
Modeling the Layer-by-Layer Growth of HKUST-1 Metal-Organic Framework Thin Films


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model and Methods
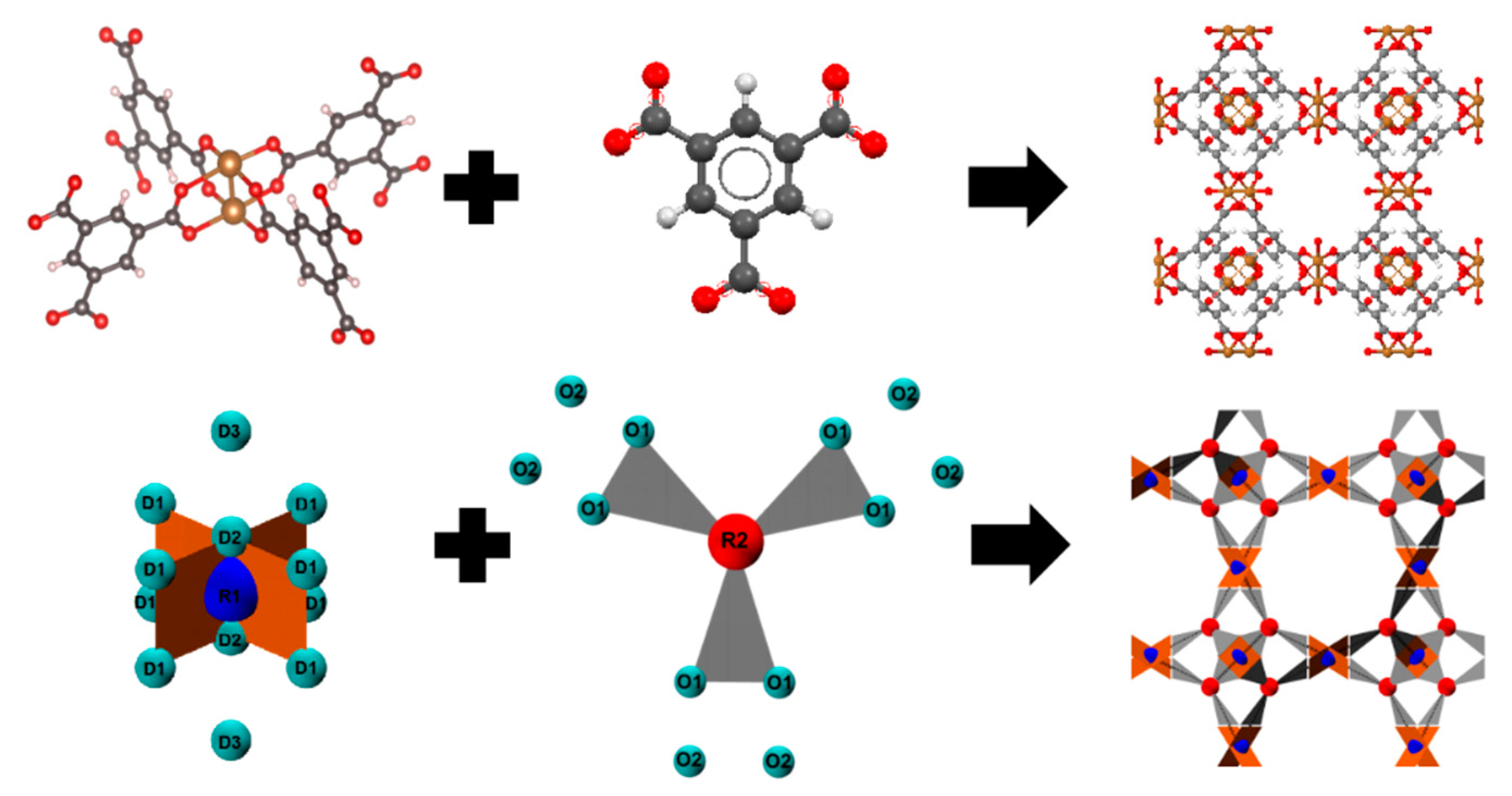
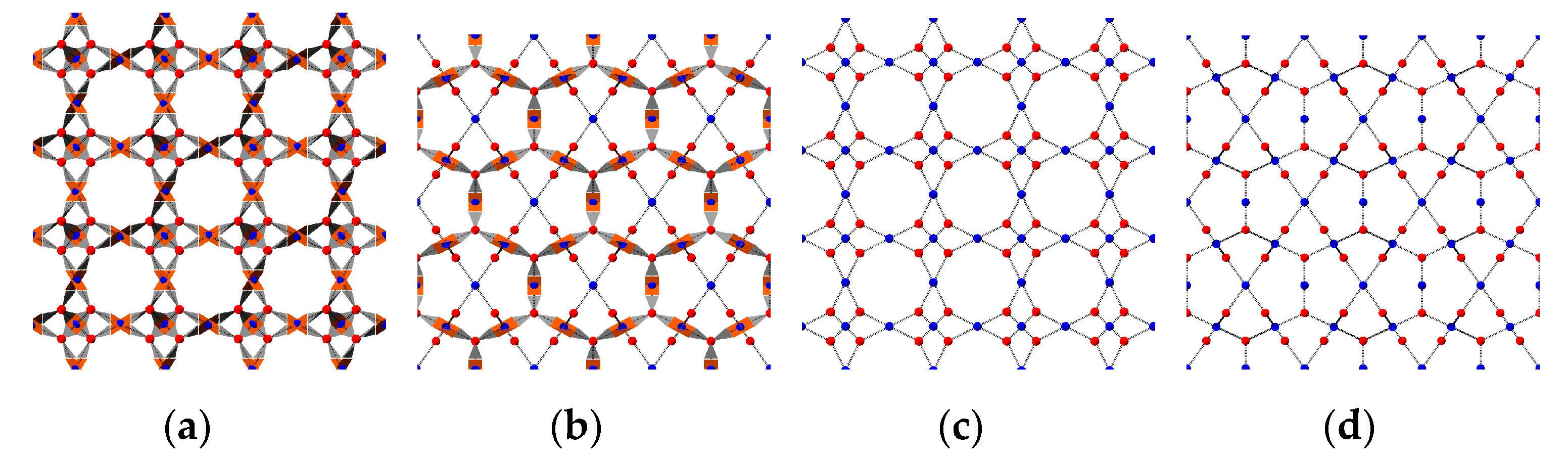
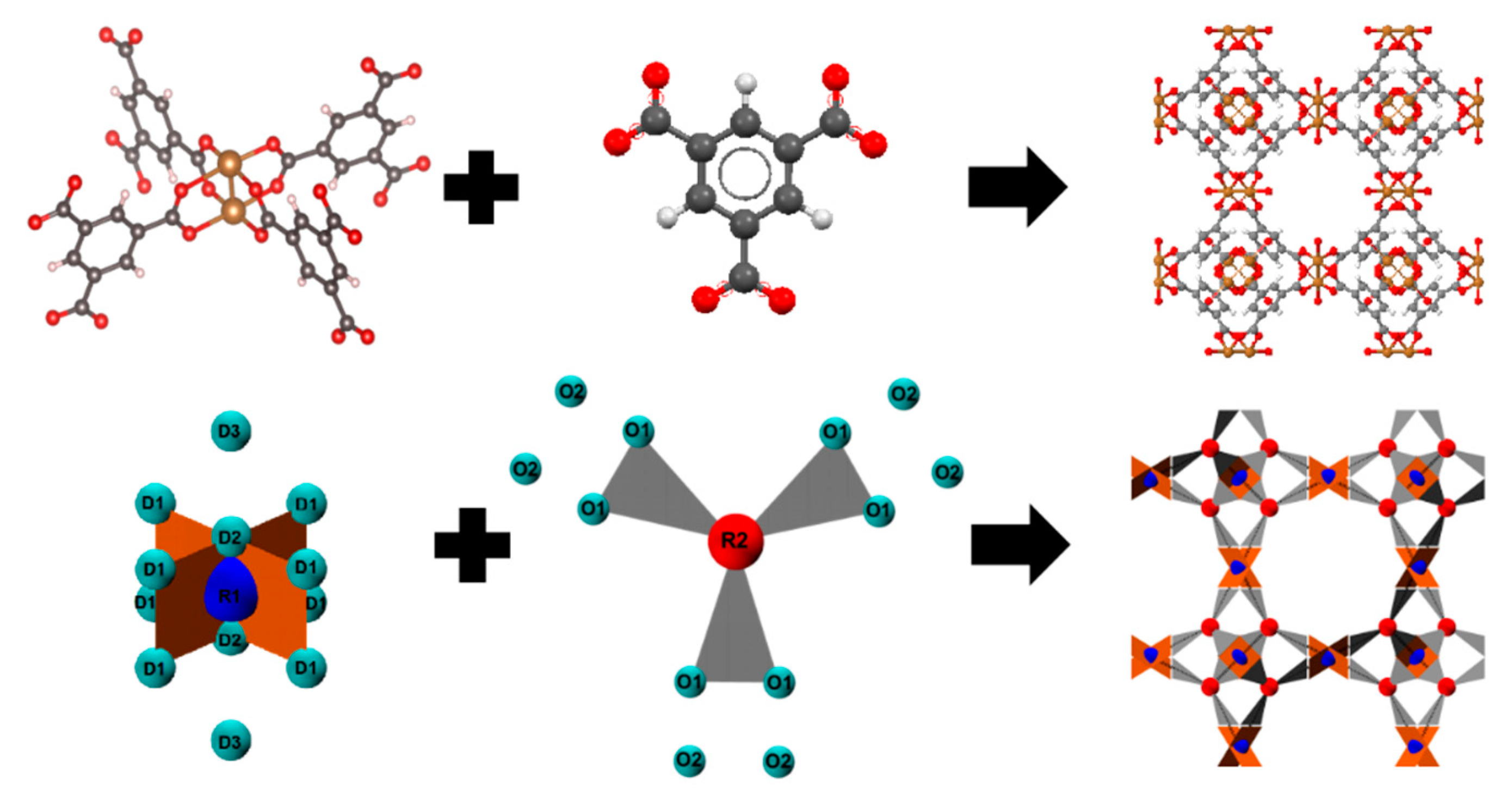
2.1. Patchy Models
2.2. Force Field
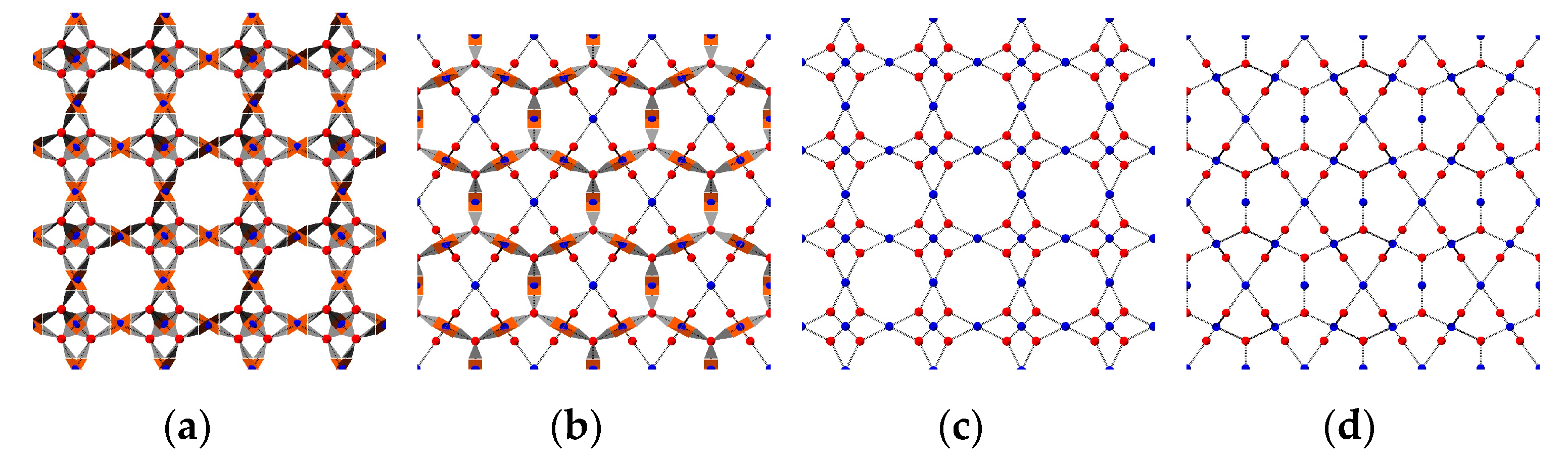
2.3. Crystallinity Quantification Algorithm
3. Results and Discussion
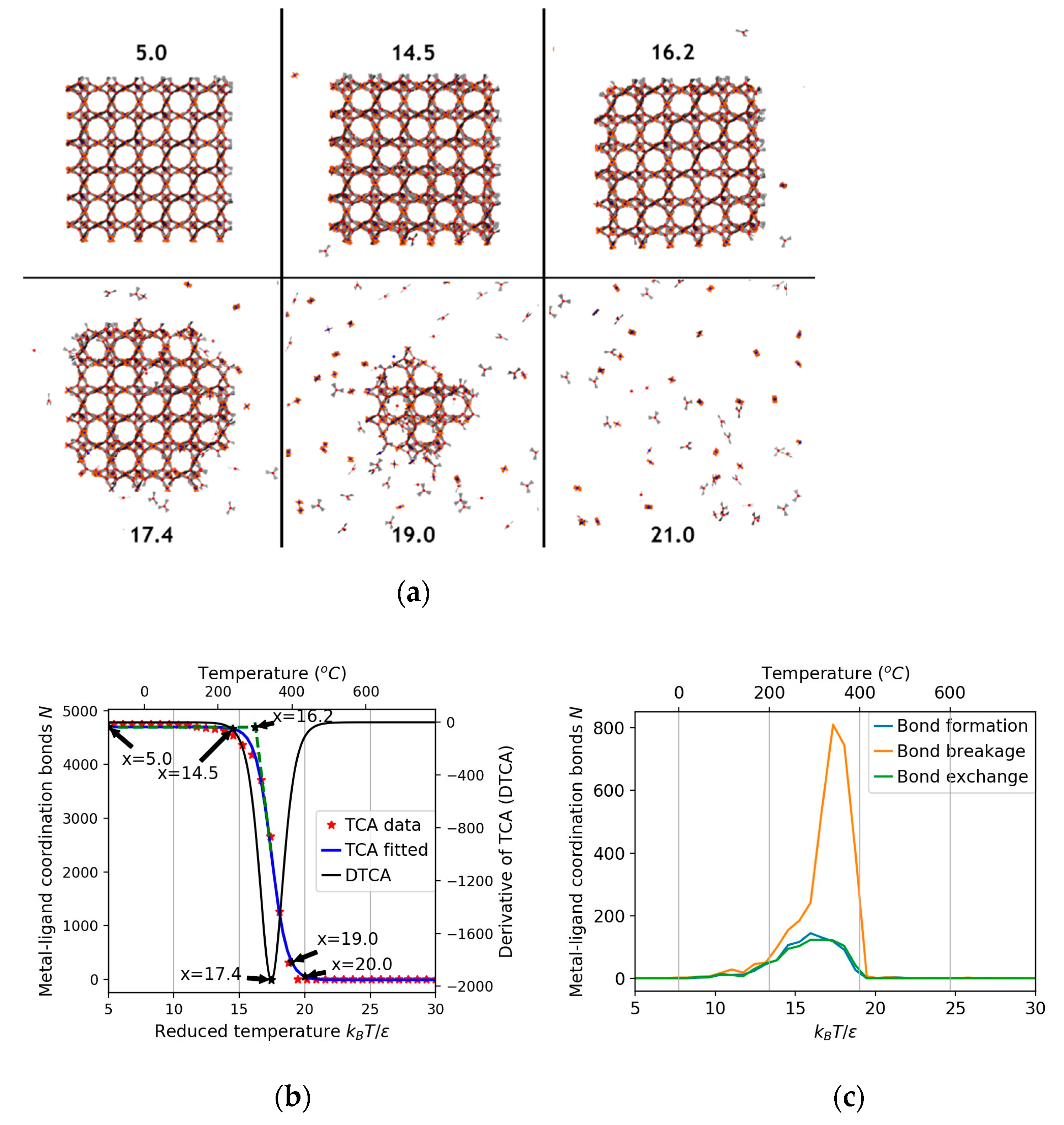
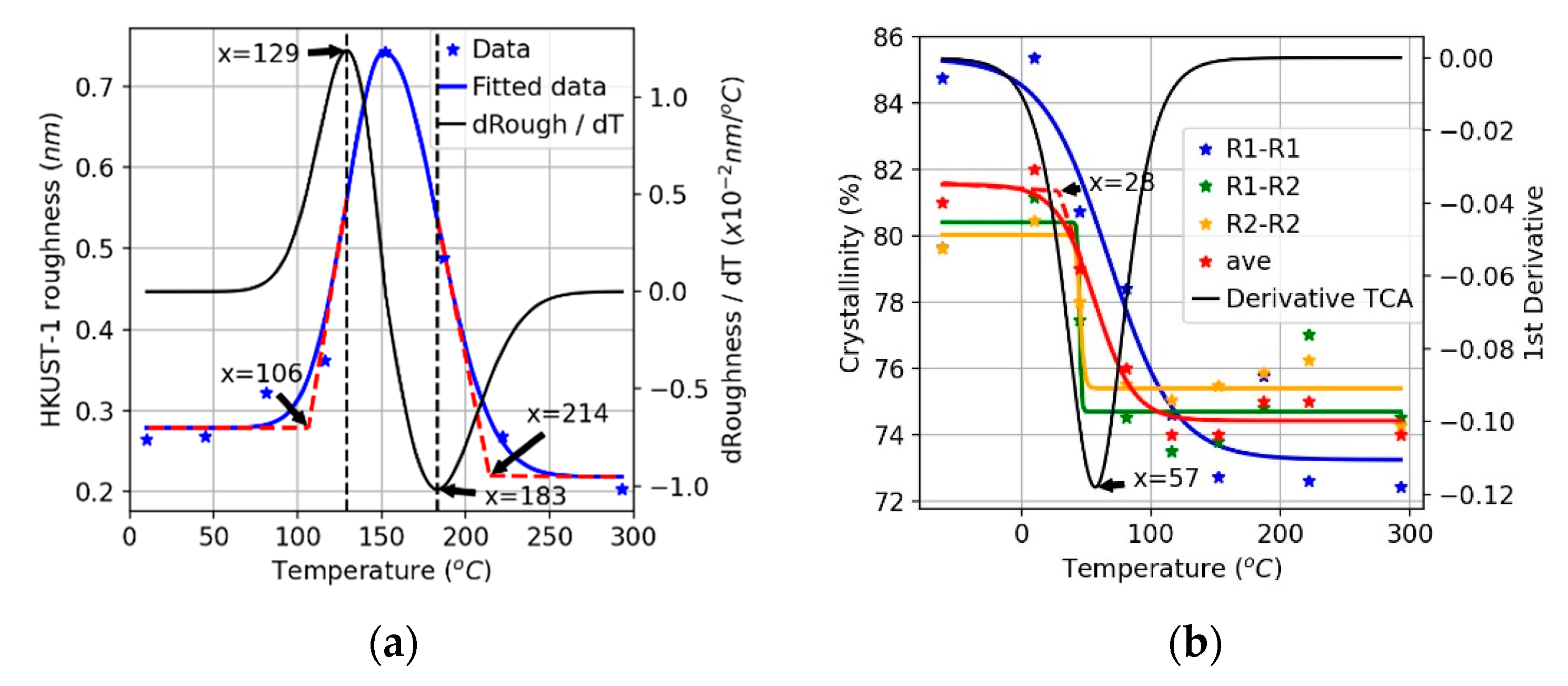
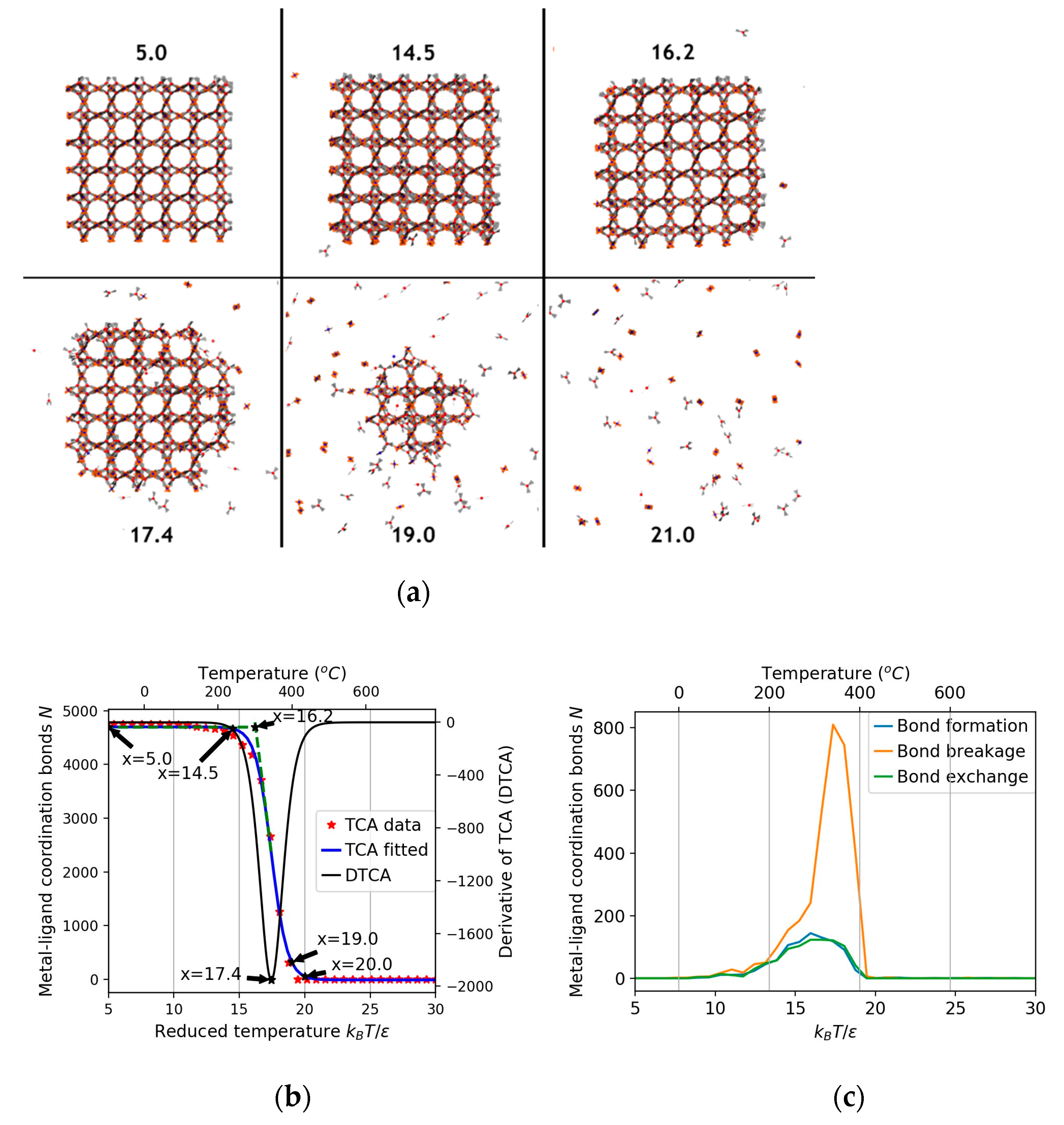
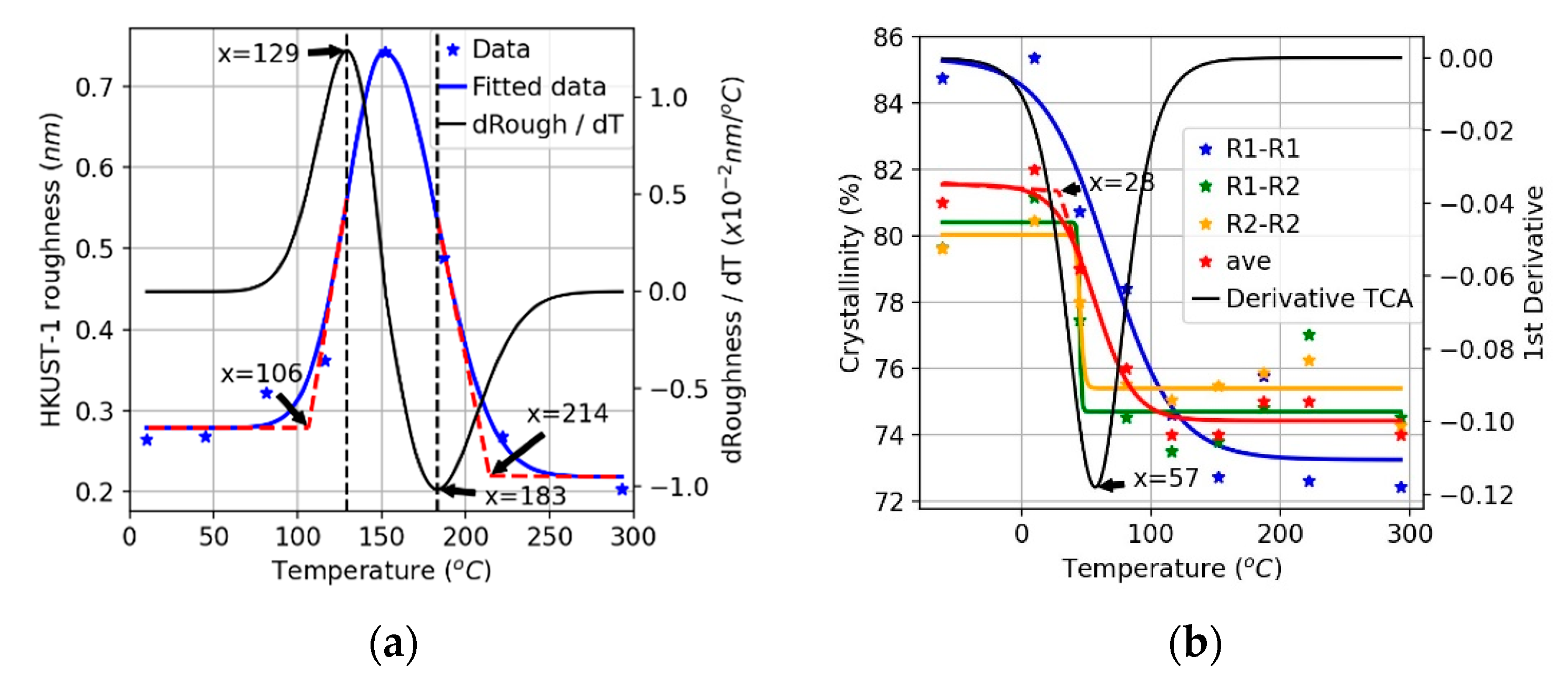
3.1. Thermal Stability Analysis
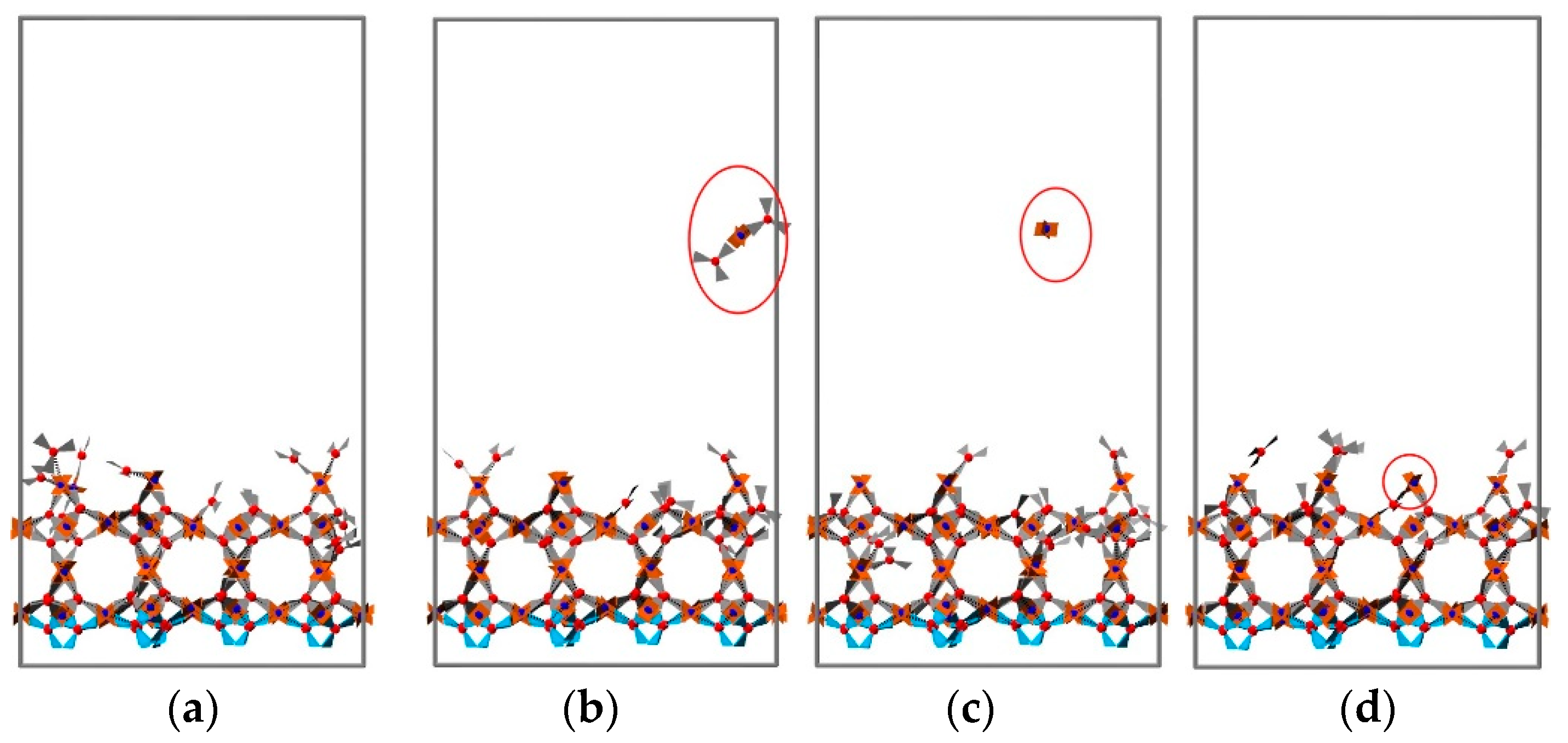
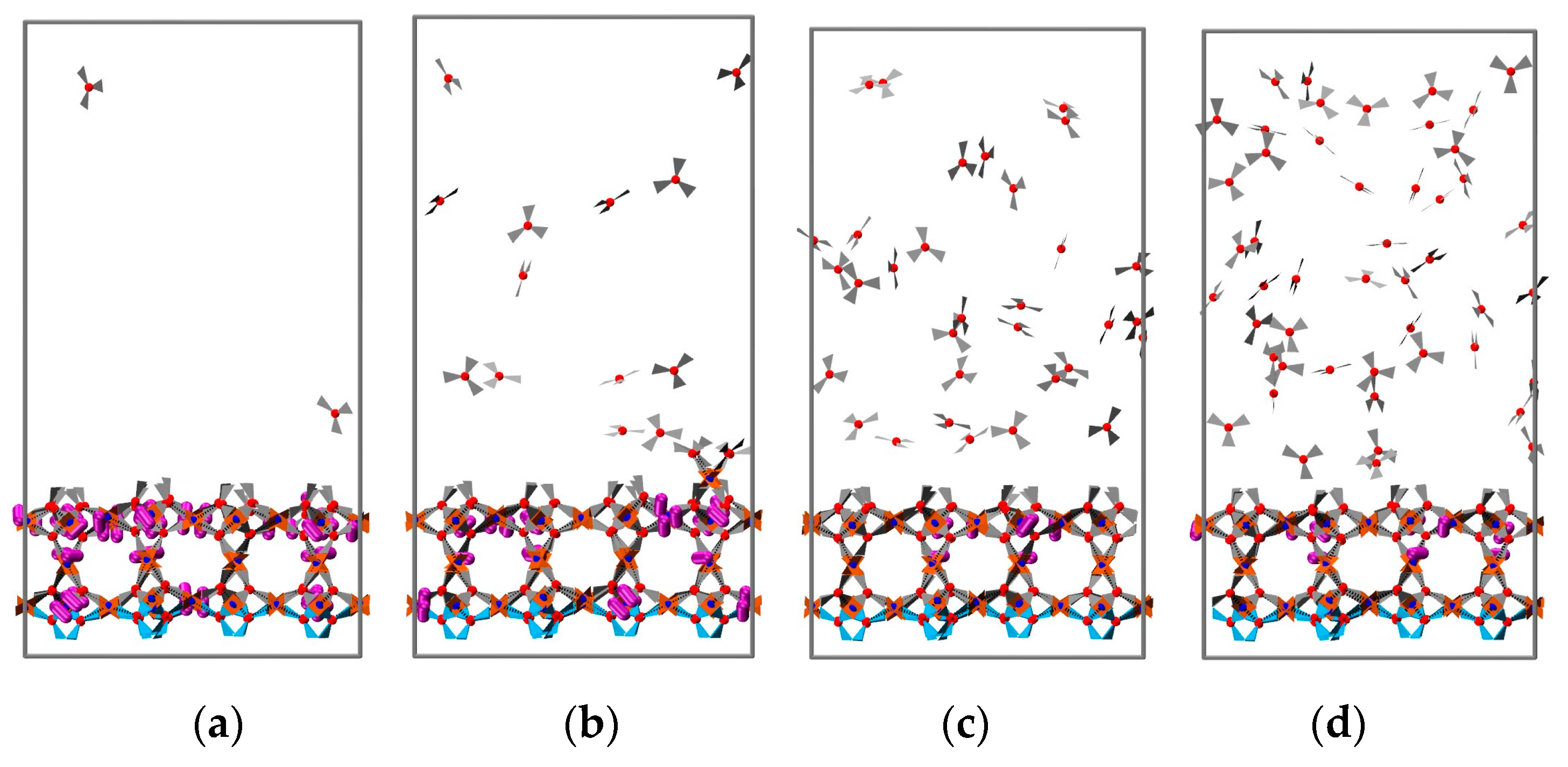
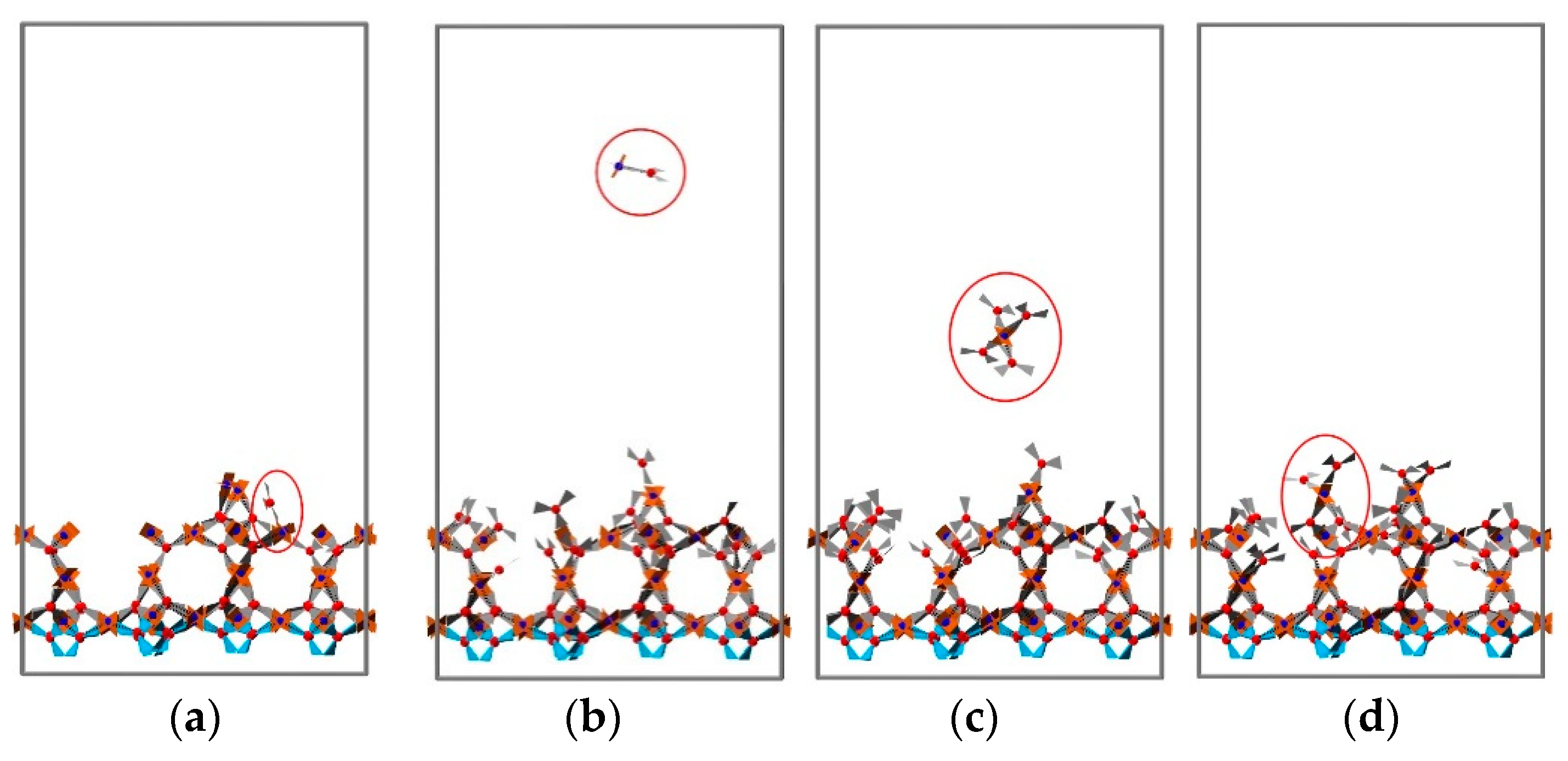
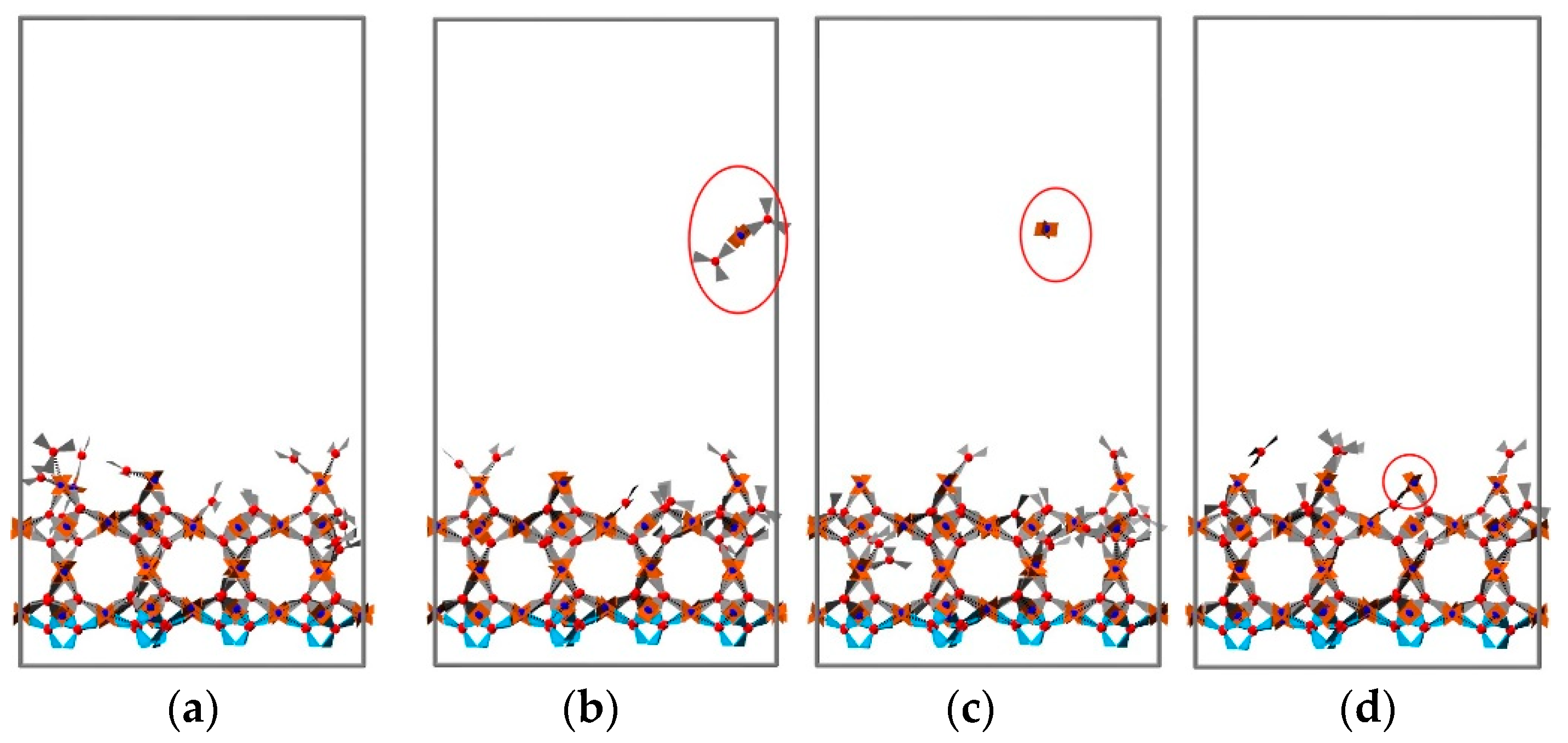
3.2. 2D Growth of HKUST-1 on Planar Substrates
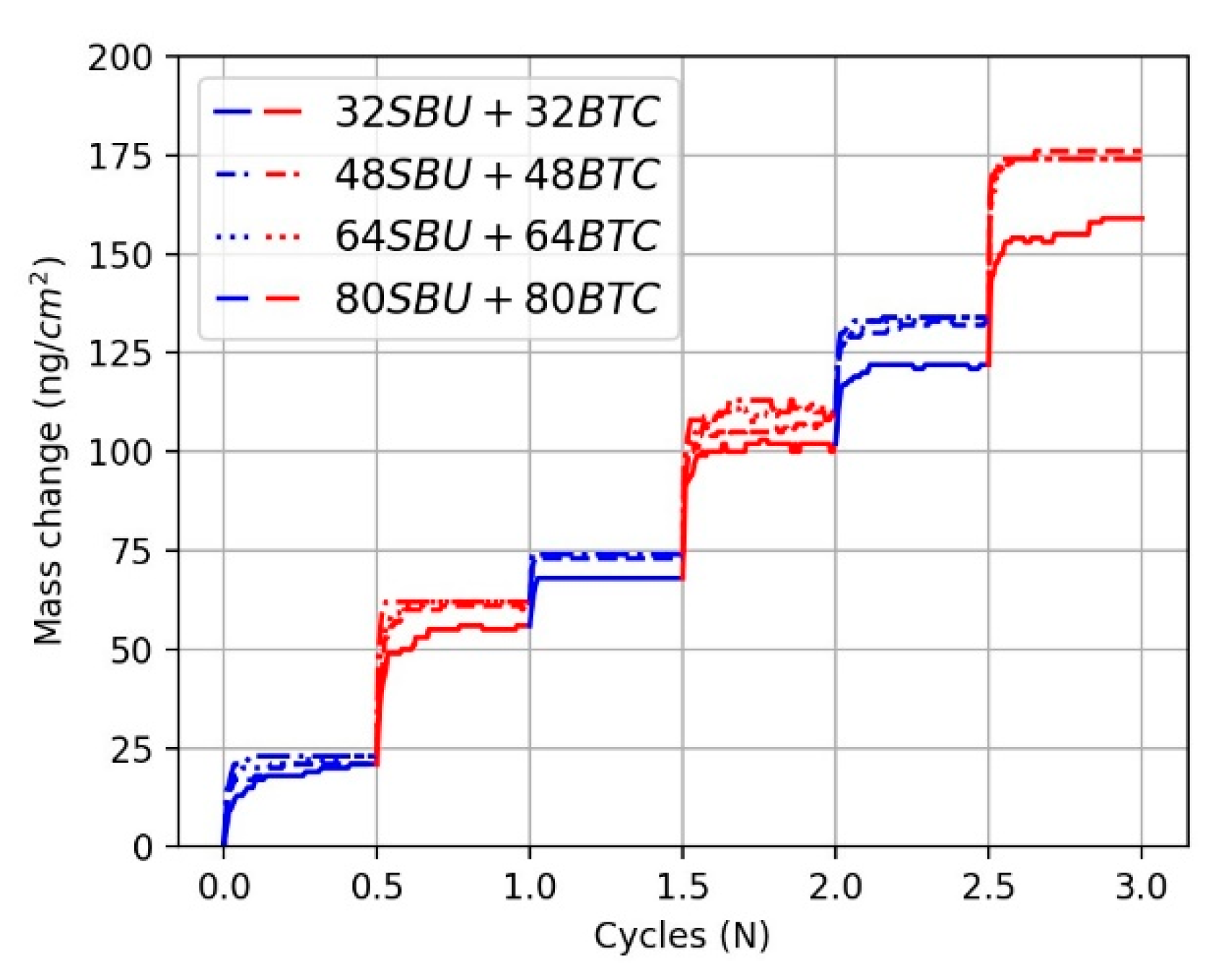
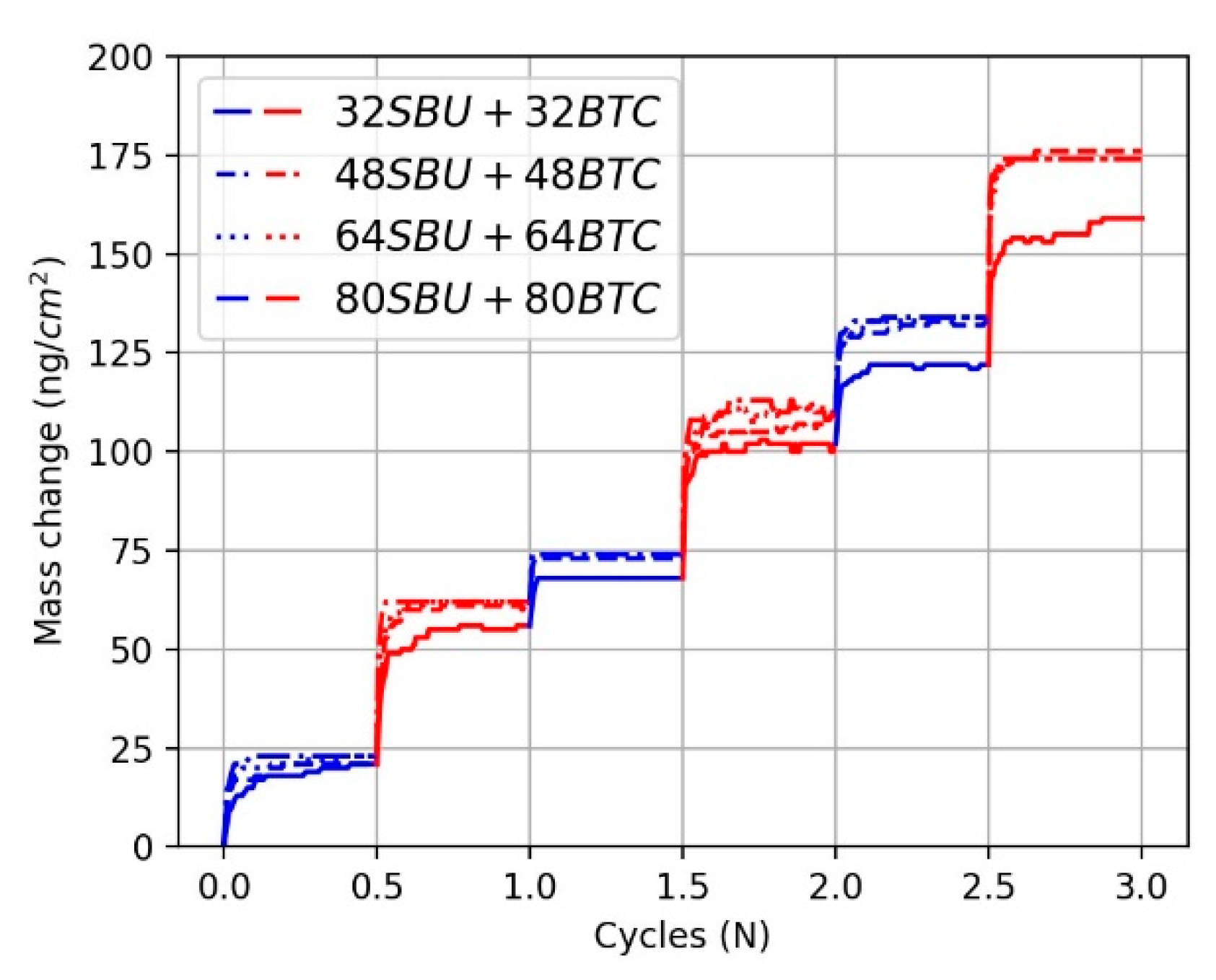
3.2.1. The Effect of Reactant Concentration
3.2.2. The Effect of Temperature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, Y.; Li, B.; He, H.; Zhou, W.; Chen, B.; Qian, G. Metal–organic frameworks as platforms for functional materials. Acc. Chem. Res. 2016, 49, 483–493. [Google Scholar] [CrossRef]
- Li, N.; Xu, J.; Feng, R.; Hu, T.L.; Bu, X.H. Governing metal–organic frameworks towards high stability. Chem. Commun. 2016, 52, 8501–8513. [Google Scholar] [CrossRef]
