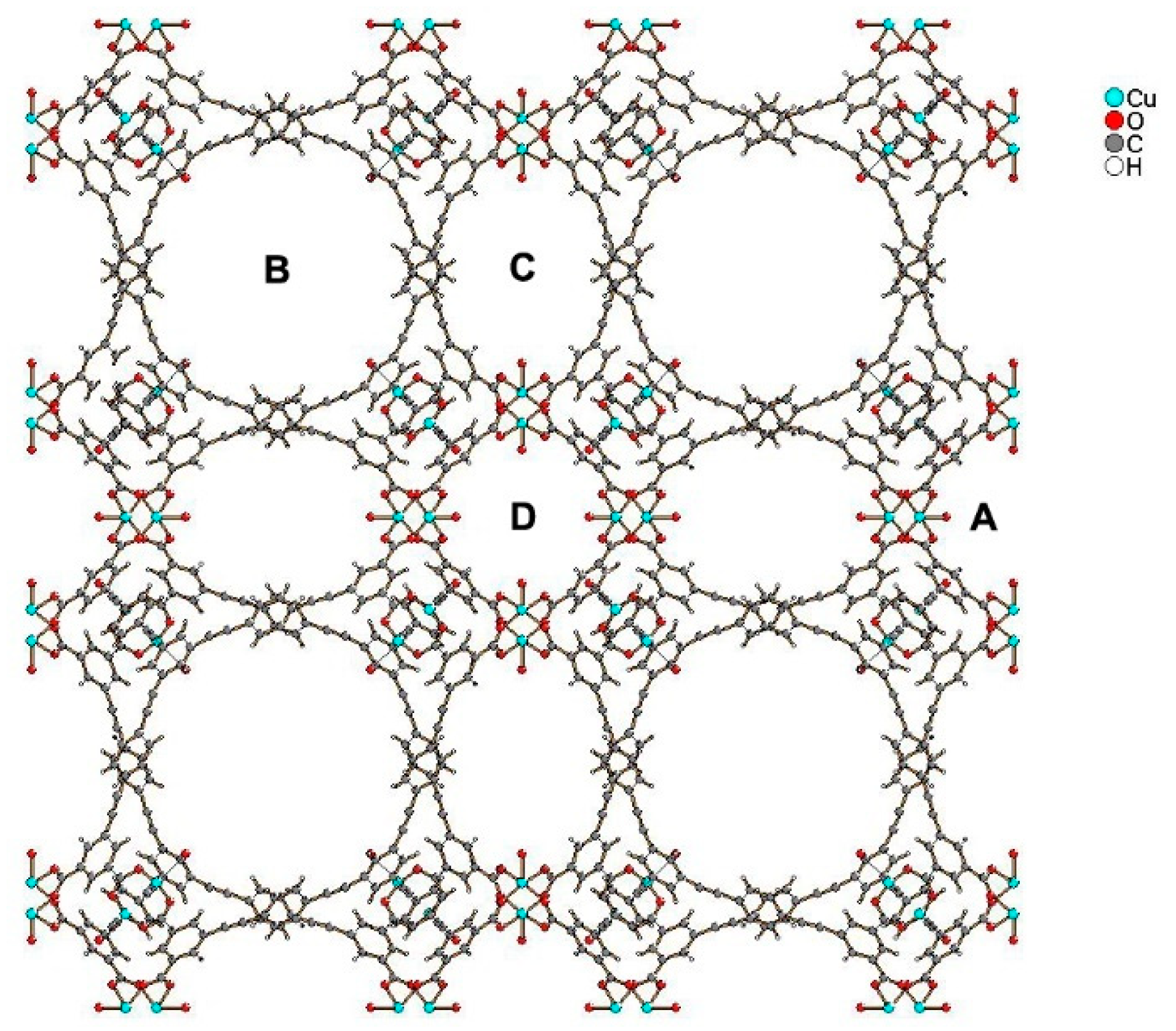
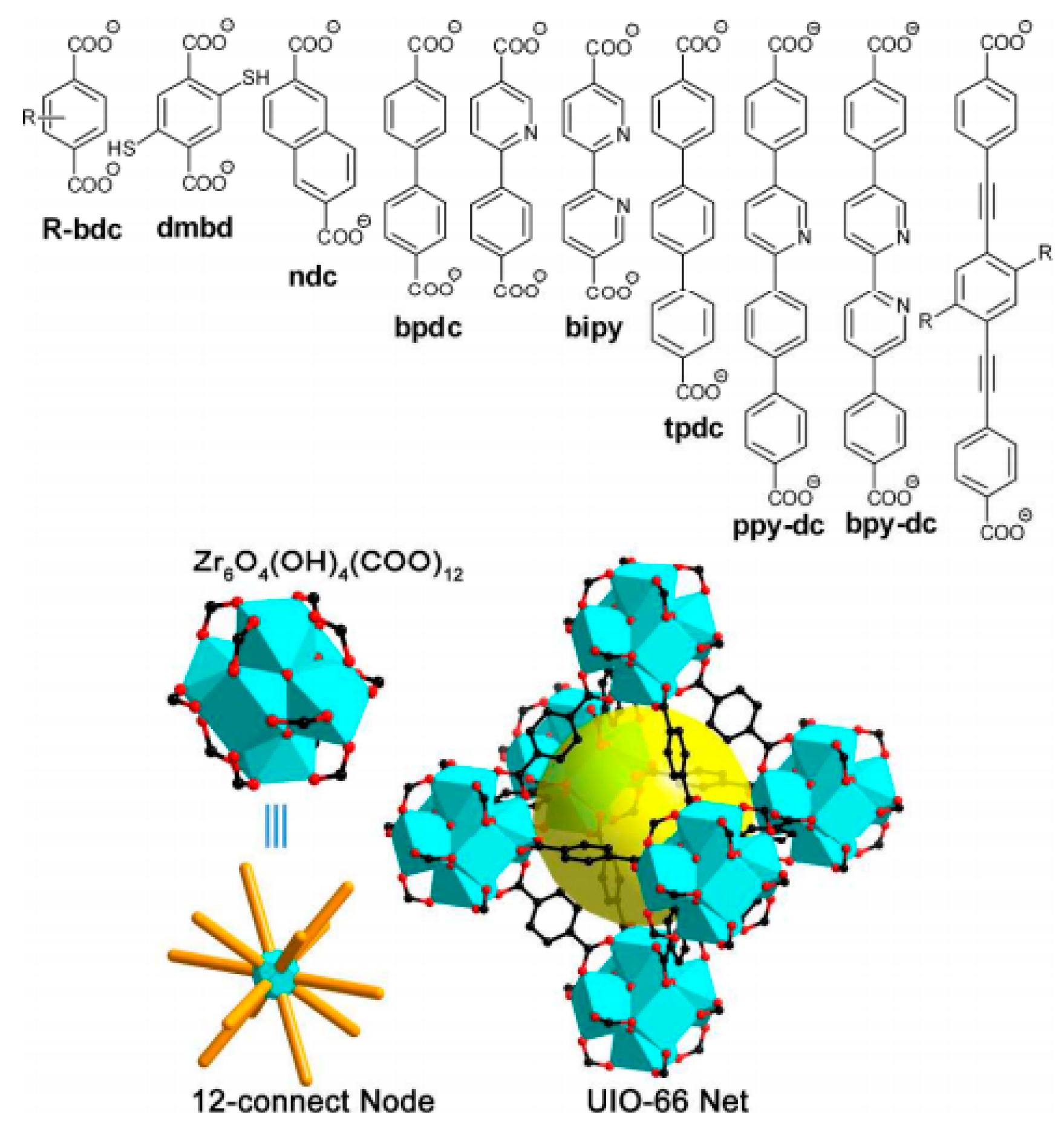
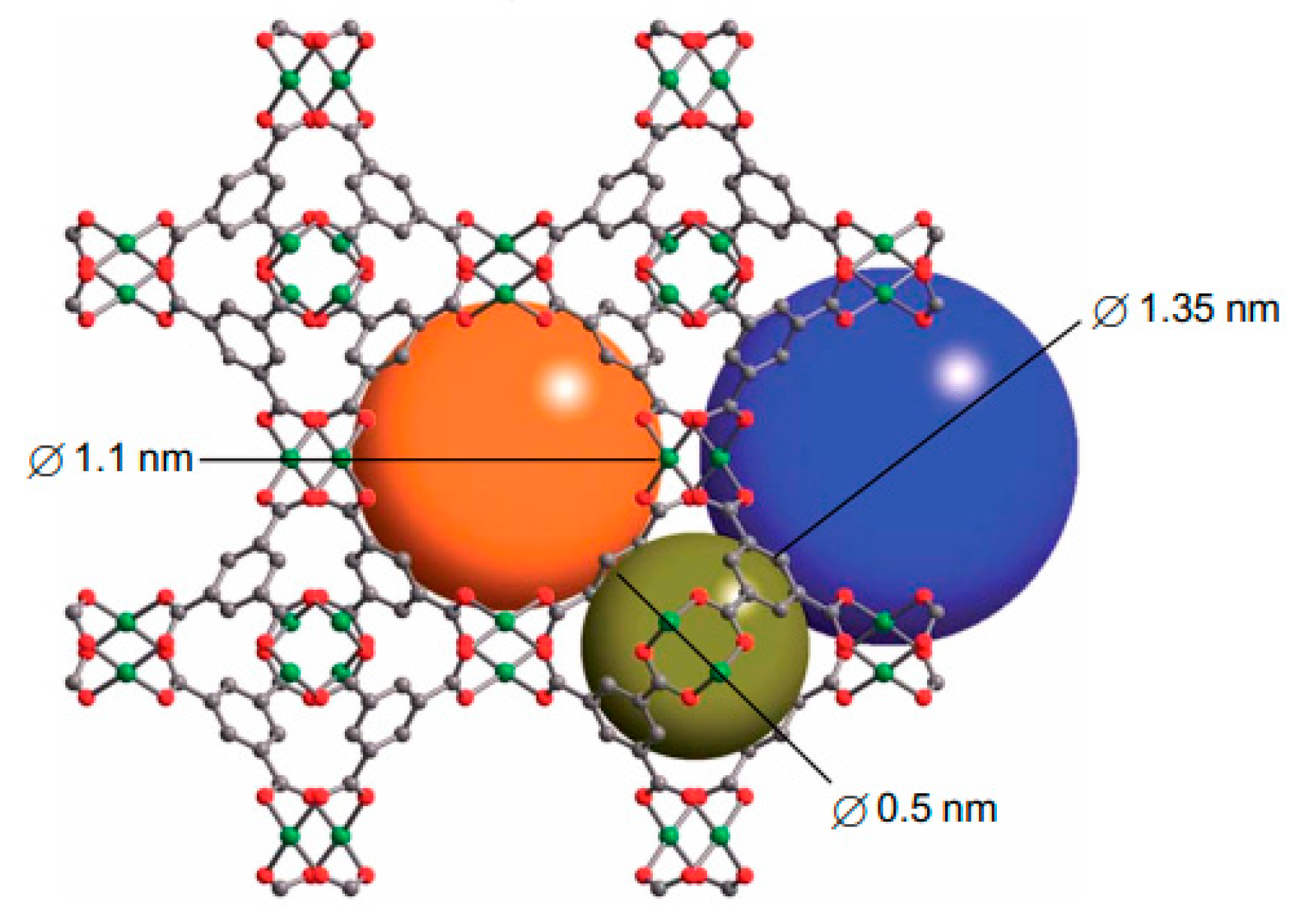
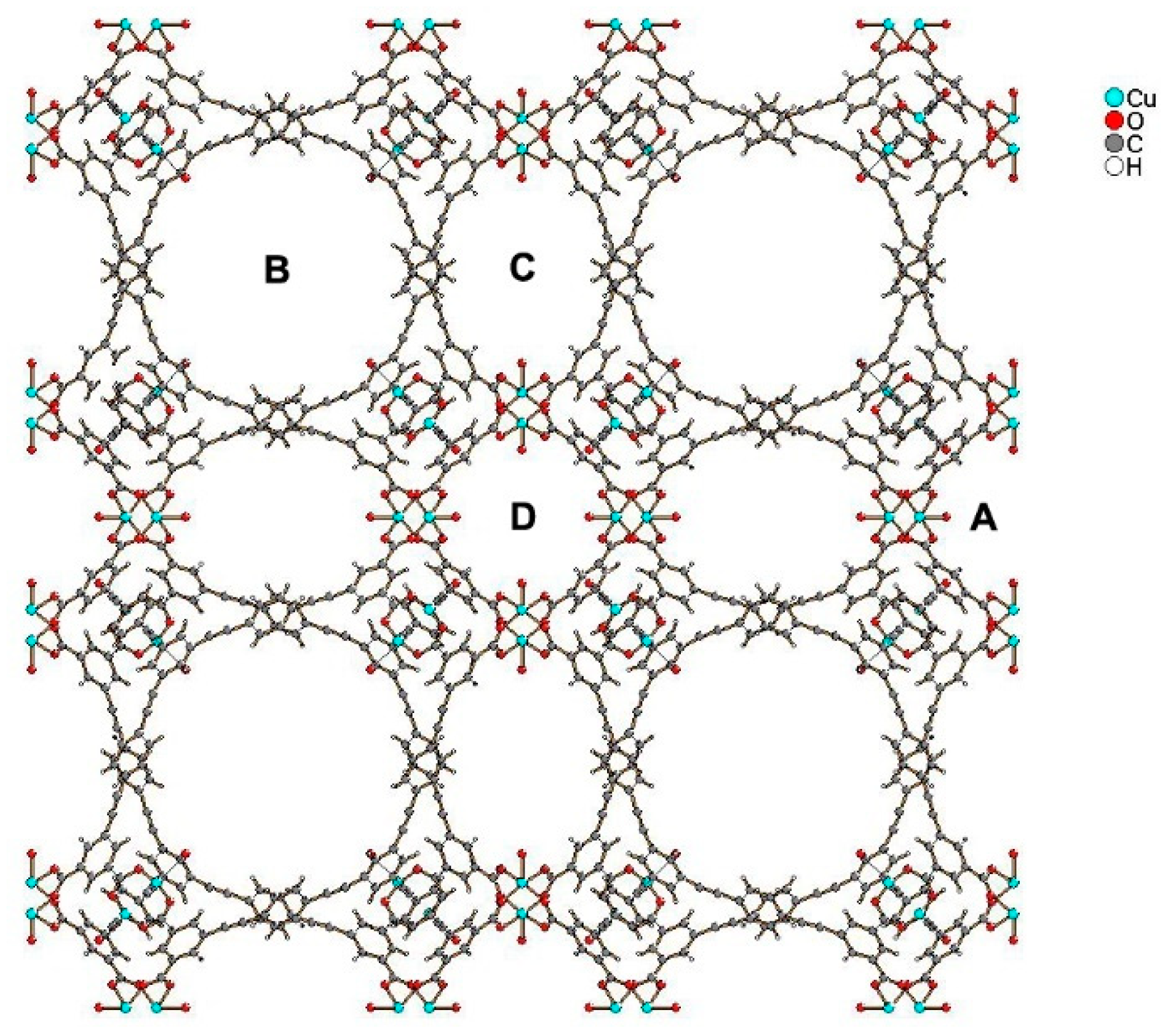
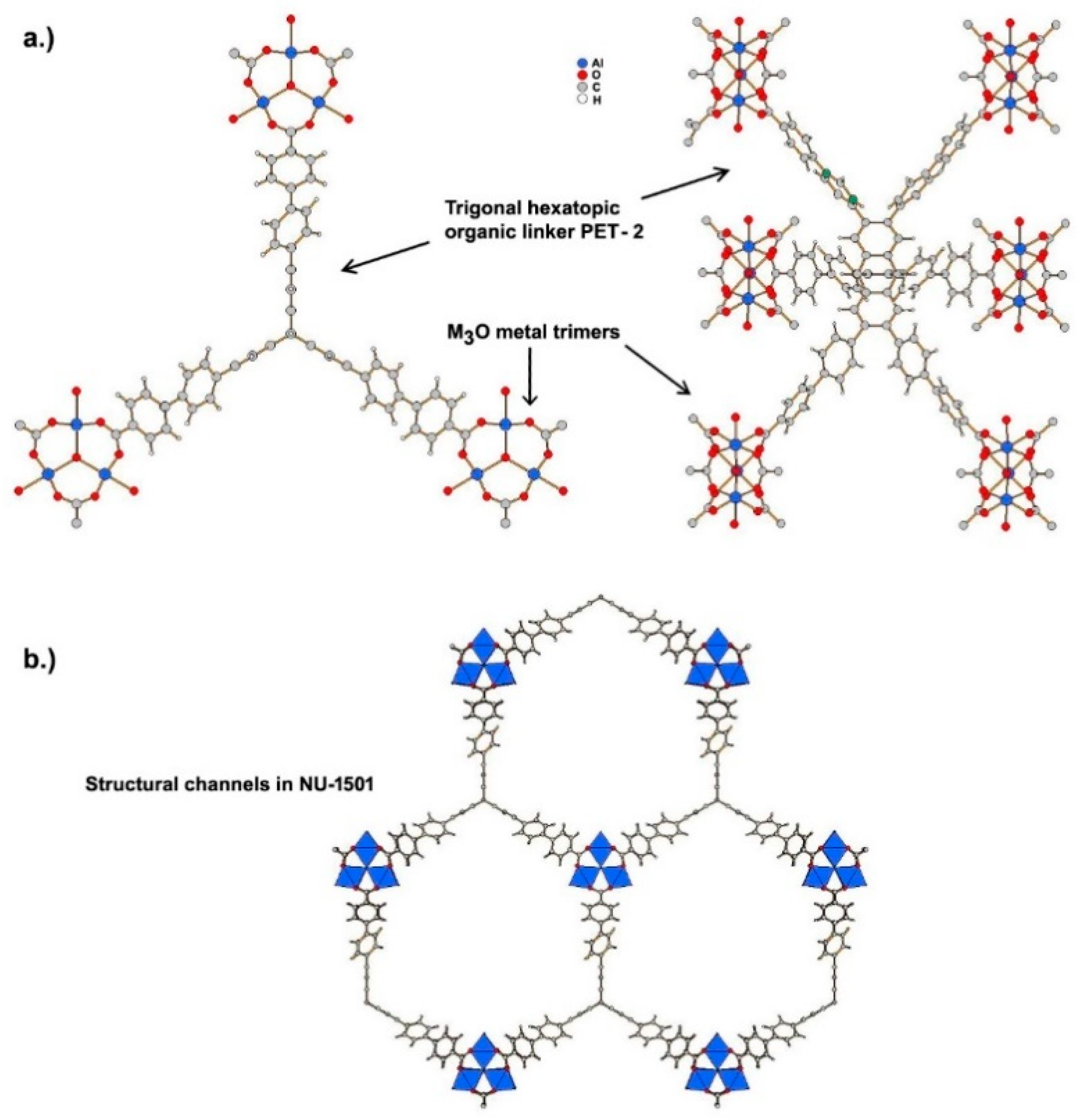
3.2. Other MOFs Formed by Metal Clusters Interconnected by Carboxylate Linkers
4. Current Approaches to Increasing Hydrogen Adsorption in MOFs
4.1. Factor of the Surface Area and Pore Volume
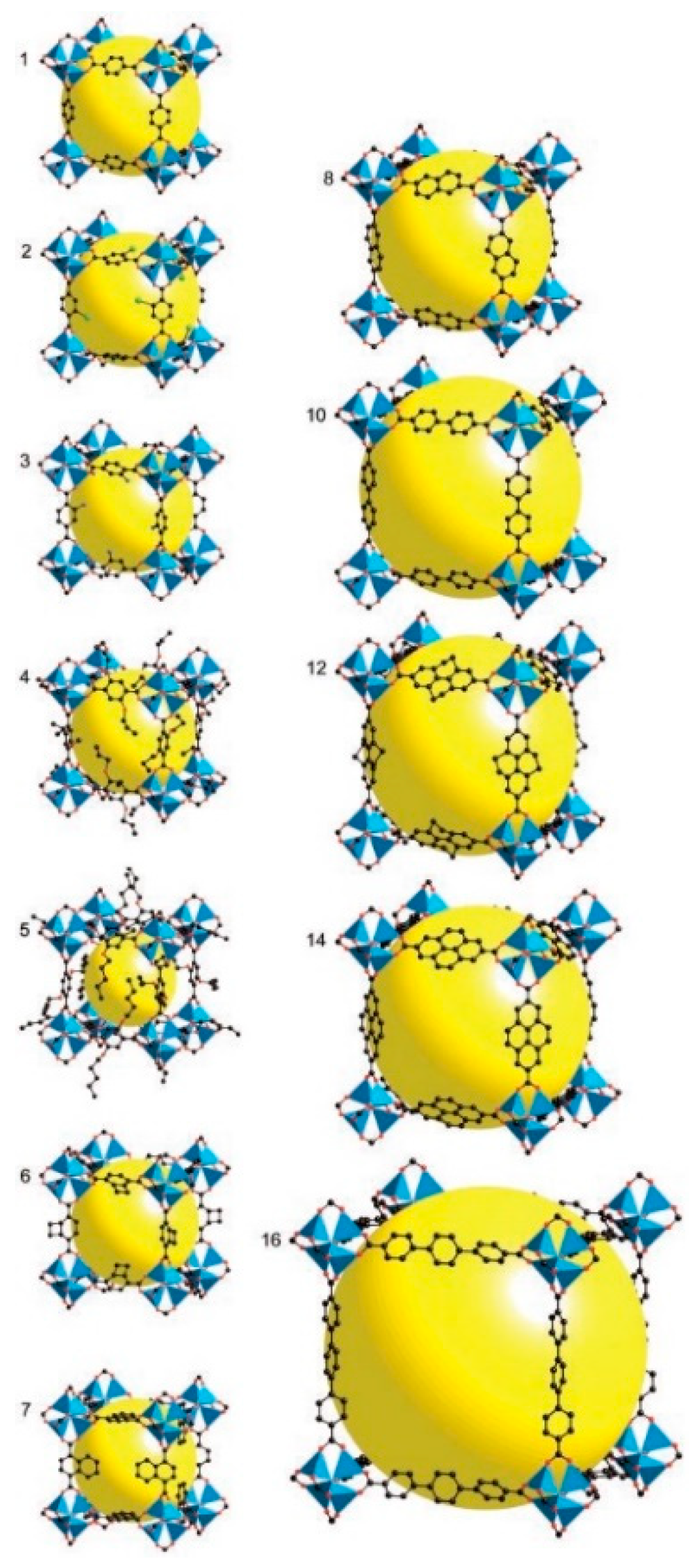
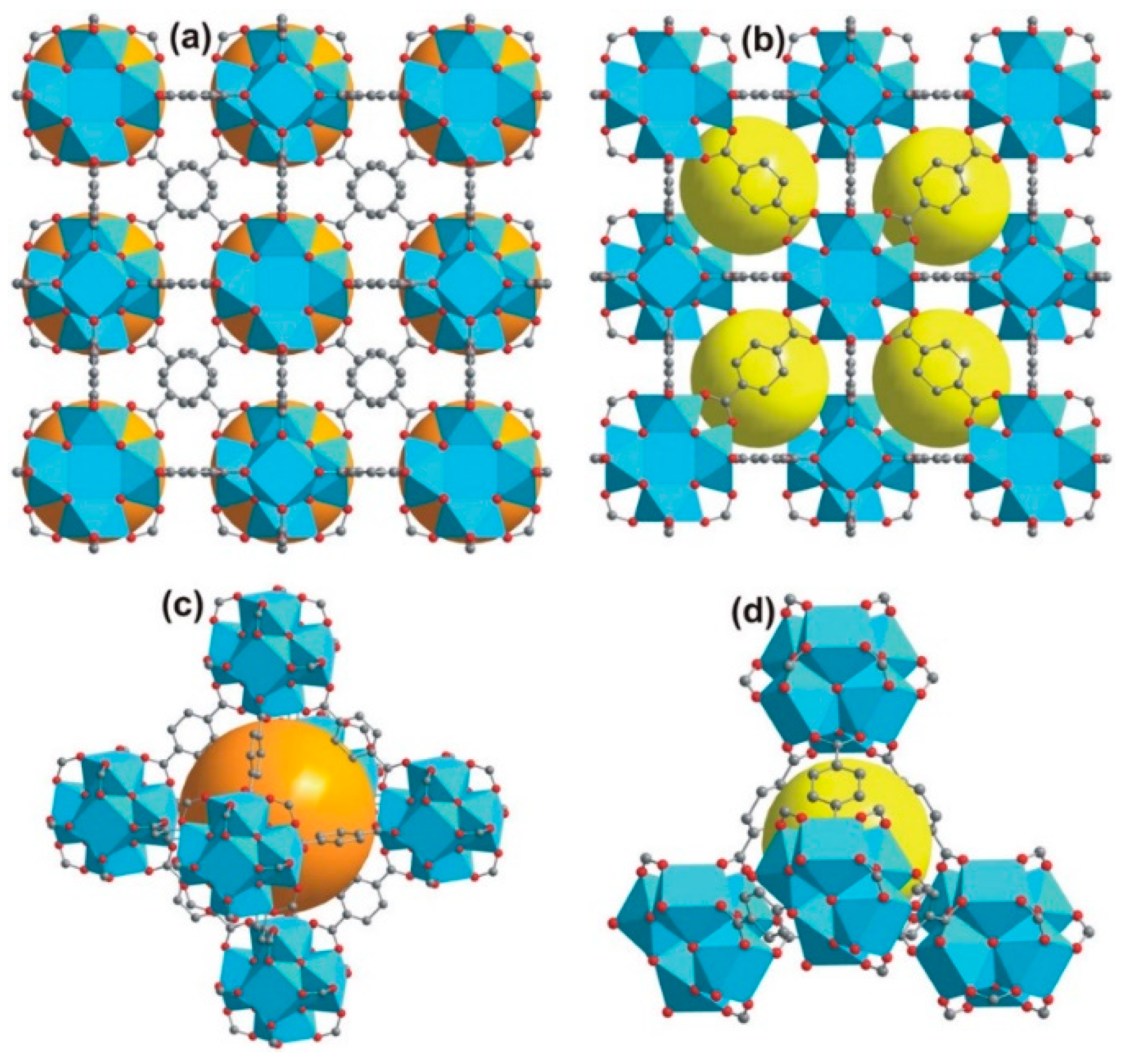
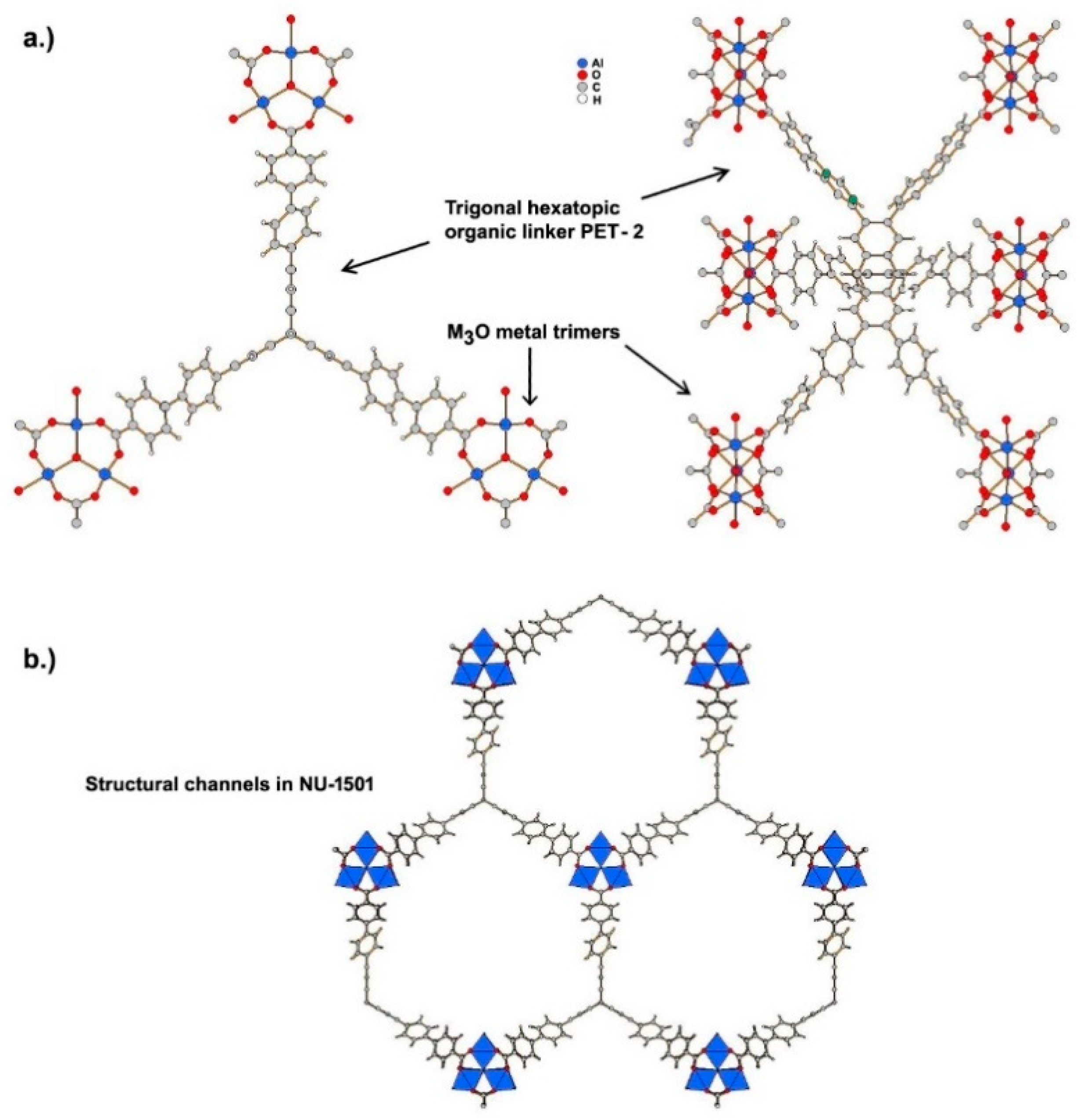
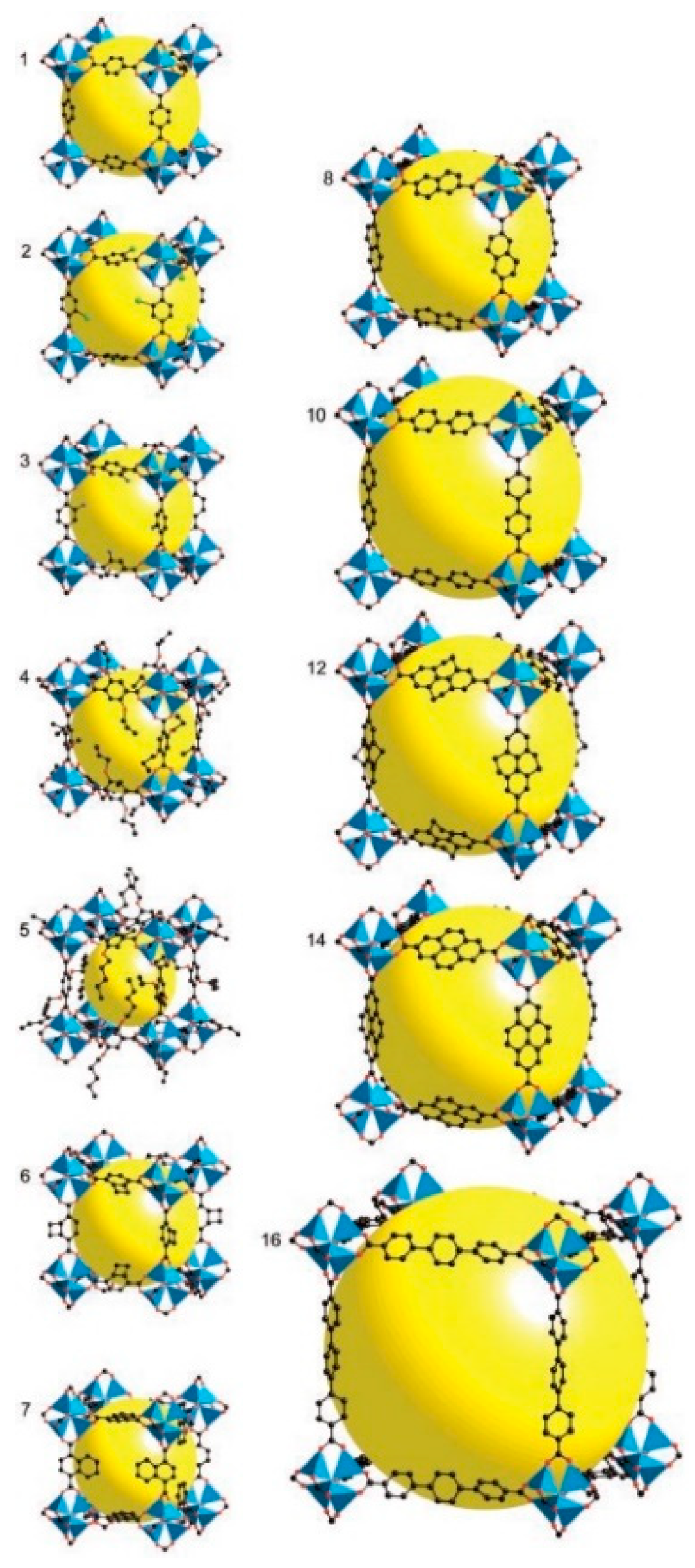
4.1.1. Length of Organic Linkers
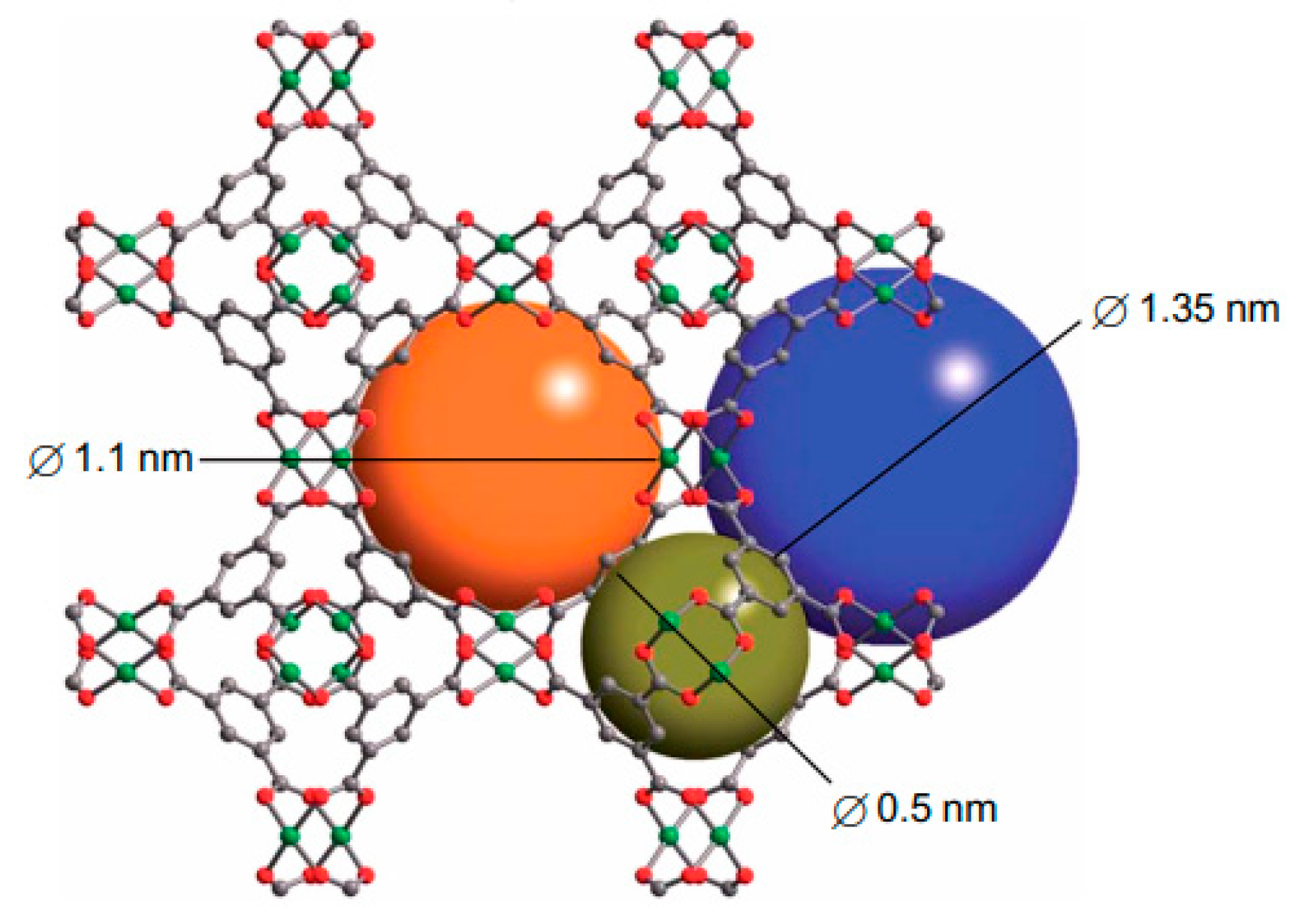
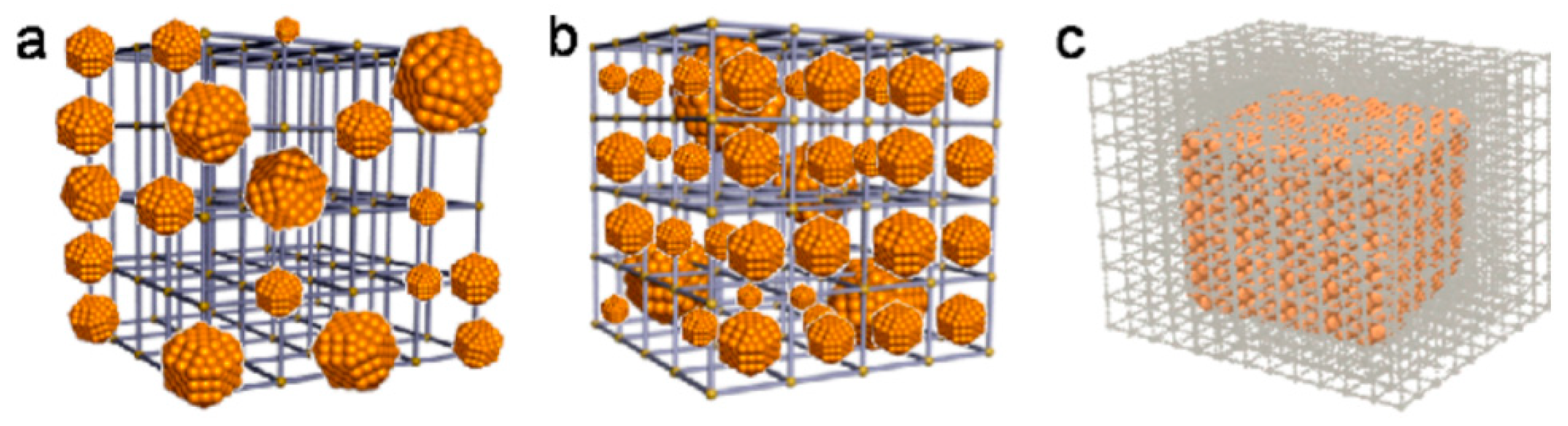
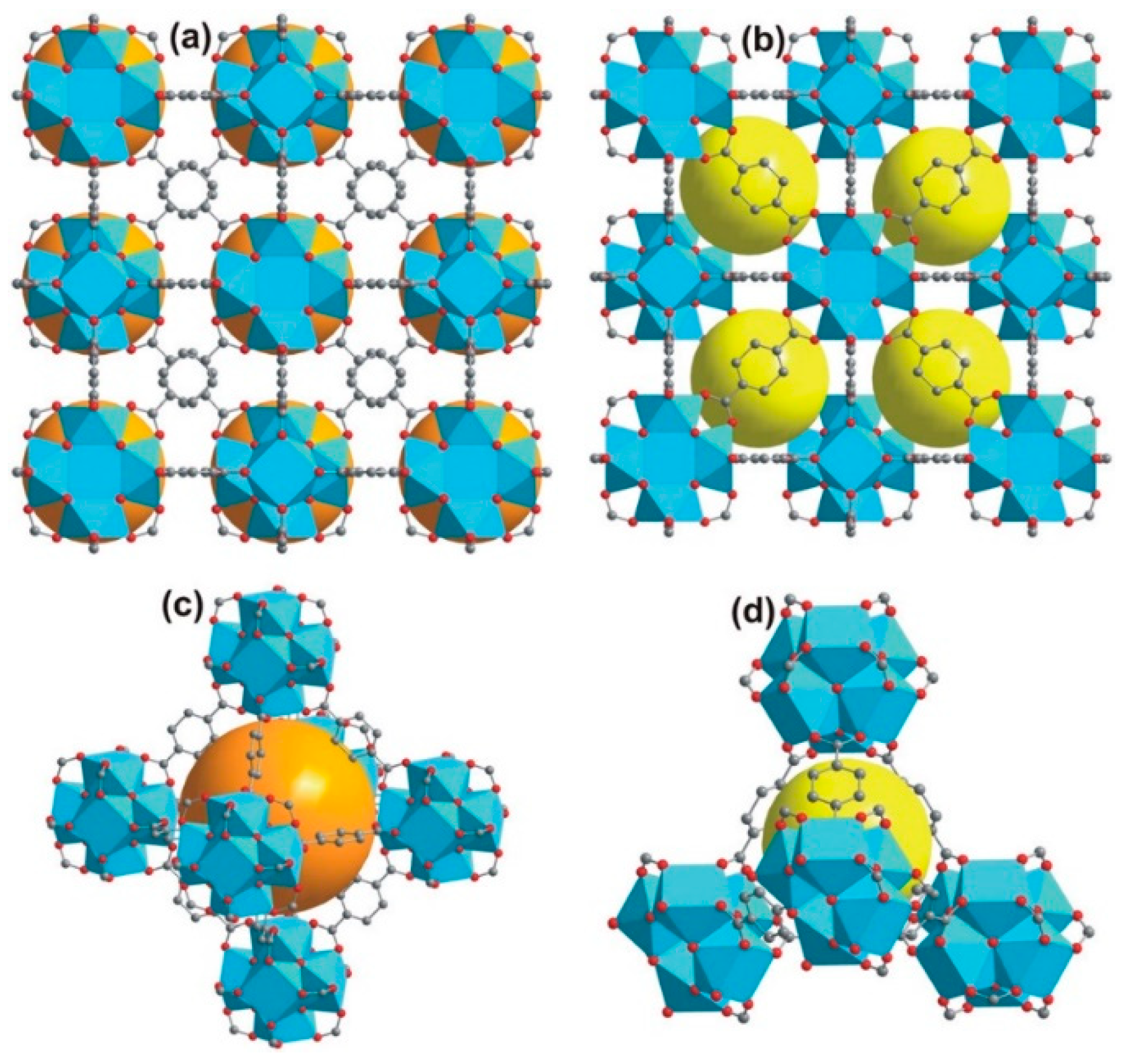
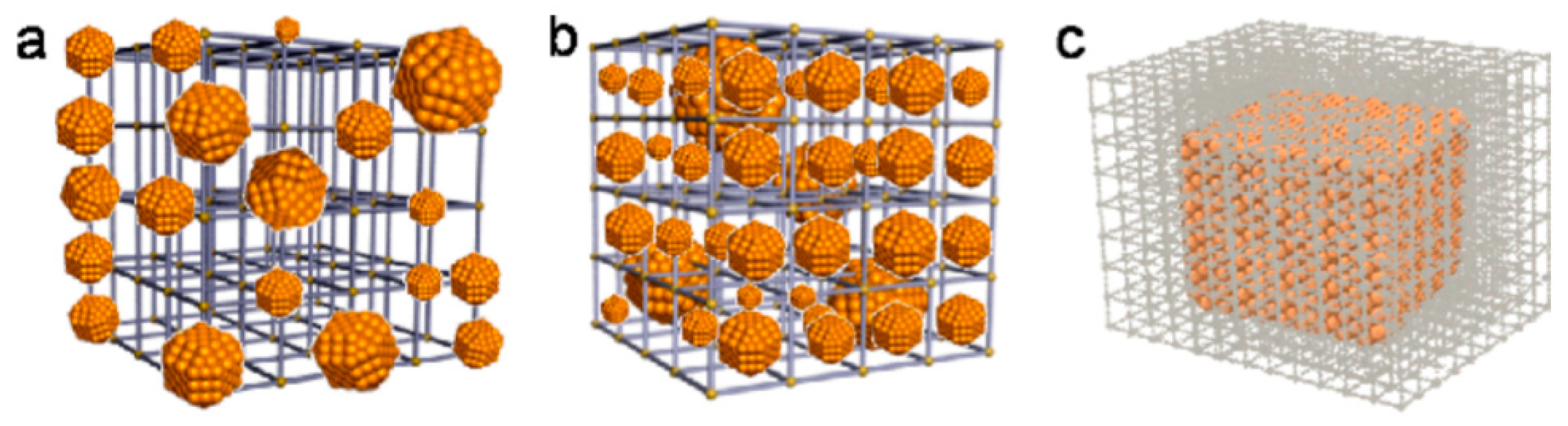
4.1.2. Catenation Process
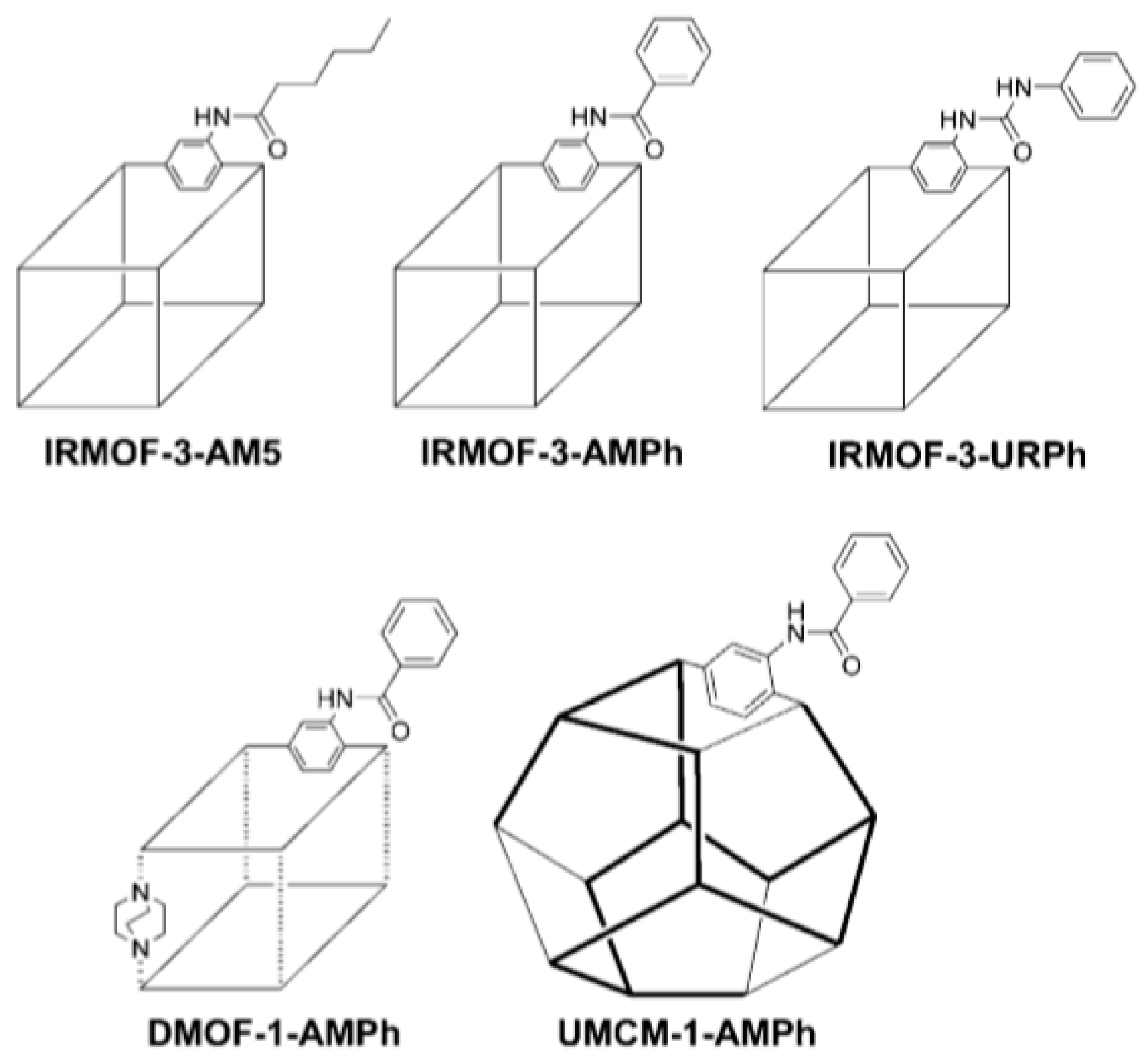
4.1.3. Different Organic Linkers
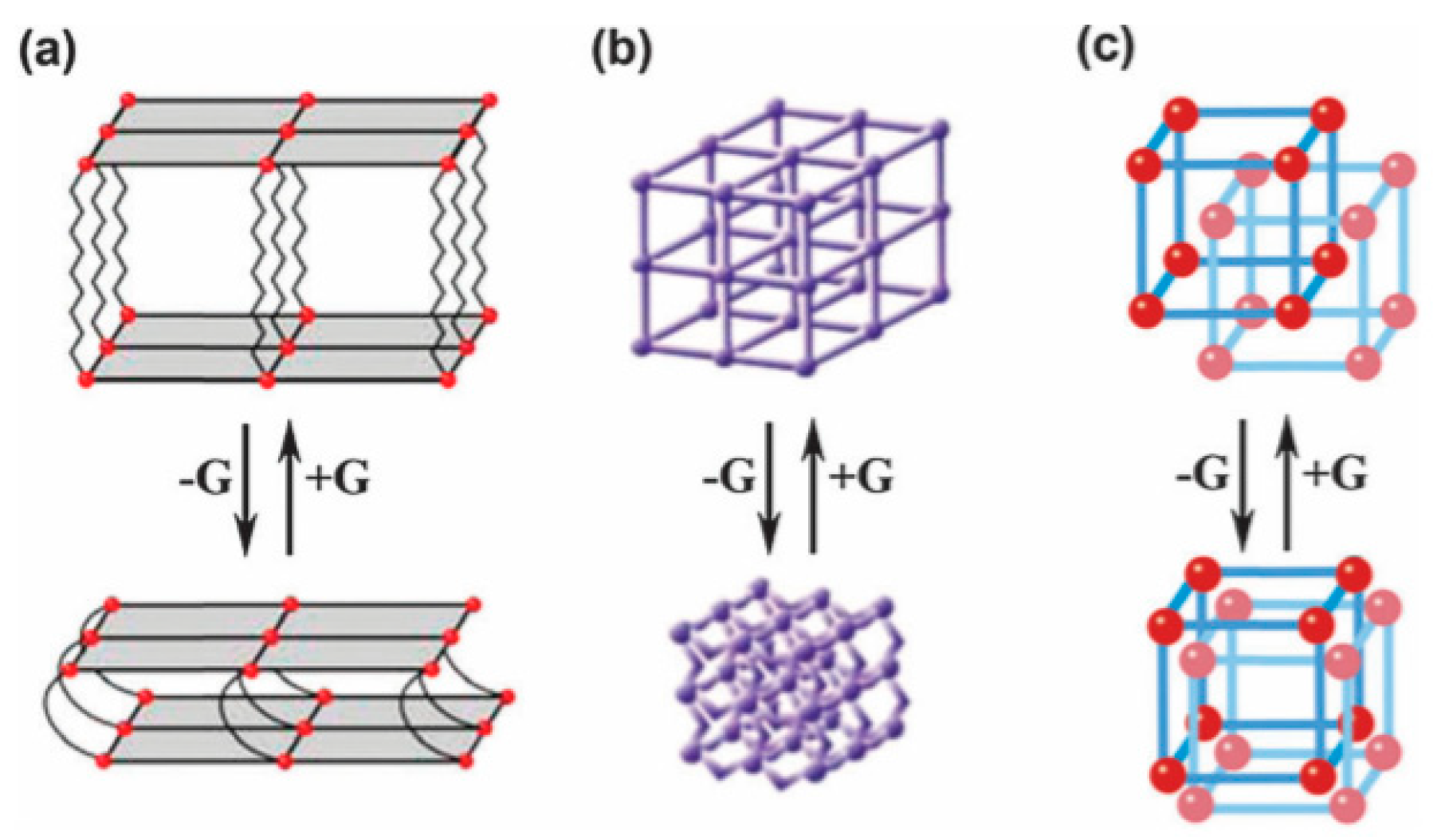
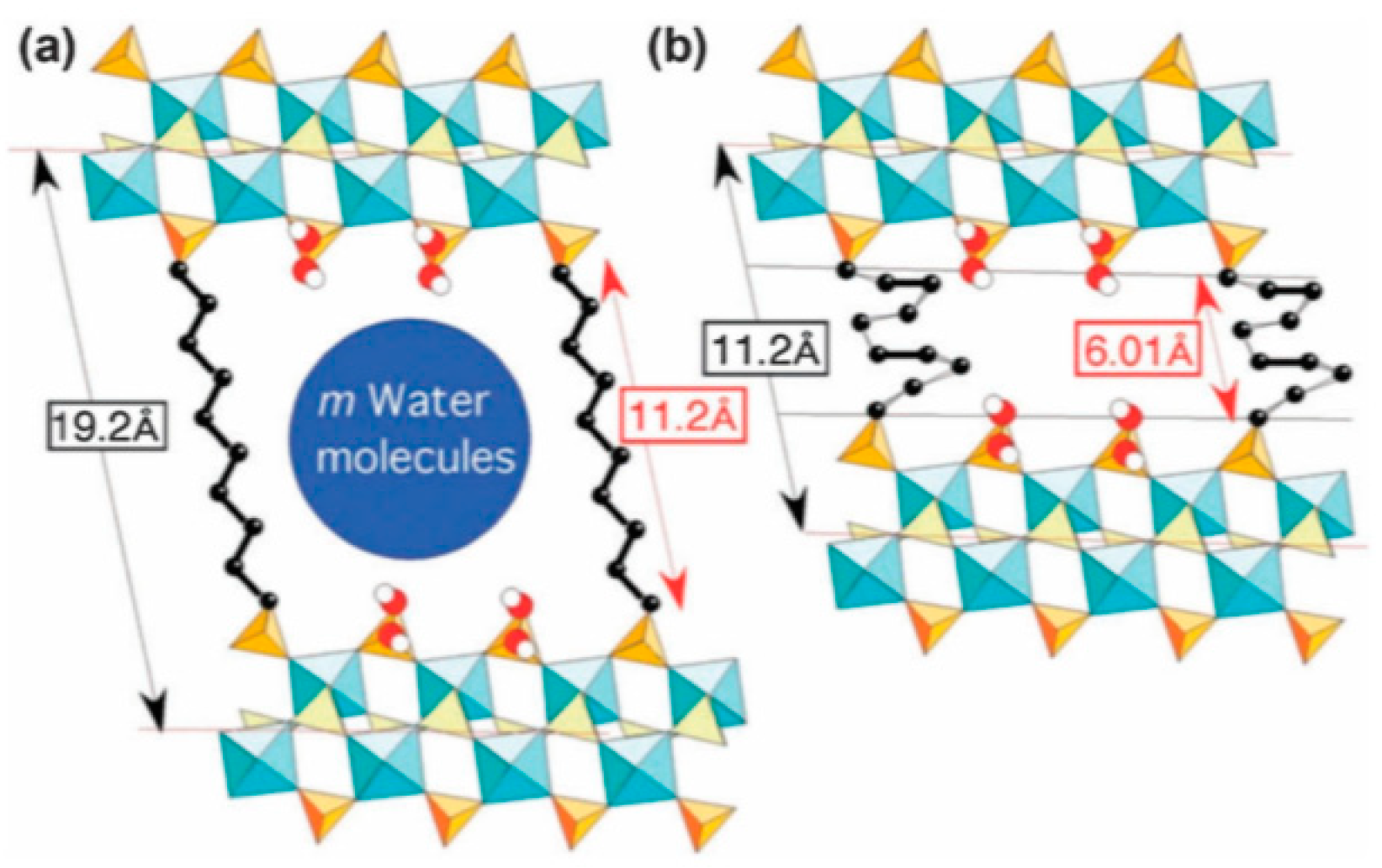
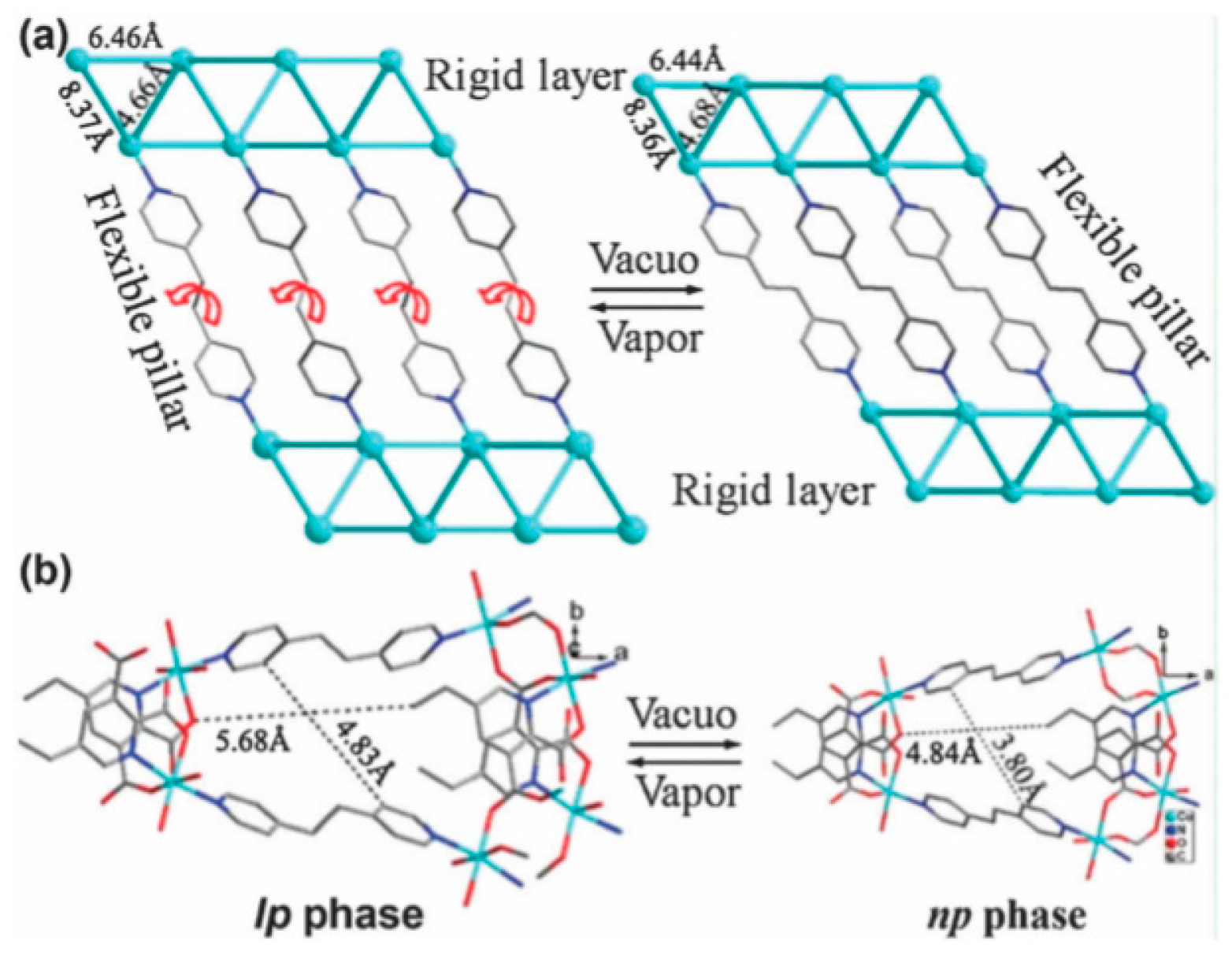
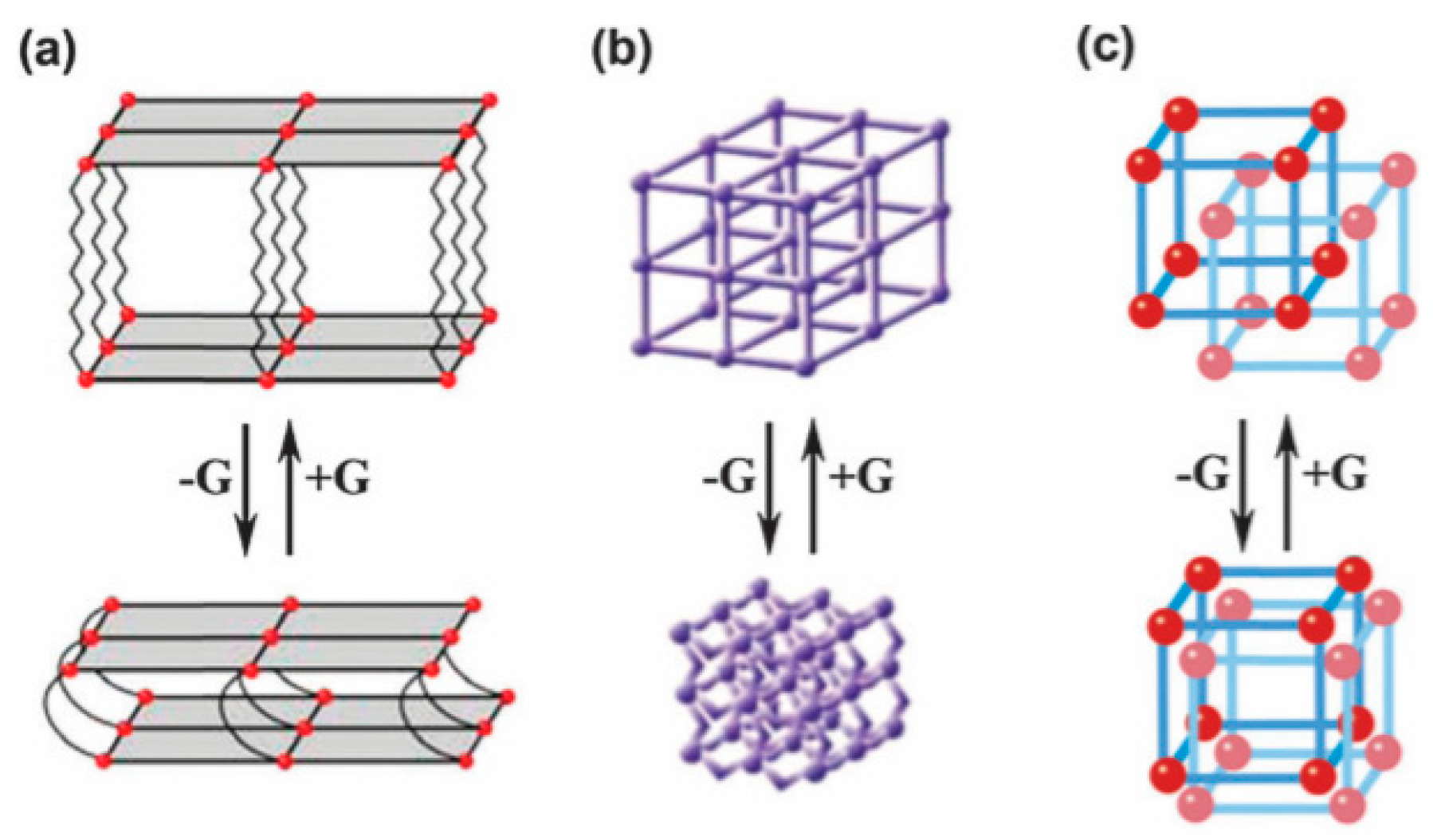
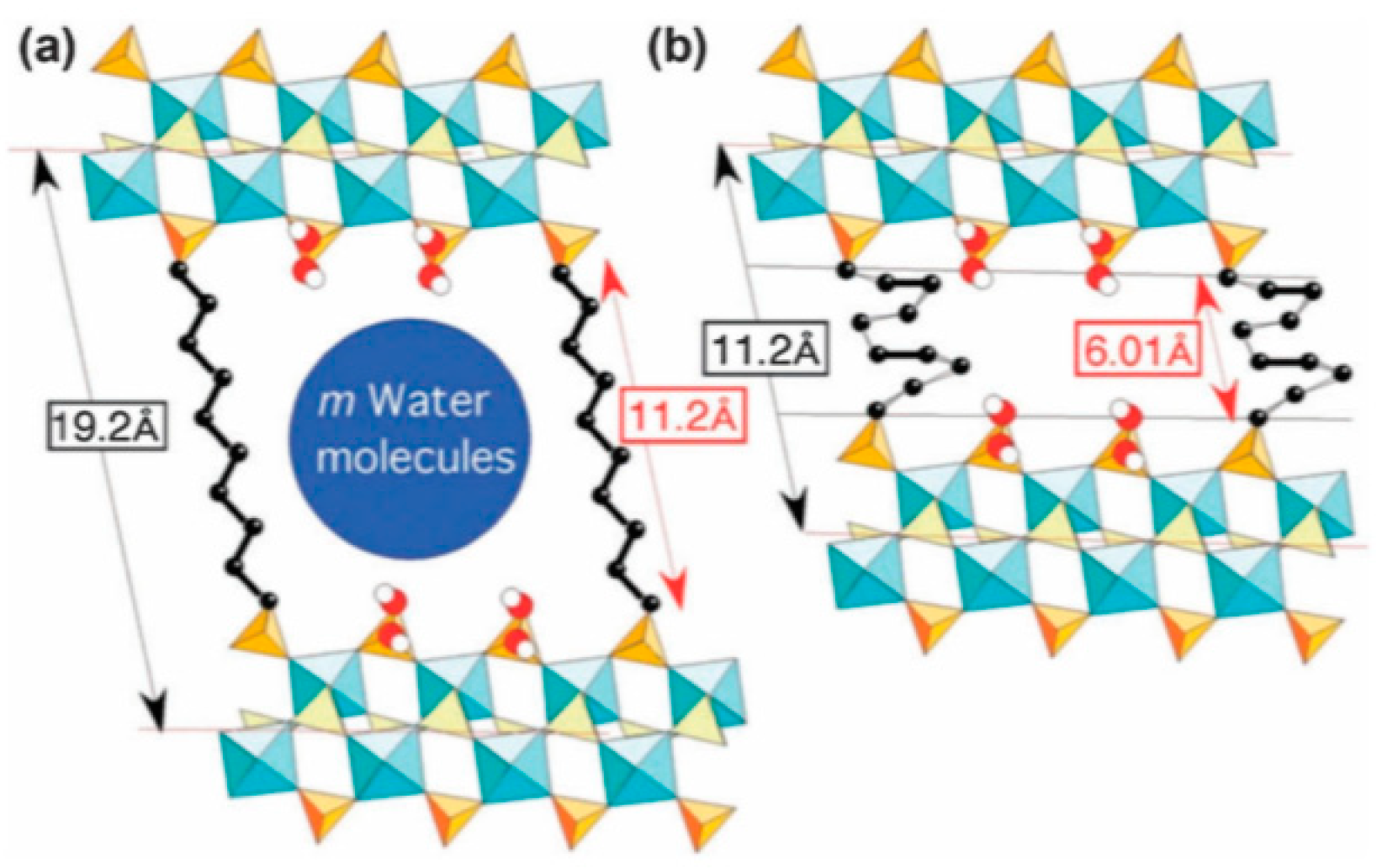
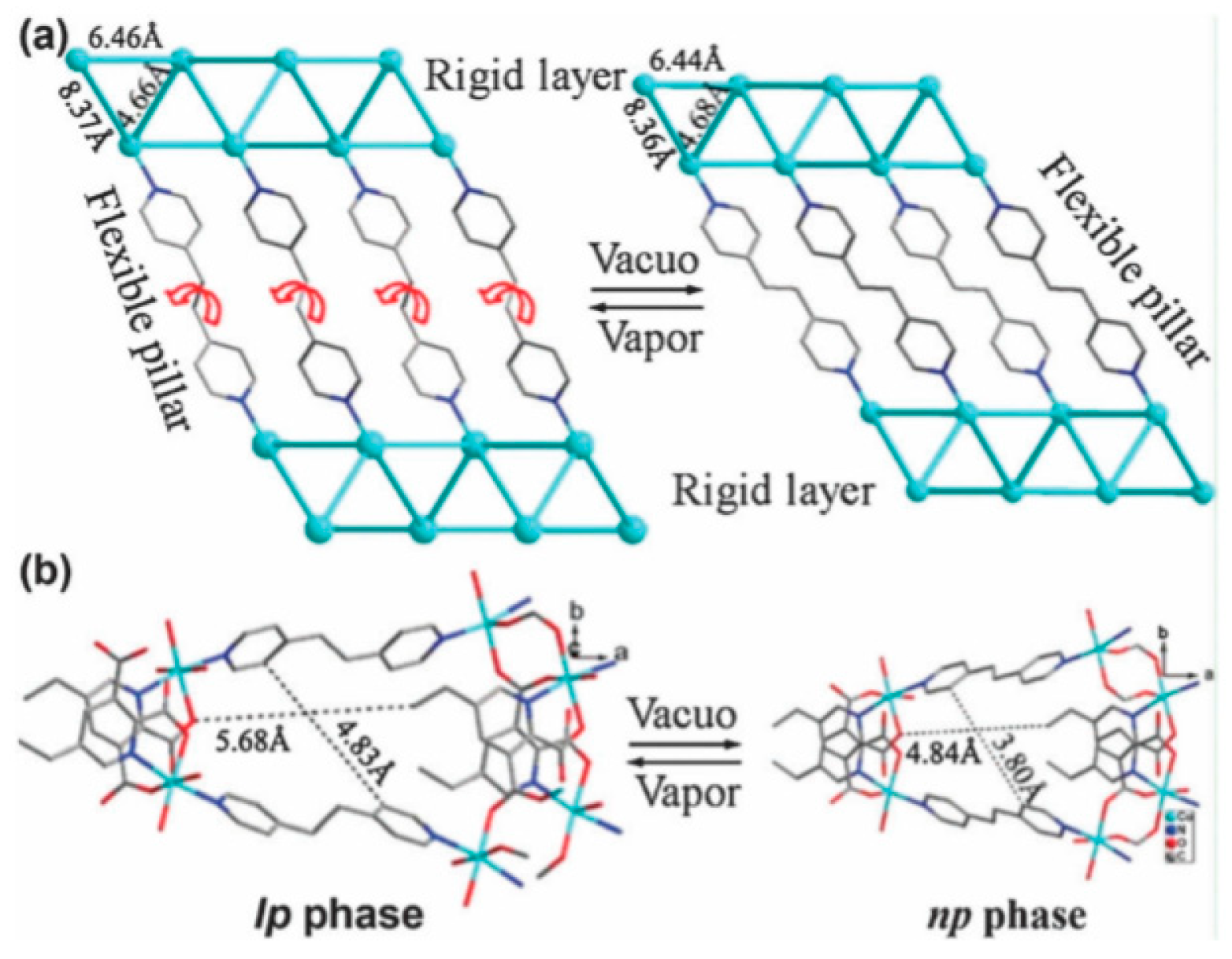
4.1.4. Flexible Organic Linkers
4.2. Factor of Isosteric Enthalpy of Hydrogen Adsorption
4.2.1. Open Metal Sites at Secondary Building Units and Organic Linkers
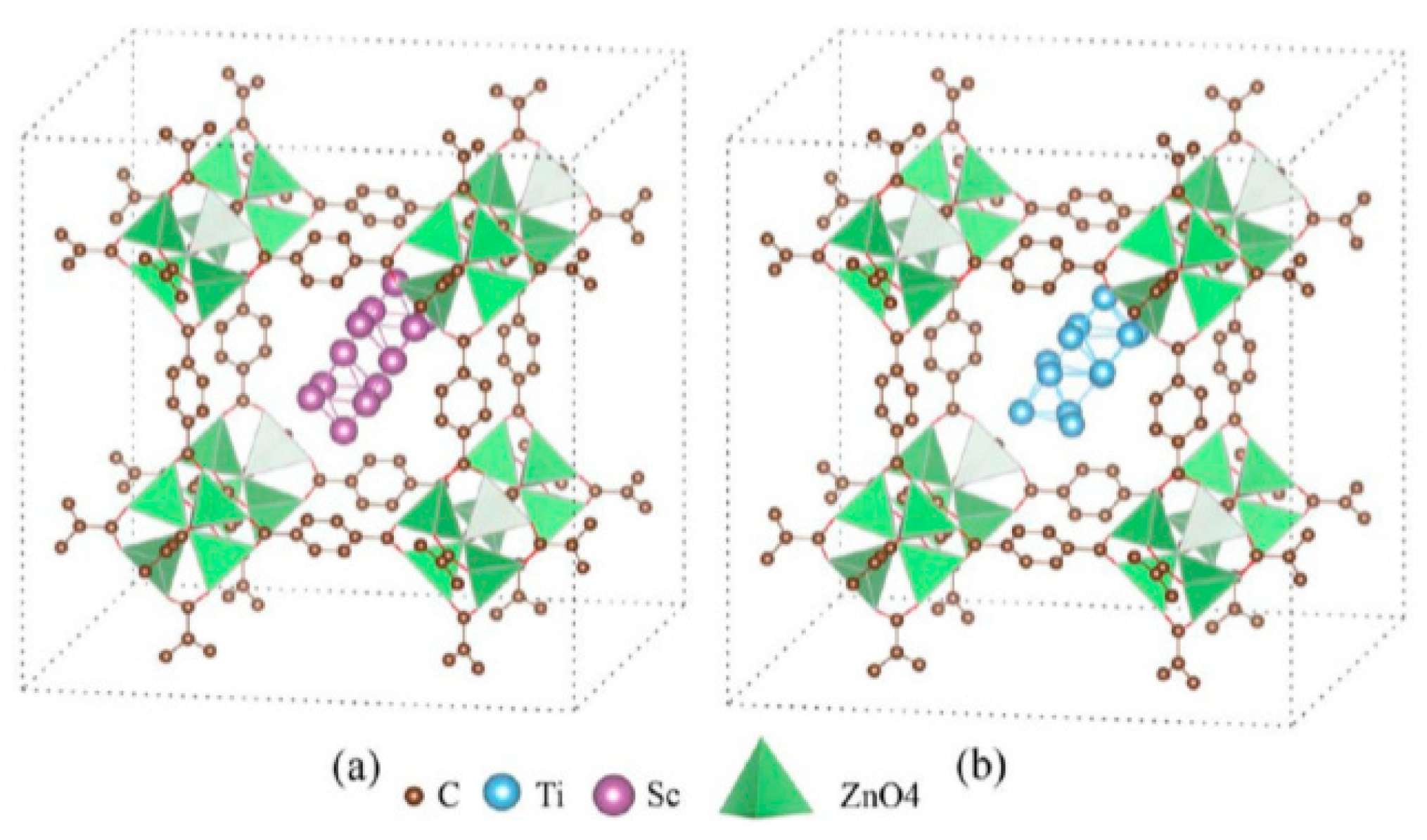
4.2.2. Metal Ions for an Electrostatic Field within the Cavities
4.3. Catalytic Effect and Pore Design
4.3.1. Incorporated Metal Nanoparticles
4.3.2. Morphology of Pores and Their Functionalization
5. Prospective Views and Outlooks
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Communication from the Commission to the European Parliament, the European Council, the Council, the European Economic and Social Committee and the Committee of the Regions. The European Green Deal, COM/2019/640 final. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=COM:2019:640:FIN (accessed on 16 April 2021).
- A European Green Deal. Striving to Be the First Climate-Neutral Continent. Available online: https://ec.europa.eu/info/strategy/priorities-2019-2024/european-green-deal (accessed on 16 April 2021).
- Communication from the Commission to the European Parliament, the European Council, the Council, the European Economic and Social Committee and the Committee of the Regions. A Hydrogen Strategy for a Climate-Neutral Europe, COM/2020/301 Final. Available online: https://ec.europa.eu/energy/sites/ener/files/hydrogen_strategy.pdf (accessed on 16 April 2021).
- Hydrogen Roadmap Europe, A Sustainable Pathway for the European energy Transition. Available online: https://www.fch.europa.eu/sites/default/files/Hydrogen%20Roadmap%20Europe_Report.pdf (accessed on 16 April 2021).
- Heynes, G. Tokyo Olympic Torch to be powered by hydrogen, In H2 View. 26 March 2021. Available online: https://www.h2-view.com/story/tokyo-olympic-torch-to-be-powered-by-hydrogen/ (accessed on 16 April 2021).
- Klein, A. Japan wants to use the Olympic games to promote hydrogen to the world. In NewScientist. 26 May 2021. Available online: https://www.newscientist.com/article/mg25033362-100-japan-wants-to-use-the-olympic-games-to-promote-hydrogen-to-the-world/ (accessed on 27 May 2021).
- Hydrogen Energy Storage Markets 2019—Global Forecast to 2024, Research and Markets, 22 October 2019. Available online: https://www.researchandmarkets.com/reports/4849112/hydrogen-energy-storage-market-by-state-liquid?utm_source=CI&utm_medium=PressRelease&utm_code=sf8vnn&utm_campaign=1310239+-+Hydrogen+Energy+Storage+Markets+2019+-+Global+Forecast+to+2024&utm_exec=chdo54prd (accessed on 16 April 2021).
- Materials-Based Hydrogen Storage, Office of Energy Efficiency & Renewable Energy, U.S. Department of Energy. Available online: https://www.energy.gov/eere/fuelcells/materials-based-hydrogen-storage (accessed on 16 April 2021).
- Semelsberger, T.A. Fuels—Hydrogen storage. Chemical carriers. In Encyclopedia of Electrochemical Power Sources; Elsevier: Oxford, UK, 2009; pp. 504–518. [Google Scholar]
- Sakintuna, B.; Lamari-Darkrim, F.; Hirscher, M. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen Energy 2007, 32, 1121–1140. [Google Scholar] [CrossRef]
- Graetz, J. New approaches to hydrogen storage. Chem. Soc. Rev. 2009, 38, 73–82. [Google Scholar] [CrossRef]
- Li, H.; Yan, Y.; Orimo, S.; Züttel, A.; Jensen, C.M. Recent progress in metal borohydrides for hydrogen storage. Energies 2011, 4, 185–214. [Google Scholar] [CrossRef]
- Paskevicius, M.; Jepsen, L.H.; Schouwink, P.; Černy, R.; Ravnsbæk, D.B.; Filinchuk, Y.; Dornheim, M.; Besenbacher, F.; Jensen, T.R. Metal borohydrides and derivatives—Synthesis, structure and properties. Chem. Soc. Rev. 2017, 46, 1565–1634. [Google Scholar] [CrossRef]
- Jain, A.; Agarwal, S.; Ichikawa, T. Catalytic Tuning of Sorption Kinetics of Lightweight Hydrides: A Review of the Materials and Mechanism. Catalysts 2018, 8, 651–687. [Google Scholar] [CrossRef] [Green Version]
- Milanese, C.; Jensen, T.R.; Hauback, B.C.; Pistidda, C.; Dornheim, M.; Yange, H.; Lombardo, L.; Züttel, A.; Filinchuk, Y.; Ngene, P.; et al. Complex hydrides for energy storage. Int. J. Hydrogen Energy 2019, 44, 7860–7874. [Google Scholar] [CrossRef] [Green Version]
- Lai, Q.; Sun, Y.; Wang, T.; Modi, P.; Cazorla, C.; Demirci, U.B.; Ares Fernandez, J.R.; Leardini, F.; Aguey-Zinsou, K.F. How to design hydrogen storage materials? Fundamentals, synthesis, and storage tanks. Adv. Sustain. Syst. 2019, 3, 1900043. [Google Scholar] [CrossRef]
- Hirscher, M.; Yartys, V.A.; Baricco, M.; von Colbe, J.B.; Blanchard, D.; Bowman, R.C., Jr.; Broom, D.P.; Buckley, C.E.; Chang, F.; Chen, P.; et al. Materials for hydrogen-based energy storage—Past, recent progress and future outlook. J. Alloys Comp. 2020, 827, 153548–153587. [Google Scholar] [CrossRef]