- Batten, S.R.; Champness, N.R.; Chen, X.M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Suh, M.P.; Reedijk, J. Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Chui, S.S.Y.; Lo, S.M.F.; Charmant, J.P.; Orpen, A.G.; Williams, I.D. A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Shen, D.; Bülow, M.; Lau, M.L.; Deng, S.; Fitch, F.R.; Lemcoff, N.O.; Semanscin, J. Metallo-organic molecular sieve for gas separation and purification. Microporous Mesoporous Mater. 2002, 55, 217–230. [Google Scholar] [CrossRef]
- Schlesinger, M.; Schulze, S.; Hietschold, M.; Mehring, M. Evaluation of synthetic methods for microporous metal–organic frameworks exemplified by the competitive formation of [Cu2(btc)3(H2O)3] and [Cu2(btc)(OH)(H2O)]. Microporous Mesoporous Mater. 2010, 132, 121–127. [Google Scholar] [CrossRef]
- Shekhah, O.; Wang, H.; Kowarik, S.; Schreiber, F.; Paulus, M.; Tolan, M.; Sternemann, C.; Evers, F.; Zacher, D.; Fischer, R.A.; et al. Step-by-step route for the synthesis of metal- organic frameworks. J. Am. Chem. Soc. 2007, 129, 15118–15119. [Google Scholar] [CrossRef] [PubMed]
- Munuera, C.; Shekhah, O.; Wang, H.; Wöll, C.; Ocal, C. The controlled growth of oriented metal–organic frameworks on functionalized surfaces as followed by scanning force microscopy. Phys. Chem. Chem. Phys. 2008, 10, 7257–7261. [Google Scholar] [CrossRef]
- Wang, W.; Sharapa, D.I.; Chandresh, A.; Nefedov, A.; Heißler, S.; Heinke, L.; Studt, F.; Wang, Y.; Wöll, C. Interplay of Electronic and Steric Effects to Yield Low-Temperature CO Oxidation at Metal Single Sites in Defect-Engineered HKUST-1. Angew. Chem. Int. Ed. 2020, 59, 10514–10518. [Google Scholar] [CrossRef] [Green Version]
- Rodenas, T.; Luz, I.; Prieto, G.; Seoane, B.; Miro, H.; Corma, A.; Kapteijn, F.; Xamena, F.X.L.I.; Gascon, J. Metal–organic framework nanosheets in polymer composite materials for gas separation. Nat. Mater. 2015, 14, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Knebel, A.; Grosjean, S.; Wagner, D.; Bräse, S.; Wöll, C.; Caro, J.; Heinke, L. Tunable molecular separation by nanoporous membranes. Nat. Commun. 2016, 7, 1–7. [Google Scholar]
- Chernikova, V.; Yassine, O.; Shekhah, O.; Eddaoudi, M.; Salama, K.N. Highly sensitive and selective SO2 MOF sensor: The integration of MFM-300 MOF as a sensitive layer on a capacitive interdigitated electrode. J. Mater. Chem. A 2018, 6, 5550–5554. [Google Scholar] [CrossRef] [Green Version]
- Baghayeri, M.; Ghanei-Motlagh, M.; Tayebee, R.; Fayazi, M.; Narenji, F. Application of graphene/zinc-based metal-organic framework nanocomposite for electrochemical sensing of As (III) in water resources. Anal. Chim. Acta 2020, 1099, 60–67. [Google Scholar] [CrossRef]
- Liu, C.S.; Li, J.; Pang, H. Metal-organic framework-based materials as an emerging platform for advanced electrochemical sensing. Coord. Chem. Rev. 2020, 410, 213222. [Google Scholar] [CrossRef]