- El Kharbachi, A.; Dematteis, E.M.; Shinzato, K.; Stevenson, S.C.; Bannenberg, L.J.; Heere, M.; Zlotea, C.; Szilágyi, P.Á.; Bonnet, J.-P.; Grochala, W.; et al. Metal hydrides and related materials. Energy carriers for novel hydrogen and electrochemical storage. J. Phys. Chem. C 2020, 124, 7599–7607. [Google Scholar] [CrossRef] [Green Version]
- Dagani, R. The bio side of organometallics. Chem. Eng. News 2002, 80, 23–29. [Google Scholar] [CrossRef]
- Froudakis, G.E. Hydrogen interaction with carbon nanotubes: A review of ab initio studies. J. Phys. Cond. Matter 2002, 14, R453. [Google Scholar] [CrossRef]
- Germain, J.; Hradil, J.; Frechet, J.M.J.; Svec, F. High Surface Area Nanoporous Polymers for Reversible Hydrogen Storage. Chem. Mater. 2006, 18, 4430–4435. [Google Scholar] [CrossRef]
- Germain, J.; Frechet, J.M.J.; Svec, F. Hypercrosslinked polyanilines with nanoporous structure and high surface area: Potential adsorbents for hydrogen storage. J. Mater. Chem. 2007, 17, 4989–4997. [Google Scholar] [CrossRef]
- Germain, J.; Svec, F.; Frechet, J.M.J. Preparation of size-selective nanoporous polymer networks of aromatic rings: Potential adsorbents for hydrogen storage. Chem. Mater. 2008, 20, 7069–7076. [Google Scholar] [CrossRef]
- Germain, J.; Frechet, J.M.J.; Svec, F. Nanoporous Polymers for Hydrogen Storage. Small 2009, 5, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Germain, J.; Frechet, J.M.J.; Svec, F. Nanoporous, hypercrosslinked polypyrroles: Effect of crosslinking moiety on pore size and selective gas adsorption. Chem. Commun. 2009, 12, 1526–1528. [Google Scholar] [CrossRef]
- Broom, D.P.; Hirscher, M. Irreproducibility in hydrogen storage material research. Energy Environ. Sci. 2016, 9, 3368–3380. [Google Scholar] [CrossRef] [Green Version]
- Fakirov, S. Editorial corner—A personal view hydrogen storage and polymers. Express Polym. Lett. 2017, 11, 162. [Google Scholar] [CrossRef]
- Rosi, N.L.; Eckert, J.; Eddaoudi, M.; Vodak, D.T.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Hydrogen storage in microporous metal-organic frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef] [Green Version]
- Rowsell, J.L.C.; Eckert, J.; Yaghi, O.M. Characterization of H2 binding sites in prototypical metal-organic frameworks by inelastic neutron scattering. J. Am. Chem. Soc. 2005, 127, 14904–14910. [Google Scholar] [CrossRef]
- Li, Y.; Yang, R.T. Hydrogen storage in metal-organic frameworks by bridged hydrogen spillover. J. Am. Chem. Soc. 2006, 128, 8136–8137. [Google Scholar] [CrossRef] [PubMed]
- Buda, C.; Dunietz, B.D. Hydrogen Physisorption on the Organic Linker in Metal Organic Frameworks: Ab Initio Computational Study. J. Phys. Chem. B 2006, 110, 10479–10484. [Google Scholar] [CrossRef]
- Li, J.; Cheng, S.; Zhao, Q.; Long, P.; Dong, J. Synthesis and hydrogen storage behavior on metal-organic framework MOF-5. Int. Ass. Hydrogen Energy 2009, 34, 1377–1382. [Google Scholar] [CrossRef]
- Murray, L.J.; Dinca, M.; Long, J.R. Hydrogen storage in metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1294–1314. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Mendoza-Cortes, J.L.; Goddard, W.A., III. Recent advances on simulation and theory of hydrogen storage in metal-organic frameworks and covalent organic frameworks. Chem. Soc. Rev. 2009, 38, 1460–1476. [Google Scholar] [CrossRef] [Green Version]
- Hirscher, M. Hydrogen storage by cryoadsorption in ultrahigh porosity metal-organic frameworks. Angew. Chem. Int. Ed. 2011, 50, 581–958. [Google Scholar] [CrossRef]
- Hirscher, M.; Panella, B.; Schmitz, B. Metal-organic frameworks for hydrogen storage. Micropor. Mesopor. Mater. 2010, 129, 335–339. [Google Scholar] [CrossRef]
- Tranchemontagne, D.J.; Park, K.S.; Furukawa, H.; Eckert, J.; Knobler, C.B.; Yaghi, O.M. Hydrogen storage in new metal-organic frameworks. J. Phys. Chem. C 2012, 116, 13143–13151. [Google Scholar] [CrossRef]
- Goldsmith, J.; Wong-Foy, A.G.; Cafarella, M.J.; Siegel, D.J. Theoretical limits of hydrogen storage in metal−organic frameworks: Opportunities and trade-offs. Chem. Mater. 2013, 25, 3373–3382. [Google Scholar] [CrossRef]
- Chavan, S.M.; Zavorotynska, O.; Lamberti, C.; Bordiga, S. H2 interaction with divalent cations in isostructural MOFs: A key study for variable temperature infrared spectroscopy. Dalton Trans. 2013, 42, 12586–12595. [Google Scholar] [CrossRef]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.-W. Hydrogen Storage in Metal-Organic Frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef]
- Kloutse, A.F.; Zacharia, R.; Cossement, D.; Chahine, R.; Balderas-Xicohtencatl, R.; Oh, H.; Streppel, B.; Schlichtenmayer, M.; Hirscher, M. Isosteric heat of hydrogen adsorption on MOFs: Comparison between adsorption calorimetry, sorption isosteric method, and analytical models. Appl. Phys. A 2015, 121, 1417–1424. [Google Scholar] [CrossRef]
- Schoedel, A.; Ji, Z.; Yaghi, O.M. The role of metal-organic frameworks in a carbon-neutral energy cycle. Nat. Energy 2016, 1, 16034. [Google Scholar] [CrossRef]
- Nandasiri, M.I.; Jambovane, S.R.; Mcgrail, B.P.; Schaef, H.T. Adsorption, separation and catalytic properties of densified metal-organic frameworks. Coord. Chem. Rev. 2016, 311, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Ullman, A.M.; Brown, J.W.; Foster, M.E.; Léonard, F.; Leong, K.; Stavila, V.; Allendorf, M.D. Transforming MOFs for energy applications using the guest@MOF concept. Inorg. Chem. 2016, 55, 7233–7249. [Google Scholar] [CrossRef]
- Maity, D.K.; Halder, A.; Pahari, G.; Haque, F.; Ghoshal, D. Hydrogen uptake by an inclined polycatenated dynamic metal-organic framework based material. Inorg. Chem. 2017, 56, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Zhang, Q.; Wang, Y.; Duan, J. Unified meso-pores and dense Cu2+ sites in porous coordination polymers for highly efficient gas storage and separation. Dalton Trans. 2018, 47, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.-J.; Tan, Y.-X.; Wang, W.; Ju, Z.; Yuan, D. Optimizing H2, D2, and C2H2 sorption properties by tuning the pore apertures in metal−organic frameworks. Inorg. Chem. 2018, 57, 13312–13317. [Google Scholar] [CrossRef]
- Ahmed, A.; Seth, S.; Purewal, J.; Wong-Foy, A.G.; Veenstra, M.; Matzger, A.J.; Siegel, D.J. Exceptional hydrogen storage achieved by screening nearly half a million metal-organic frameworks. Nat. Commun. 2019, 10, 1568. [Google Scholar] [CrossRef] [Green Version]
- Molefe, L.Y.; Musyoka, N.M.; Ren, J.; Langmi, H.W.; Mathe, M. Polymer-based shaping strategy for zeolite templated carbons (ZTC) and their metal organic framework (MOF) composites for improved hydrogen storage properties. Front. Chem. 2019, 7, 864. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, R.-B.; Wang, J.; Wang, B.; Liang, B.; Yildirim, T.; Zhang, J.; Zhou, W.; Chen, B. Optimization of the pore structures of mofs for record high hydrogen volumetric working capacity. Adv. Mater. 2020, 32, 1907995. [Google Scholar] [CrossRef]
- Yurdus, A.; Yürüm, A.; Yürüm, Y. Engineering MIL-88B crystallites for enhanced H2 uptake capacity: The role of ultramicropores. Int. J. Energy Res. 2020, 44, 2875–2888. [Google Scholar] [CrossRef]
- Gupta, M.; Chatterjee, N.; De, D.; Saha, R.; Kumar Chattaraj, P.; Oliver, C.L.; Bharadwaj, P.K. Metal-organic frameworks of Cu(II) constructed from functionalized ligands for high capacity H2 and CO2 gas adsorption and catalytic studies. Inorg. Chem. 2020, 59, 1810–1822. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, D.; Cheng, M.; Liu, Z.; Lai, C.; Zhang, C.; Zhou, C.; Xiong, W.; Qin, L.; Shao, B.; et al. Metal sulfide/MOF-based composites as visible-light-driven photocatalysts for enhanced hydrogen production from water splitting. Coord. Chem. Rev. 2020, 409, 213220. [Google Scholar] [CrossRef]
- Yu, S.; Jing, G.; Li, S.; Li, Z.; Ju, X. Tuning the hydrogen storage properties of MOF-650: A combined DFT and GCMC simulations study. Int. J. Hydrogen Energy 2020, 45, 6757–6764. [Google Scholar] [CrossRef]
- Liu, J.; Mukherjee, S.; Wang, F.; Fischer, R.A.; Zhang, J. Homochiral metal–organic frameworks for enantioseparation. Chem. Soc. Rev. 2021, 50, 5706–5745. [Google Scholar] [CrossRef]
- Tan, Y.X.; Wang, F.; Zhang, J. Design and synthesis of multifunctional metal-organic zeolites. Chem. Soc. Rev. 2018, 47, 2130–2144. [Google Scholar] [CrossRef]
- Zhang, H.X.; Hong, Q.L.; Li, J.; Wang, F.; Huang, X.; Chen, S.; Tu, W.; Yu, D.; Xu, R.; Zhou, T.; et al. Isolated square-planar copper center in boron imidazolate nanocages for photocatalytic reduction of CO2 to CO. Angew. Chem. Int. Ed. 2019, 58, 11752–11756. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, D.F.; Sun, Y.; Gao, M.Y.; Zheng, N.; Gu, C.; Wang, F.; Zhang, J. Large titanium-oxo clusters as precursors to synthesize the single crystals of Ti-MOFs. ACS Mater. Lett. 2021, 3, 64–68. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Rosi, N.L.; Eddaoudi, M.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Infinite secondary building units and forbidden catenation in metal-organic frameworks. Angew. Chem. Int. Ed. 2002, 41, 284–287. [Google Scholar] [CrossRef]
- Llewellyn, P.L.; Rodriquez-Reinoso, F.; Rouqerol, J.; Seaton, N. Characterization of Porous Solids VII; Elsevier: Oxford, UK, 2007; pp. 750. [Google Scholar]
- Senkovska, I.; Kaskel, S. Ultrahigh porosity in mesoporous MOFs: Promises and limitations. Chem. Commun. 2014, 50, 7089–7098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouquerol, J.; Rouquerol, F.; Sing, K.S.W.; Llewellyn, P.; Maurin, G. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, 2nd ed.; Elsevier: Oxford, UK, 2014; 630p. [Google Scholar]
- Parilla, P.A.; Gross, K.; Hurst, K.; Gennett, T. Recommended volumetric capacity definitions and protocols for accurate, standardized and unambiguous metrics for hydrogen storage materials. Appl. Phys. A 2016, 122, 201. [Google Scholar] [CrossRef]
- Dang, S.; Zhu, Q.-L.; Xu, Q. Nanomaterials Derived from Metal-Organic Frameworks. Nat. Rev. Mater. 2018, 3, 17045. [Google Scholar] [CrossRef]
- Giraldo, L.; Rodriguez-Estupiñán, P. Moreno-Piraján, J.C. Isosteric Heat: Comparative study between Clausius–Clapeyron, CSK and adsorption calorimetry methods. Processes 2019, 7, 203. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Saha, B.B.; Koyama, S.; Ng, K.C. Specific heat capacity of a single component adsorbent-adsorbate system. App. Phys. Lett. 2006, 89, 171902. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; Groy, T.L.; Yaghi, O.M. Establishing microporosity in open metal−organic frameworks: gas sorption isotherms for Zn(BDC) (BDC = 1,4-Benzenedicarboxylate). J. Am. Chem. Soc. 1998, 120, 8571–8572. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Flaig, R.W.; Jiang, H.L.; Yaghi, O.M. Carbon capture and conversion using metal–organic frameworks and MOF-based materials. Chem. Soc. Rev. 2019, 48, 2783–2828. [Google Scholar] [CrossRef]
- Gangu, K.K.; Maddila, S.; Mukkamala, S.B.; Jonnalagadda, S.B. Characteristics of MOF, MWCNT and graphene containing materials for hydrogen storage: A review. J. Energy Chem. 2019, 30, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Rojas, S.; Arenas-Vivo, A.; Horcajada, P. Metal-organic frameworks: A novel platform for combined advanced therapies. Cord. Chem. Rev. 2019, 388, 202–226. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Liu, Y.; Purewal, J.; Tran, L.D.; Wong-Foy, A.G.; Veenstra, M.; Matzger, A.J.; Siegel, D.J. Balancing gravimetric and volumetric hydrogen density in MOFs. Energy Environ. Sci. 2017, 10, 2459–2471. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Biswas, S.; Zhang, J.; Li, Z.; Liu, Y.Y.; Grzywa, M.; Sun, L.; Volkmer, D.; Van Der Voort, P. Enhanced selectivity of CO2 over CH4 in sulphonate-, carboxylate- and iodo-functionalized UiO-66 frameworks. Dalton Trans. 2013, 42, 4730–4737. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wei, Z.; Gu, Z.Y.; Liu, T.F.; Park, J.; Park, J.; Tian, J.; Zhang, M.; Zhang, Q.; Gentle, T.; et al. Tuning the structure and function of metal–organic frameworks via linker design. Chem. Soc. Rev. 2014, 43, 5561–5593. [Google Scholar] [CrossRef] [PubMed]