- Teplensky, M.H.; Fantham, M.; Li, P.; Wang, T.C.; Mehta, J.P.; Young, L.J.; Moghadam, P.Z.; Hupp, J.T.; Farha, O.K.; Kaminski, C.F.; et al. Temperature treatment of highly porous zirconium-containing metal–organic frameworks extends drug delivery release. J. Am. Chem. Soc. 2017, 139, 7522–7532. [Google Scholar] [CrossRef] [Green Version]
- Giliopoulos, D.; Zamboulis, A.; Giannakoudakis, D.; Bikiaris, D.; Triantafyllidis, K. Polymer/metal organic framework (MOF) nanocomposites for biomedical applications. Molecules 2020, 25, 185. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yan, J.; Wen, N.; Xiong, H.; Cai, S.; He, Q.; Hu, Y.; Peng, D.; Liu, Z.; Liu, Y. Metal-organic frameworks for stimuliresponsive drug delivery. Biomaterials 2020, 230, 119619. [Google Scholar] [CrossRef]
- Moghadam, P.Z.; Li, A.; Wiggin, S.B.; Tao, A.; Maloney, A.G.; Wood, P.A.; Ward, S.C.; Fairen-Jimenez, D. Development of a Cambridge Structural Database subset: A collection of metal–organic frameworks for past, present, and future. Chem. Mater. 2017, 29, 2618–2625. [Google Scholar] [CrossRef]
- Reinsch, H.; Stock, N. Synthesis of MOFs: A personal view on rationalisation, application and exploration. Dalton Trans. 2017, 46, 8339–8349. [Google Scholar] [CrossRef]
- Schäfer, P.; Kapteijn, F.; van der Veen, M.A.; Domke, K.F. Understanding the inhibiting effect of BTC on CuBTC growth through experiment and modeling. Cryst. Growth Des. 2017, 17, 5603–5607. [Google Scholar] [CrossRef]