- Chui, S.S.; Los, S.M.F.; Charmant, J.P.H.; Open, A.G.; Williams, I.D. A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 1999, 38, 1148–1150. [Google Scholar] [CrossRef]
- Mason, J.A.; Veenstra, M.; Long, J.R. Evaluating metal-organic frameworks for natural gas storage. Chem. Sci. 2014, 5, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, P.; Anderson, R.; Wang, X.; Zhang, X.; Robison, L.; Redfern, L.R.; Moribe, S.; Islamoglu, T.; Gómez-Gualdrón, D.A.; et al. Balancing volumetric and gravimetric uptake in highly porous materials for clean energy. Science 2020, 368, 297–303. [Google Scholar] [CrossRef]
- Frost, H.; Duren, T.; Snurr, R.Q. Effects of surface area, free volume, and heat of adsorption on hydrogen uptake in metal-organic frameworks. J. Phys. Chem. B 2006, 110, 9565–9570. [Google Scholar] [CrossRef]
- Furukawa, H.; Ko, N.; Go, Y.B.; Aratani, N.; Choi, S.B.; Choi, E.; Yazaydin, A.O.; Snurr, R.Q.; O’Keeffe, M.; Kim, J.; et al. Ultrahigh porosity in metal-organic frameworks. Science 2010, 329, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Panella, B.; Hirscher, M.; Pütter, H.; Müller, U. Hydrogen adsorption in metal-organic frameworks: Cu-MOFs and Zn-MOFs compared. Adv. Funct. Mater. 2006, 16, 520–524. [Google Scholar] [CrossRef]
- Yang, S.J.; Jung, H.; Kim, T.; Im, J.H.; Park, C.R. Effects of structural modifications on the hydrogen storage capacity of MOF-5. Int. J. Hydrogen Energy 2012, 37, 5777–5783. [Google Scholar] [CrossRef]
- Hönicke, I.M.; Senkovska, I.; Bon, V.; Baburin, I.A.; Bönisch, N.; Raschke, S.; Evans, J.D.; Kaskel, S. Balancing mechanical stability and ultrahigh porosity in crystalline framework materials. Angew. Chem. Int. Ed. 2018, 57, 13780–13783. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Miller, M.A.; Yaghi, O.M. Independent verification of the saturation hydrogen uptake in MOF-177 and establishment of a benchmark for hydrogen adsorption in metal-organic frameworks. J. Mater. Chem. 2007, 17, 3197–3204. [Google Scholar] [CrossRef]
- Chavan, S.; Vitillo, J.G.; Gianolio, D.; Zavorotynska, O.; Civalleri, B.; Jakobsen, S.; Nilsen, M.H.; Valenzano, L.; Lamberti, C.; Lillerud, K.P.; et al. H2 storage in isostructural UiO-67 and UiO-66 MOFs. Phys. Chem. Chem. Phys. 2012, 14, 1614–1626. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, W.; Luo, Z.; Li, B.; Yuan, D. Microporous metal-organic framework based on ligand-truncation strategy with high performance for gas adsorption and separation. Inorg. Chem. 2017, 56, 10215–10219. [Google Scholar] [CrossRef]
- Rowsell, J.L.C.; Millward, A.R.; Park, K.S.; Yaghi, O.M. Hydrogen sorption in functionalized metal-organic frameworks. J. Am. Chem. Soc. 2004, 126, 5666–5667. [Google Scholar] [CrossRef]
- Wong-Foy, A.G.; Matzger, A.J.; Yaghi, O.M. Exceptional H2 saturation uptake in microporous metal-organic frameworks. J. Am. Chem. Soc. 2006, 128, 3494–3495. [Google Scholar] [CrossRef]
- Kaskel, S. Metal-Organic Frameworks: Design and Application; MacGillivray, L.R., Ed.; Wiley: Hoboken, NJ, USA, 2010; 349p. [Google Scholar]
- Batten, S.R. Topology of interpenetration. CrystEngComm 2001, 3, 67–72. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Ma, S.Q.; Sun, D.F.; Ambrogio, M.; Fillinger, J.A.; Parkin, S. Framework-catenation isomerism in metal−organic frameworks and its impact on hydrogen uptake. J. Am. Chem. Soc. 2007, 129, 1858–1859. [Google Scholar]
- Farha, O.K.; Malliakas, C.D.; Kanatzidis, M.G.; Hupp, J.T. Control over catenation in metal−organic frameworks via rational design of the organic building block. J. Am. Chem. Soc. 2010, 132, 950–952. [Google Scholar] [CrossRef]
- Zhang, J.; Wojtas, L.; Larsen, R.W.; Eddaoudi, M.; Zaworotko, M.J. Temperature and concentration control over interpenetration in a metal−organic material. J. Am. Chem. Soc. 2009, 131, 17040–17041. [Google Scholar] [CrossRef]
- Shekhah, O.; Wang, H.; Paradinas, M.; Ocal, C.; Schupbach, B.; Terfort, A.; Zacher, D.; Fischer, R.A.; Wӧll, C. Controlling interpenetration in metal-organic frameworks by liquid-phase epitaxy. Nat. Mater. 2009, 8, 481–484. [Google Scholar] [CrossRef]
- Rowsell, J.L.; Yaghi, O.M. Strategies for hydrogen storage in metal–organic frameworks. Angew. Chem. Int. Ed. 2005, 44, 4670–4679. [Google Scholar] [CrossRef]
- Kanoo, P.; Matsuda, R.; Higuchi, M.; Kitagawa, S.; Maji, T.K. New interpenetrated copper coordination polymer frameworks having porous properties. Chem. Mater. 2009, 21, 5860–5866. [Google Scholar] [CrossRef]
- Ryan, P.; Broadbelt, L.J.; Snurr, R.Q. Is catenation beneficial for hydrogenstorage in metal-organic frameworks? Chem. Commun. 2008, 44, 4132–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Suh, M.P. Mixed-ligand metal-organic frameworks with large pores: Gas sorption properties and single-crystal-to-single-crystal transformation on guest exchange. Chem. Eur. J. 2008, 14, 8812–8821. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.; Wong-Foy, A.G.; Matzger, A.J. Coordination copolymerization mediated by Zn4O(CO2R)6 metal clusters: A balancing act between statistics and geometry. J. Am. Chem. Soc. 2010, 132, 15005–15010. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keefe, M.; Yaghi, O.M. The Chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chernikova, V.; Liu, Y.; Zhang, K.; Belmabkhout, Y.; Shekhah, O.; Zhang, C.; Yi, S.; Eddaoudi, M.; Koros, W.J. Mixed matrix formulations with MOF molecular sieving for key energy-intensive separations. Nat. Mater. 2018, 17, 283–289. [Google Scholar] [CrossRef]