- Dighe, A.V.; Nemade, R.Y.; Singh, M.R. Modeling and simulation of crystallization of metal–organic frameworks. Processes 2019, 7, 527. [Google Scholar] [CrossRef] [Green Version]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Bureekaew, S.; Amirjalayer, S.; Tafipolsky, M.; Spickermann, C.; Roy, T.K.; Schmid, R. MOF-FF–A flexible first-principles derived force field for metal-organic frameworks. Physica Status Solidi (b) 2013, 250, 1128–1141. [Google Scholar] [CrossRef]
- Addicoat, M.A.; Vankova, N.; Akter, I.F.; Heine, T. Extension of the universal force field to metal–organic frameworks. J. Chem. Theory Comput. 2014, 10, 880–891. [Google Scholar] [CrossRef]
- Coupry, D.E.; Addicoat, M.A.; Heine, T. Extension of the universal force field for metal–organic frameworks. J. Chem. Theory Comput. 2016, 12, 5215–5225. [Google Scholar] [CrossRef] [Green Version]
- Vanduyfhuys, L.; Vandenbrande, S.; Verstraelen, T.; Schmid, R.; Waroquier, M.; Van Speybroeck, V. QuickFF: A program for a quick and easy derivation of force fields for metal-organic frameworks from ab initio input. J. Comput. Chem. 2015, 36, 1015–1027. [Google Scholar] [CrossRef] [Green Version]
- Bristow, J.K.; Tiana, D.; Walsh, A. Transferable force field for metal–organic frameworks from first-principles: BTW-FF. J. Chem. Theory Comput. 2014, 10, 4644–4652. [Google Scholar] [CrossRef] [Green Version]
- Van Duin, A.C.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef] [Green Version]
- Yoneya, M.; Tsuzuki, S.; Aoyagi, M. Simulation of metal–organic framework self-assembly. Phys. Chem. Chem. Phys. 2015, 17, 8649–8652. [Google Scholar] [CrossRef]