- Schneemann, A.; Bon, V.; Schwedler, I.; Senkovska, I.; Kaskel, S.; Fischer, R.A. Flexible metal-organic frameworks. Chem. Soc. Rev. 2014, 43, 6062–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Yang, S.; Blake, A.J.; Lewis, W.; Poirier, E.; Barnett, S.A.; Champness, N.R.; Schrӧder, M. A mesoporous metal–organic framework constructed from a nanosized C3-symmetric linker and [Cu24(isophthalate)24] cuboctahedra. Chem. Commun. 2011, 47, 9995–9997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.-S.; Chen, K.-J.; Liao, P.-Q.; Zhu, B.-Y.; Lin, R.-B.; Zhou, H.-L.; Wang, B.-Y.; Xue, W.; Zhang, J.-P.; Chen, X.-M. Turning on the flexibility of isoreticular porous coordination frameworks for drastically tunable framework breathing and thermal expansion. Chem. Sci. 2013, 4, 1539–1546. [Google Scholar] [CrossRef]
- Yang, S.; Liu, L.; Sun, J.; Thomas, K.M.; Davies, A.J.; George, M.W.; Blake, A.J.; Hill, A.H.; Fitch, A.N.; Tang, C.C.; et al. Irreversible network transformation in a dynamic porous host catalyzed by sulfur dioxide. J. Am. Chem. Soc. 2013, 135, 4954–4957. [Google Scholar] [CrossRef]
- Ferey, G.; Serre, C. Large breathing effects in three-dimensional porous hybrid matter: Facts, analyses, rules and consequences. Chem. Soc. Rev. 2009, 38, 1380–1399. [Google Scholar] [CrossRef]
- Ferey, G. Nanoporous materials: A selective magnetic sponge. Nat. Mater. 2003, 2, 136–137. [Google Scholar] [CrossRef]
- Maspoch, D.; Ruiz-Molina, D.; Wurst, K.; Domingo, N.; Cavallini, M.; Biscarini, F.; Tejada, J.; Rovira, C.; Veciana, J. A nanoporous molecular magnet with reversible solvent-induced mechanical and magnetic properties. Nat. Mater. 2003, 2, 190–195. [Google Scholar] [CrossRef]
- Lin, Z.-J.; Lü, J.; Hong, M.; Cao, R. Metal–organic frameworks based on flexible ligands (FL-MOFs): Structures and applications. Chem. Soc. Rev. 2014, 43, 5867–5895. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.; Zhang, D.-S.; Chen, Q.; Li, R.-F.; Hu, T.-L.; Bu, X.-H. Rational construction of 3D pillared metal-organic frameworks: Synthesis, structures, and hydrogen adsorption properties. Inorg. Chem. 2011, 50, 7555–7562. [Google Scholar] [CrossRef]
- Montes-Andrés, H.; Leo, P.; Orcajo, G.; Rodríguez-Diéguez, A.; Choquesillo-Lazarte, D.; Martos, C.; Botas, J.Á.; Calleja, G. Synthesis, structural features, and hydrogen adsorption properties of three new flexible sulfur-containing metal–organic frameworks. Cryst. Growth Des. 2020, 20, 6707–6714. [Google Scholar] [CrossRef]
- Li, X.-L.; Liu, G.-Z.; Xin, L.-Y.; Wang, L.-Y. A novel metal–organic framework displaying reversibly shrinking and expanding pore modulation. CrystEngComm 2012, 14, 5757–5760. [Google Scholar] [CrossRef]
- Kitagawa, S.; Uemura, K. Dynamic porous properties of coordination polymers inspired by hydrogen bonds. Chem. Soc. Rev. 2005, 34, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Barthelet, K.; Marrot, J.; Riou, D.; Ferey, G. A breathing hybrid organic-inorganic solid with very large pores and high magnetic characteristics. Angew. Chem. Int. Ed. 2002, 41, 281–284. [Google Scholar] [CrossRef]
- Serre, C.; Millange, F.; Thouvenot, C.; Noguès, M.; Marsolier, G.; Louër, D.; Ferey, G. Very large breathing effect in the first nanoporous chromium (III)-based solids: MIL-53 or CrIII(OH)·{O2C-C6H4-CO2}·{HO2C-C6H4-CO2H}x·H2Oy. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef] [PubMed]
- Surble, S.; Serre, C.; Mellot-Draznieks, C.; Millangeand, F.; Ferey, G. A new isoreticular class of metal-organic-frameworks with the MIL-88 topology. Chem. Commun. 2006, 3, 284–286. [Google Scholar] [CrossRef]
- Serre, C.; Mellot-Draznieks, C.; Surble, S.; Audebrand, N.; Filinchuk, Y.; Ferey, G. Role of solvent-host interactions that lead to very large swelling of hybrid frameworks. Science 2007, 315, 1828–1831. [Google Scholar] [CrossRef] [Green Version]
- Broom, D.P.; Webb, C.J.; Fanourgakis, G.S.; Froudakis, G.E.; Trikalitis, P.N.; Hirscher, M. Concepts for improving hydrogen storage in nanoporous materials. Int. J. Hydrogen Energy 2019, 44, 7768–7779. [Google Scholar] [CrossRef]
- Lochan, R.C.; Head-Gordon, M. Computational studies of molecular hydrogen binding affinities: The role of dispersion forces, electrostatics, and orbital interactions. Phys. Chem. Chem. Phys. 2006, 8, 1357–1370. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Myers, A.L. Optimum Conditions for Adsorptive Storage. Langmuir 2006, 22, 1688–1700. [Google Scholar] [CrossRef]
- Frost, H.; Snurr, R.Q. Design Requirements for Metal-Organic Frameworks as Hydrogen Storage Materials. J. Phys. Chem. C 2007, 111, 18794–18803. [Google Scholar] [CrossRef]
- Bae, Y.-S.; Snurr, R.Q. Optimal isosteric heat of adsorption for hydrogen storage and delivery using metal-organic frameworks. Microp. Mesopor. Mater. 2010, 132, 300–303. [Google Scholar] [CrossRef]
- Li, B.; Chrzanowski, M.; Zhang, Y.; Ma, S. Application of metal-organic frameworks featuring multi-functional sites. Coord. Chem. Rev. 2016, 307, 106–129. [Google Scholar] [CrossRef] [Green Version]
- Dinca, M.; Long, J.R. High-enthalpy hydrogen adsorption in cation-exchanged variants of the microporous metal-organic framework Mn3[(Mn4Cl)3(BTT)8(CH3OH)10]2. J. Am. Chem. Soc. 2007, 129, 11172–11176. [Google Scholar] [CrossRef]
- Nouar, F.; Eckert, J.; Eubank, J.F.; Forster, P.; Eddaoudi, M.J. Zeolite-like metal−organic frameworks (zmofs) as hydrogen storage platform: Lithium and magnesium ion-exchange and H2-(rho-ZMOF) interaction studies. Am. Chem. Soc. 2009, 131, 2864–2870. [Google Scholar] [CrossRef]
- Cheon, Y.E.; Suh, M.P. Selective gas adsorption in a microporous metal–organic framework constructed of CoII4 clusters. Chem. Commun. 2009, 45, 2296–2298. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhou, H.-C. A metal−organic framework with entatic metal centers exhibiting high gas adsorption affinity. J. Am. Chem. Soc. 2006, 128, 11734–11735. [Google Scholar] [CrossRef]
- Kapelewski, M.T.; Runčevski, T.; Tarver, J.D.; Jiang, H.Z.H.; Hurst, K.E.; Parilla, P.A.; Ayala, A.; Gennett, T.; FitzGerald, S.A.; Brown, C.M.; et al. Record high hydrogen storage capacity in the metal-organic framework ni2(m-dobdc) at near-ambient temperatures. Chem. Mater. 2018, 30, 8179–8189. [Google Scholar] [CrossRef] [PubMed]
- Kosal, M.E.; Chou, J.-H.; Wilson, S.R.; Suslick, K.S. A functional zeolite analogue assembled from metalloporphyrins. Nat. Mater. 2002, 1, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, B.; Hutin, M.; Bulach, V.; Hosseini, M.W.; Cian, A.D.; Kyritsakas, N. Coordination polymers based on porphyrin and copper: The influence of the crystallization solvents on the dimensionality of the network. New J. Chem. 2002, 26, 1532–1535. [Google Scholar] [CrossRef]
- Deiters, E.; Bulach, V.; Hosseini, M.W. Reversible single-crystal-to-single-crystal guest exchange in a 3-D coordination network based on a zinc porphyrin. Chem. Commun. 2005, 31, 3906–3908. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Bulach, V.; Hosseini, M.W. Molecular tectonics: Control of pore size and polarity in 3-D hexagonal coordination networks based on porphyrins and a zinc cation. Chem. Commun. 2008, 41, 5104–5106. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, T.; Usuki, A.; Fukumori, K.; Ohta, T.; Ito, M.; Tatsumi, K. New porphyrin-based metal−organic framework with high porosity: 2-d infinite 22.2-Å square-grid coordination network. Inorg. Chem. 2006, 45, 7988–7990. [Google Scholar] [CrossRef]
- Alkordi, M.H.; Liu, Y.L.; Larsen, R.W.; Eubank, J.F.; Eddaoudi, M. Zeolite-like metal−organic frameworks as platforms for applications: On metalloporphyrin-based catalysts. J. Am. Chem. Soc. 2008, 130, 12639–12641. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, Y.; Jiang, D.L. CMPs as scaffolds for constructing porous catalytic frameworks: A built-in heterogeneous catalyst with high activity and selectivity based on nanoporous metalloporphyrin polymers. J. Am. Chem. Soc. 2010, 132, 9138–9143. [Google Scholar] [CrossRef] [PubMed]
- Farha, O.K.; Shultz, A.M.; Sarjeant, A.A.; Nguyen, S.T.; Hupp, J.T. Active-site-accessible, porphyrinic metal-organic framework materials. J. Am. Chem. Soc. 2011, 133, 5652–5655. [Google Scholar] [CrossRef]
- Song, F.; Wang, C.; Falkowski, J.M.; Ma, L.; Lin, W.J. Isoreticular chiral metal−organic frameworks for asymmetric alkene epoxidation: Tuning catalytic activity by controlling framework catenation and varying open channel sizes. Am. Chem. Soc. 2010, 132, 15390–15398. [Google Scholar] [CrossRef]
- Kitaura, R.; Onoyama, G.; Sakamoto, H.; Matsuda, R.; Noro, S.; Kitagawa, S. Immobilization of a metallo schiff base into a microporous coordination polymer. Angew. Chem. Int. Ed. 2004, 43, 2684–2687. [Google Scholar] [CrossRef]
- Wu, H.-B.; Wang, Q.-M. Construction of heterometallic cages with tripodal metalloligands. Angew. Chem. Int. Ed. 2009, 48, 7343–7345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, H.; Matsuda, R.; Bureekaew, S.; Tanaka, D.; Kitagawa, S. A porous coordination polymer with accessible metal sites and its complementary coordination action. Chem. Eur. J. 2009, 15, 4985–4989. [Google Scholar] [CrossRef]
- Roesky, P.W.; Bhunia, A.; Lan, Y.; Powell, A.K.; Kureti, S. Salen-based metal-organic frameworks of nickel and the lanthanides. Chem. Commun. 2011, 47, 2035–2037. [Google Scholar] [CrossRef] [PubMed]
- Shultz, A.M.; Farha, O.K.; Adhikari, D.; Sarjeant, A.A.; Hupp, J.T.; Nguyen, S.T. Selective surface and near-surface modification of a noncatenated, catalytically active metal-organic framework material based on Mn(salen) struts. Inorg. Chem. 2011, 50, 3174–3176. [Google Scholar] [CrossRef] [PubMed]
- Shultz, A.M.; Sarjeant, A.A.; Farha, O.K.; Hupp, J.T.; Nguyen, S.T. Post-synthesis modification of a metalorganic framework to form metallosalen-containing mof materials. J. Am. Chem. Soc. 2011, 133, 13252–13255. [Google Scholar] [CrossRef]
- Wu, C.-D.; Hu, A.; Zhang, L.; Lin, W.J. A homochiral porous metal-organic framework for highly enantioselective heterogeneous asymmetric catalysis. J. Am. Chem. Soc. 2005, 127, 8940–8941. [Google Scholar] [CrossRef] [PubMed]
- Oisaki, K.; Li, Q.; Furukawa, H.; Czaja, A.U.; Yaghi, O.M. A metal-organic framework with covalently bound organometallic complexes. J. Am. Chem. Soc. 2010, 132, 9262–9264. [Google Scholar] [CrossRef] [PubMed]
- Bloch, E.D.; Britt, D.; Lee, C.; Doonan, C.; Uribe-Romo, F.J.; Furukawa, H.; Long, J.R.; Yaghi, O.M. Metal insertion in a microporous metal−organic framework lined with 2,2′-bipyridine. J. Am. Chem. Soc. 2010, 132, 14382–14384. [Google Scholar] [CrossRef]
- Gadzikwa, T.; Farha, O.K.; Mulfort, K.L.; Hupp, J.T.; Nguyen, S.T. A Zn-based, pillared paddlewheel MOF containing free carboxylic acids via covalent post-synthesis elaboration. Chem. Commun. 2009, 45, 3720–3722. [Google Scholar] [CrossRef]
- Doonan, C.J.; Morris, W.; Furukawa, H.; Yaghi, O.M. Isoreticular metalation of metal-organic frameworks. J. Am. Chem. Soc. 2009, 131, 9492–9493. [Google Scholar] [CrossRef]
- Kaye, S.S.; Long, J.R. Matrix isolation chemistry in a porous metal−organic framework: photochemical substitutions of N2 and H2 in Zn4O [(η6-1,4-benzenedicarboxylate)Cr(CO)3]3. J. Am. Chem. Soc. 2008, 130, 806–807. [Google Scholar] [CrossRef]
- Dinca, M.; Long, J.R. Structure and charge control in metal–organic frameworks based on the tetrahedral ligand tetrakis(4-tetrazolylphenyl)methane. Chem. Eur. J. 2008, 14, 10280–10285. [Google Scholar] [CrossRef]
- Allendorf, M.D.; Hulvey, Z.; Gennett, T.; Ahmed, A.; Autrey, T.; Camp, J.; Cho, E.S.; Furukawa, H.; Haranczyk, M.; Head-Gordon, M.; et al. An assessment of strategies for the development of solid-state adsorbents for vehicular hydrogen storage. Energy Environ. Sci. 2018, 11, 2784–2812. [Google Scholar] [CrossRef] [Green Version]
- Botas, J.A.; Calleja, G.; Sanchez, M.; Orcajo, M.G. Cobalt doping of the MOF-5 framework and its effect on gas-adsorption properties. Langmuir 2010, 26, 5300–5303. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ma, Y.; Song, W.-C.; Chang, Z.; Li, J.-R.; Zhang, J.; Sun, H.-W.; Balbuena, P.B.; Bu, X.-H. Why porous materials have selective adsorptions: A rational aspect from electrodynamics. Inorg. Chem. 2017, 56, 2614–2620. [Google Scholar] [CrossRef]