- Biswal, D.; Kusalik, P.G. Molecular simulations of self-assembly processes in metal-organic frameworks: Model dependence. J. Chem. Phys. 2017, 147, 044702. [Google Scholar] [CrossRef]
- Biswal, D.; Kusalik, P.G. Probing Molecular Mechanisms of Self-Assembly in Metal–Organic Frameworks. ACS Nano 2017, 11, 258–268. [Google Scholar] [CrossRef]
- Yoneya, M.; Yamaguchi, T.; Sato, S.; Fujita, M. Simulation of metal–ligand self-assembly into spherical complex M6L8. J. Am. Chem. Soc. 2012, 134, 14401–14407. [Google Scholar] [CrossRef] [PubMed]
- Yoneya, M.; Tsuzuki, S.; Yamaguchi, T.; Sato, S.; Fujita, M. Coordination-directed self-assembly of M12L24 nanocage: Effects of kinetic trapping on the assembly process. ACS Nano 2014, 8, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.P. Novel zinc protein molecular dynamics simulations: Steps toward antiangiogenesis for cancer treatment. Mol. Modeling Annu. 1999, 5, 196–202. [Google Scholar] [CrossRef]
- Pang, Y.P.; Xu, K.; El Yazal, J.; Prendergast, F.G. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 2000, 9, 1857–1865. [Google Scholar]
- Maula, T.A.; Hatch, H.W.; Shen, V.K.; Rangarajan, S.; Mittal, J. Designing molecular building blocks for the self-assembly of complex porous networks. Mol. Syst. Des. Eng. 2019, 4, 644–653. [Google Scholar] [CrossRef]
- Anderson, J.A.; Lorenz, C.D.; Travesset, A. General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys. 2008, 227, 5342–5359. [Google Scholar] [CrossRef]
- Glaser, J.; Nguyen, T.D.; Anderson, J.A.; Lui, P.; Spiga, F.; Millan, J.A.; Morse, D.C.; Glotzer, S.C. Strong scaling of general-purpose molecular dynamics simulations on GPUs. Comput. Phys. Commun. 2015, 192, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Addicoat, M.A.; Coupry, D.E.; Heine, T. AuToGraFS: Automatic topological generator for framework structures. J. Phys. Chem. A 2014, 118, 9607–9614. [Google Scholar] [CrossRef]
- Delgado-Friedrichs, O.; O’Keeffe, M.; Yaghi, O.M. Three-periodic nets and tilings: Edge-transitive binodal structures. Acta Crystallogr. Sect. A Found. Crystallogr. 2006, 62, 350–355. [Google Scholar] [CrossRef]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of repulsive forces in determining the equilibrium structure of simple liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Zhang, J.Z. Theory and Application of Quantum Molecular Dynamics; World Scientific: Singapore, 1999. [Google Scholar]
- Shekhah, O.; Wang, H.; Zacher, D.; Fischer, R.A.; Wöll, C. Growth mechanism of metal–organic frameworks: Insights into the nucleation by employing a step-by-step route. Angew. Chem. Int. Ed. 2009, 48, 5038–5041. [Google Scholar] [CrossRef] [PubMed]