- Zhu, Q.-L.; Xu, Q. Metal–organic framework composites. Chem. Soc. Rev. 2014, 43, 5468–5512. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Sumby, C.J.; Doonan, C.J. Post-synthetic metalation of metal-organic frameworks. Chem. Soc. Rev. 2014, 43, 5933–5951. [Google Scholar] [CrossRef]
- Kobayashi, H.; Mitsuka, Y.; Kitagawa, H. Metal nanoparticles covered with a metal-organic framework: From one-pot synthetic methods to synergistic energy storage and conversion functions. Inorg. Chem. 2016, 55, 7301–7310. [Google Scholar] [CrossRef]
- Railey, P.; Song, Y.; Liu, T.; Li, Y. Metal organic frameworks with immobilized nanoparticles: Synthesis and applications in photocatalytic hydrogen generation and energy storage. Mater. Res. Bull. 2017, 96, 385–394. [Google Scholar] [CrossRef]
- Ishida, T.; Nagaoka, M.; Akita, T.; Haruta, M. Deposition of gold clusters on porous coordination polymers by solid grinding and their catalytic activity in aerobic oxidation of alcohols. Chem. Eur. J. 2008, 14, 8456–8460. [Google Scholar] [CrossRef]
- Pan, H.; Li, X.; Zhang, D.; Guan, Y.; Wu, P. Pt nanoparticles entrapped in mesoporous metal-organic frameworks MIL-101 as an efficient and recyclable catalyst for the asymmetric hydrogenation of α-ketoesters. J. Mol. Catal. A Chem. 2013, 377, 108–114. [Google Scholar] [CrossRef]
- Khajavi, H.; Stil, H.A.; Kuipers, H.; Gascon, J.; Kapteijn, F. Shape and transition state selective hydrogenations using egg-shell Pt-MIL-101(Cr) catalyst. ACS Catal. 2013, 3, 2617–2626. [Google Scholar] [CrossRef]
- Du, W.; Chen, G.; Nie, R.; Li, Y.; Hou, Z. Highly dispersed Pt in MIL-101: An efficient catalyst for the hydrogenation of nitroarenes. Catal. Commun. 2013, 41, 56–59. [Google Scholar] [CrossRef]
- Yadav, M.; Xu, Q. Catalytic chromium reduction using formic acid and metal nanoparticles immobilized in a metal-organic framework. Chem. Commun. 2013, 49, 3327–3329. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Xu, Q. Metal-organic framework supported bimetallic Ni–Pt nanoparticles as high-performance catalysts for hydrogen generation from hydrazine in aqueous solution. ChemCatChem 2013, 5, 3000–3004. [Google Scholar] [CrossRef]
- Hermes, S.; Schrӧter, M.-K.; Schmid, R.; Khodeir, L.; Muhler, M.; Tissler, A.; Fischer, R.W.; Fischer, R.A. Metal@MOF: Loading of highly porous coordination polymers host lattices by metal organic chemical vapor deposition. Angew. Chem. Int. Ed. 2005, 44, 6237–6241. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Zhuang, J.; Chou, L.Y.; Lee, H.K.; Ling, X.Y.; Chuang, Y.C.; Tsung, C.K. Surfactant-directed atomic to mesoscale alignment: Metal nanocrystals encased individually in single-crystalline porous nanostructures. J. Am. Chem. Soc. 2014, 136, 10561–10564. [Google Scholar] [CrossRef] [PubMed]
- Ohhashi, T.; Tsuruoka, T.; Matsuyama, T.; Takashima, Y.; Nawafune, H.; Minami, H.; Akamatsu, K.J. Metal nanocrystal/metal-organic framework core/shell nanostructure from selective self-assembly induced by localization of metal ion precursors on nanocrystal surface. Colloid Interface Sci. 2015, 451, 212–215. [Google Scholar] [CrossRef]
- Li, G.; Kobayashi, H.; Taylor, J.M.; Ikeda, R.; Kubota, Y.; Kato, K.; Takata, M.; Yamamoto, T.; Toh, S.; Matsumura, S.; et al. Hydrogen storage in Pd nanocrystals covered with a metal-organic framework. Nat. Mater. 2014, 13, 802–806. [Google Scholar] [CrossRef]
- Wang, Q.; Astruc, D. State of the art and prospects in metal-organic framework (MOF)based and MOF-derived nanocatalysis. Chem. Rev. 2020, 120, 1438–1511. [Google Scholar] [CrossRef]
- Zlotea, C.; Campesi, R.; Cuevas, F.; Leroy, E.; Dibandjo, P.; Volkringer, C.; Loiseau, T.; Ferey, G.; Latroche, M. Pd nanoparticles embedded into a metal-organic framework: Synthesis, structural characteristics, and hydrogen sorption properties. J. Am. Chem. Soc. 2010, 132, 2991–2997. [Google Scholar] [CrossRef]
- Malouche, A.; Blanita, G.; Lupu, D.; Bourgon, J.; Nelayah, J.; Zlotea, C. Hydrogen absorption in 1 nm Pd clusters confined in MIL-101(Cr). J. Mater. Chem. A 2017, 5, 23043–23052. [Google Scholar] [CrossRef]
- Lestari, W.W.; Afifah, E.N.; Mohammed, O.; Saraswati, T.E.; Al-Adawiyah, R.; Kadja, G.T.M.; Widiastuti, N. DUT-5 modified Pd metal-nanoparticles: Synthesis, chemical stability, and hydrogen sorption studies. Mater. Res. Express 2019, 6, 1250d4. [Google Scholar] [CrossRef]
- King, J.; Zhang, L.; Doszczeczko, S.; Sambalova, O.; Luo, H.; Rohman, F.; Phillips, O.; Borgschulte, A.; Hirscher, M.; Addicoat, M.; et al. How to functionalise metal–organic frameworks to enable guest nanocluster embedment. J. Mater. Chem. A 2020, 8, 4889–4897. [Google Scholar] [CrossRef]
- Luzan, S.M.; Talyzin, A.V. Hydrogen adsorption in Pt catalyst/MOF-5 materials. Microporous Mesoporous Mater. 2010, 135, 201–205. [Google Scholar] [CrossRef]
- Campesi, R.; Cuevas, F.; Latroche, M.; Hirscher, M. Hydrogen spillover measurements of unbridged and bridged metal-organic frameworks—revisited. Phys. Chem. Chem. Phys. 2010, 12, 10457–10459. [Google Scholar] [CrossRef]
- Wang, L.; Stuckert, N.R.; Chen, H.; Yang, R.T. Effects of Pt particle size on hydrogen storage on Pt-doped metal−organic framework IRMOF-8. J. Phys. Chem. C 2011, 115, 4793–4799. [Google Scholar] [CrossRef]
- Proch, S.; Herrmannsdofer, J.; Kempe, R.; Kern, C.; Jess, A.; Seyfarth, L.; Senker, J. Pt@MOF-177: Synthesis, room-temperature hydrogen storage and oxidation catalysis. Chem. Eur. J. 2008, 14, 8204–8212. [Google Scholar] [CrossRef]
- Dixit, M.; Adit Maark, T.; Ghatak, K.; Ahuja, R.; Pal, S. Scandium-decorated MOF-5 as potential candidates for room temperature hydrogen storage: A solution for the clustering problem in MOFs. J. Phys. Chem. C 2012, 116, 17336–17342. [Google Scholar] [CrossRef]