- Delen, G.; Ristanovic´, Z.; Mandemaker, L.D.; Weckhuysen, B.M. Mechanistic Insights into Growth of Surface-Mounted MetalOrganic Framework Films Resolved by Infrared (Nano-) Spectroscopy. Chemistry 2018, 24, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavila, V.; Volponi, J.; Katzenmeyer, A.M.; Dixon, M.C.; Allendorf, M.D. Kinetics and mechanism of metal–organic framework thin film growth: Systematic investigation of HKUST-1 deposition on QCM electrodes. Chem. Sci. 2012, 3, 1531–1540. [Google Scholar] [CrossRef]
- Chandresh, A.; Liu, X.; Wöll, C.; Heinke, L. Programmed Molecular Assembly of Abrupt Crystalline Organic/Organic Heterointerfaces Yielding Metal-Organic Framework Diodes with Large On-Off Ratios. Adv. Sci. 2021, 8, 2001884. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Pramudya, Y.; Wenzel, W.; Wöll, C. Modeling the Layer-by-Layer Growth of HKUST-1 Metal-Organic Framework Thin Films. Nanomaterials 2021, 11, 1631. https://doi.org/10.3390/nano11071631
Zhang Q, Pramudya Y, Wenzel W, Wöll C. Modeling the Layer-by-Layer Growth of HKUST-1 Metal-Organic Framework Thin Films. Nanomaterials. 2021; 11(7):1631. https://doi.org/10.3390/nano11071631
Chicago/Turabian StyleZhang, Qiang, Yohanes Pramudya, Wolfgang Wenzel, and Christof Wöll. 2021. "Modeling the Layer-by-Layer Growth of HKUST-1 Metal-Organic Framework Thin Films" Nanomaterials 11, no. 7: 1631. https://doi.org/10.3390/nano11071631
APA StyleZhang, Q., Pramudya, Y., Wenzel, W., & Wöll, C. (2021). Modeling the Layer-by-Layer Growth of HKUST-1 Metal-Organic Framework Thin Films. Nanomaterials, 11(7), 1631. https://doi.org/10.3390/nano11071631