- Mukoyoshi, M.; Kobayashi, H.; Kusada, K.; Hayashi, M.; Yamada, T.; Maesato, M.; Taylor, J.M.; Kubota, Y.; Kato, K.; Takata, M.; et al. Hybrid materials of Ni NP@MOF prepared by a simple synthetic method. Chem. Commun. 2015, 51, 12463–12466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.L.; Wang, R.; Zhang, M.Y.; Yuan, Y.P.; Xue, C. Dye-sensitized MIL-101 metal organic frameworks loaded with Ni/NiOx nanoparticles for efficient visible-light-driven hydrogen generation. Appl. Mater. 2015, 3, 104403. [Google Scholar] [CrossRef]
- Zhen, W.L.; Gao, H.B.; Tian, B.; Ma, J.T.; Lu, G.X. Fabrication of low adsorption energy Ni–Mo cluster cocatalyst in metal–organic frameworks for visible photocatalytic hydrogen evolution. ACS Appl. Mater. Interfaces 2016, 8, 10808–10819. [Google Scholar] [CrossRef] [PubMed]
- Zhen, W.L.; Ma, J.T.; Lu, G.X. Small-sized Ni(111) particles in metal-organic frameworks with low over-potential for visible photocatalytic hydrogen generation. Appl. Catal. B Environ. 2016, 190, 12–25. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, A.-F.; Cao, C.-S.; Zhao, B. Applications of MOFs: Recent advances in photocatalytic hydrogen production from water. Coord. Chem. Rev. 2019, 390, 50–75. [Google Scholar] [CrossRef]
- Zhao, D.; Yan, D.; Zhou, H.-C. The current status of hydrogenstorage in metal-organic frameworks. Energy Environ. Sci. 2008, 1, 222–235. [Google Scholar] [CrossRef]
- Li, Y.; Xie, L.; Liu, Y.; Yang, R.; Li, X. Favorable hydrogen storage properties of M(HBTC)(4,4′-bipy)·3DMF (M = Ni and Co). Inorg. Chem. 2008, 47, 10372–10377. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Jung, H.; Koo, G.; Jeong, H.; Kim, D.-K. Efficient hydrogen sorption in 8-connected MOFs based on trinuclear pinwheel motifs. Inorg. Chem. 2008, 47, 5355–5359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tanabe, K.K.; Cohen, S.M. Tuning hydrogen sorption properties of metal–organic frameworks by postsynthetic covalent modification. Chem. Eur. J. 2010, 16, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.S.; Chang, Z.; Li, Y.F.; Jiang, Z.Y.; Xuan, Z.H.; Zhang, Y.H.; Li, J.R.; Chen, Q.; Hu, T.L.; Bu, X.H. Fluorous metal-organic frameworks with enhanced stability and high H2/CO2 storage capacities. Sci. Rep. 2013, 3, 3312. [Google Scholar] [CrossRef]
- Rostami, S.; Pour, A.N.; Salimi, A.; Abolghasempour, A. Hydrogen adsorption in metal- organic frameworks (MOFs): Effects of adsorbent architecture. Int. J. Hydrogen Energy 2018, 43, 7072–7080. [Google Scholar] [CrossRef]
- Stavila, V.; Bhakta, R.K.; Alam, T.M.; Majzoub, E.H.; Allendorf, M.D. Reversible hydrogen storage by NaAlH4 confined within a titanium-functionalized MOF-74(Mg) nanoreactor. ACS Nano 2012, 6, 9807–9817. [Google Scholar] [CrossRef]
- Callini, E.; Szilágyi, P.Á.; Paskevicius, M.; Stadie, N.P.; Réhault, J.; Buckley, C.E.; Borgschulte, A.; Züttel, A. Stabilization of volatile Ti(BH4)3 by nano-confinement in a metal-organic framework. Chem. Sci. 2016, 7, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Hadjiivanov, K.I.; Panayotov, D.A.; Mihaylov, M.Y.; Ivanova, E.Z.; Chakarova, K.K.; Andonova, S.M.; Drenchev, N.L. Power of infrared and raman spectroscopies to characterize metal-organic frameworks and investigate their interaction with guest molecules. Chem. Rev. 2021, 121, 1286–1424. [Google Scholar] [CrossRef] [PubMed]
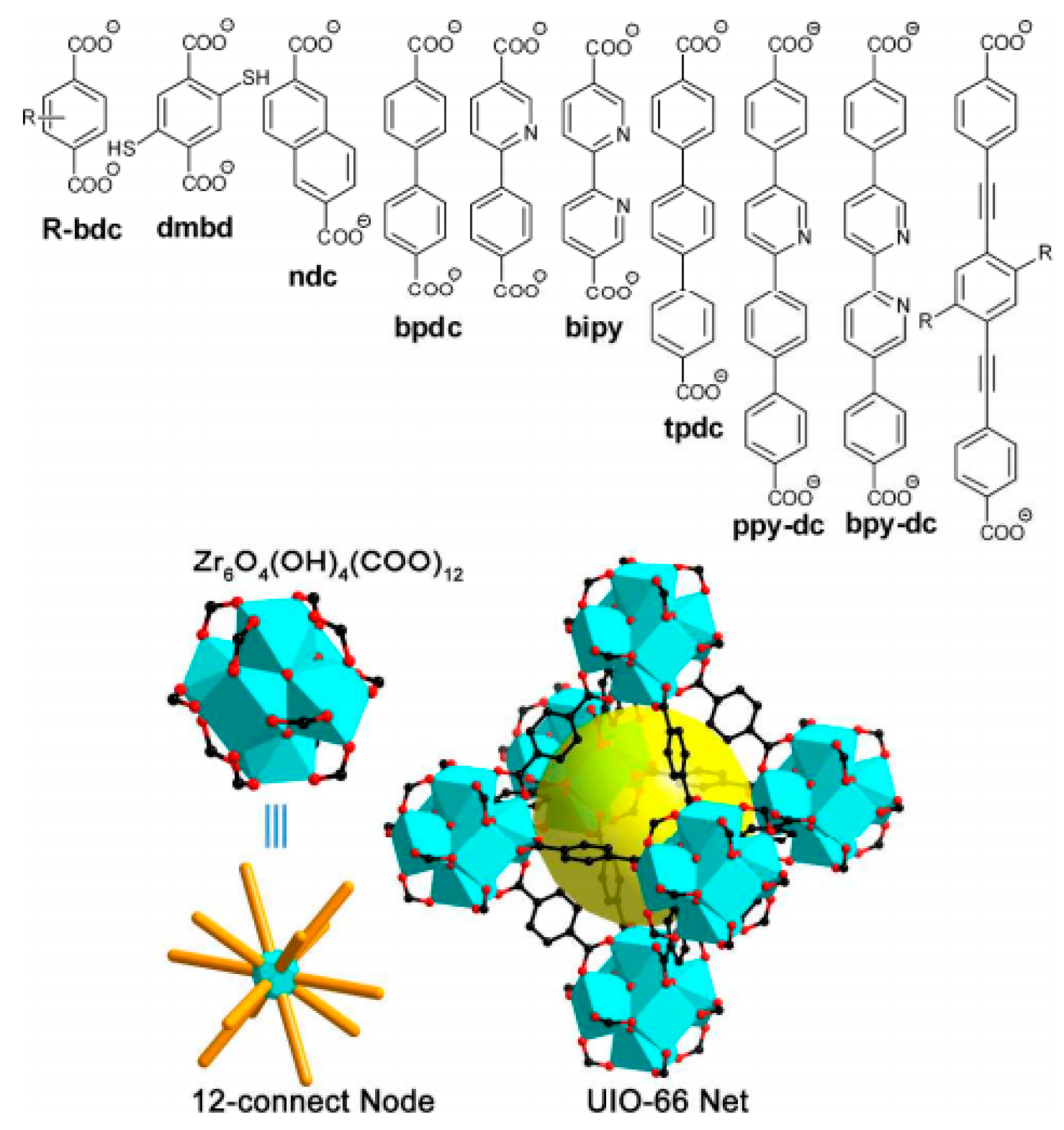


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
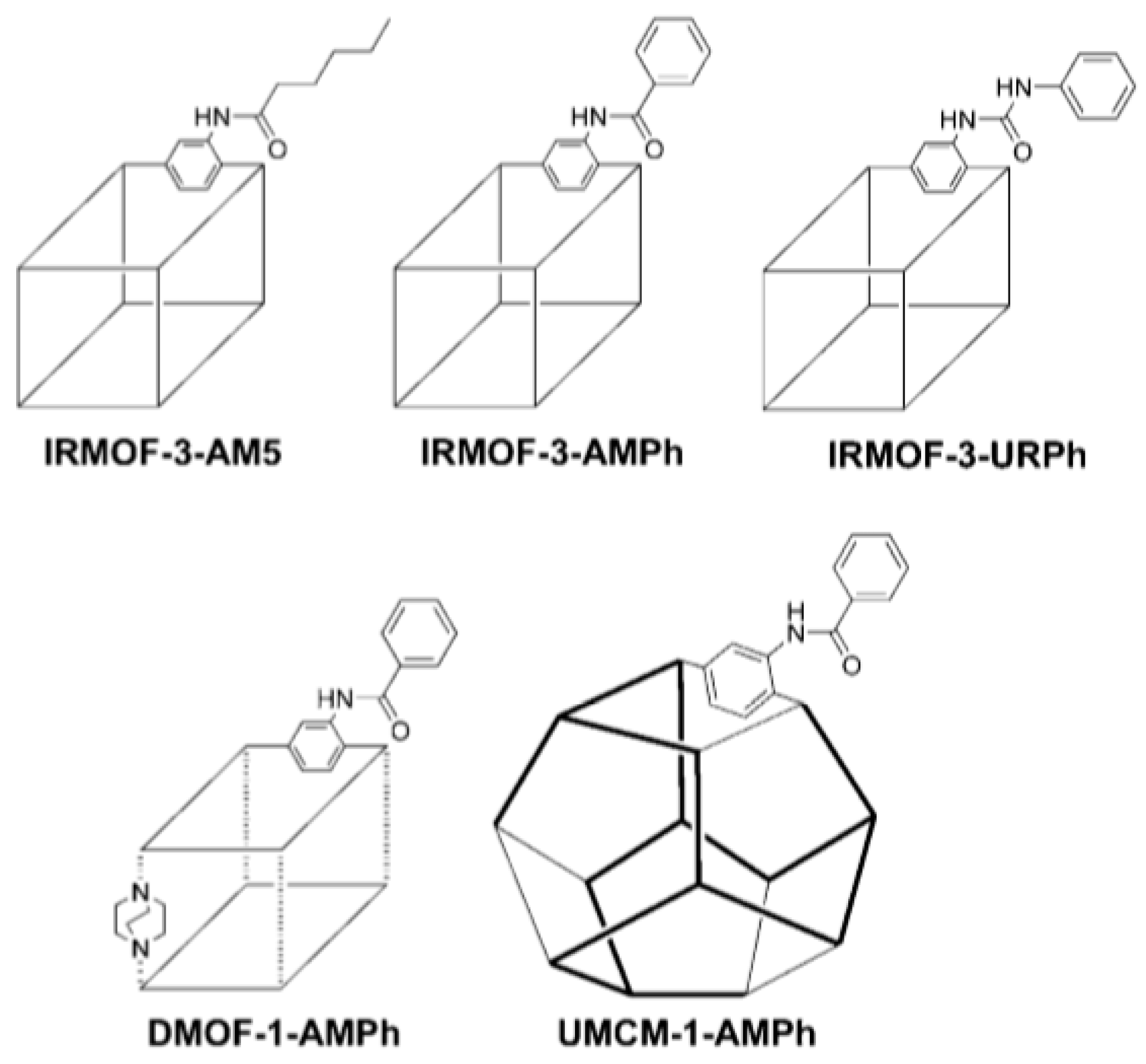{kind=link}
| MOF-5 Sample | As, m2·g–1 | Va, cm3·g–1 | Pore Size, Å | wt% of H2 | Tdec, K |
|---|---|---|---|---|---|
| Low-crystalline | 2050 | 0.88 | ~6 and ~13 | 1.2 | 711 |
| High-crystalline | 3400 | 1.42 | ~7 and ~14 | 1.3 | 755 |
| Interwoven | 1010 | 0.51 | ~6 | 1.7 | 773 |
| Interwoven with incorporated MWCNTs | 1170 | 0.57 | ~6 | 2.0 | 783 |
| MOF Sample | Organic Linker | Pore Characteristics | H2 Adsorption, wt% | |
|---|---|---|---|---|
| As, m2·g–1 | Va, cm3·g–1 | |||
| IRMOF-1 | 1,4-benzenedicarboxylate | 3362 | ---- | 1.32 |
| IRMOF-2 | 2-bromobenzene-1,4-dicarboxylate | 1722 | 0.88 | 1.21 |
| IRMOF-3 | 2-aminobenzene-1,4-dicarboxylate | 2446 | 1.07 | 1.42 |
| IRMOF-6 | 1,2-dihydrocyclobutabenzene-3,6-dicarboxylate | 2476 | 1.14 | 1.48 |
| IRMOF-8 | Naphthalenedicarboxylate | 890 | 0.45 | 1.45 |
| IRMOF-9 | 4,4′-biphenyldicarboxylate | 1904 | 0.9 | 1.17 |
| IRMOF-11 | 4,5,9,10-tetrahydropyrene-2,7-dicarboxylate | ---- | ---- | 1.62 |
| IRMOF-13 | pyrene-2,7-dicarboxylate | 1551 | 0.73 | 1.73 |
| IRMOF-16 | p-terphenyl-4,4′-dicarboxylate | ---- | ---- | ---- |
| IRMOF-18 | 2,3,5,6-tetramethylbenzene-1,4-dicarboxylate | 1501 | ---- | 0.89 |
| IRMOF-20 | thieno[3,2-b]thiophen-2,5-dicarboxylate | 3409 | 1.53 | 1.35 |
| Temperature, K | Catenated PCN-6 | Non-Catenated PCN-60 | ||
|---|---|---|---|---|
| na, wt% of H2 | nσ, wt% of H2 | na, wt% of H2 | nσ, wt% of H2 | |
| 77 | 8.7 | 6.7 | 5.5 | 4.0 |
| 298 | 1.5 | 0.9 | 0.8 | 0.4 |
| NaAlH4 | Amount of Ti, mol% | Ea, kJ·mol–1 | H2 Adsorption, wt% |
|---|---|---|---|
| Bulk | 0 | 118.1 | 5.12 |
| Bulk | 2 | 79.5 | 4.25 |
| Infiltrated in MOF-74(Mg) | 3 | 57.4 | 4.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeleňák, V.; Saldan, I. Factors Affecting Hydrogen Adsorption in Metal–Organic Frameworks: A Short Review. Nanomaterials 2021, 11, 1638. https://doi.org/10.3390/nano11071638
Zeleňák V, Saldan I. Factors Affecting Hydrogen Adsorption in Metal–Organic Frameworks: A Short Review. Nanomaterials. 2021; 11(7):1638. https://doi.org/10.3390/nano11071638
Chicago/Turabian StyleZeleňák, Vladimír, and Ivan Saldan. 2021. "Factors Affecting Hydrogen Adsorption in Metal–Organic Frameworks: A Short Review" Nanomaterials 11, no. 7: 1638. https://doi.org/10.3390/nano11071638
APA StyleZeleňák, V., & Saldan, I. (2021). Factors Affecting Hydrogen Adsorption in Metal–Organic Frameworks: A Short Review. Nanomaterials, 11(7), 1638. https://doi.org/10.3390/nano11071638