
Targeting NF-κB with Nanotherapy in a Mouse Model of Adult T-Cell Leukemia/Lymphoma

Abstract
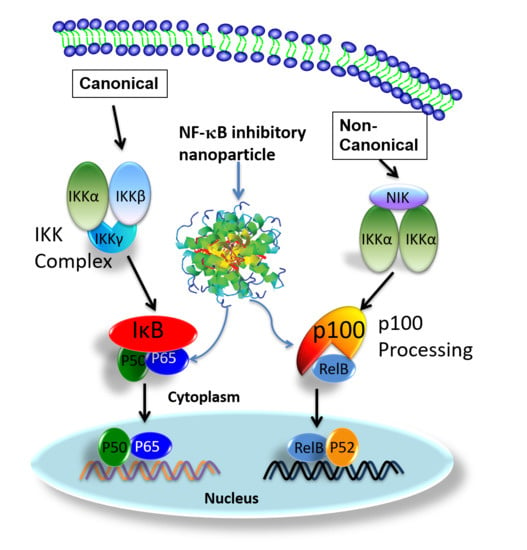
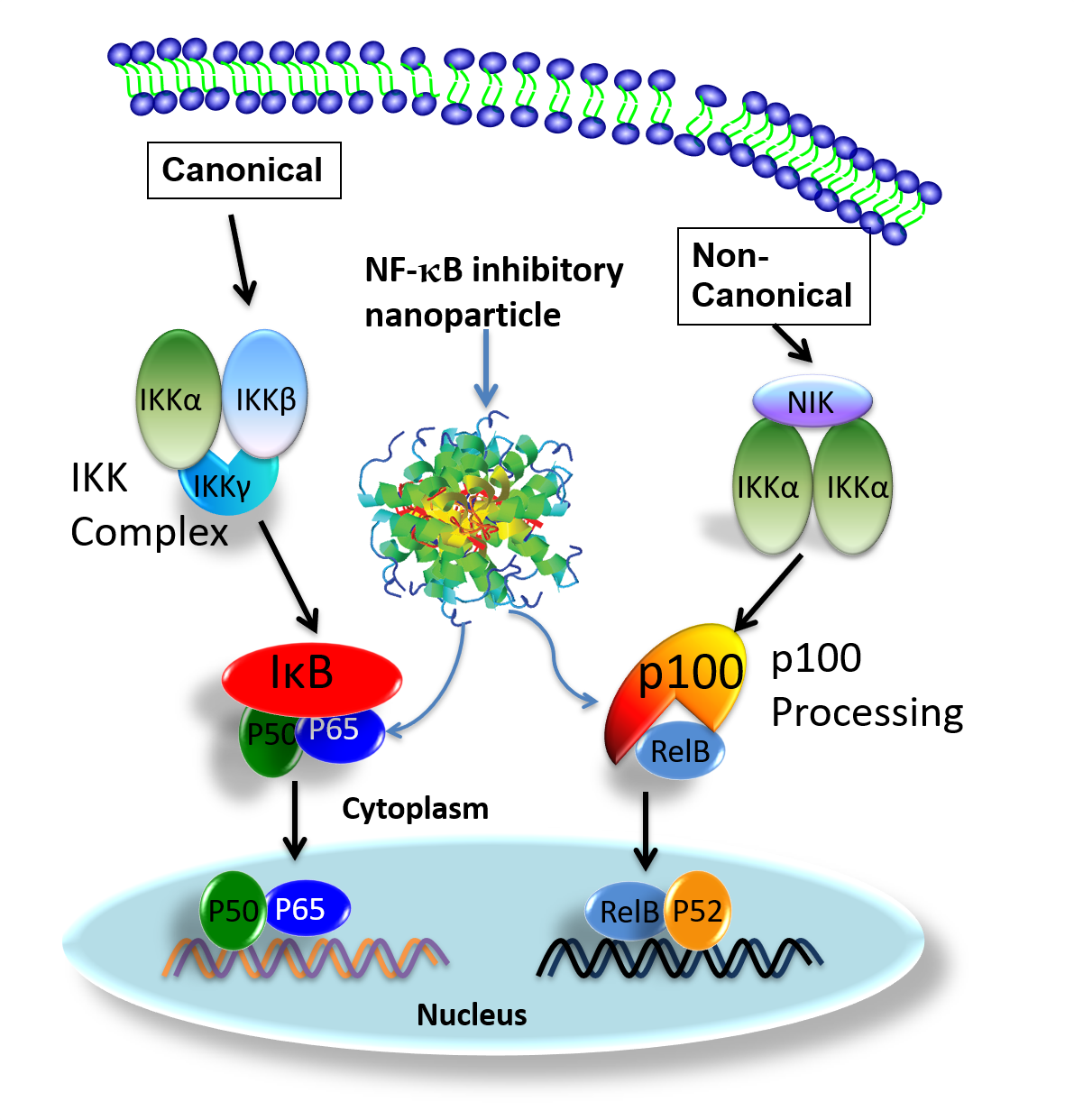{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of p5RHH-NF-kB Inhibitory Polyplex Nanoparticles
2.2. Real-Time RT-PCR
2.3. Western Blot
2.4. Histology
2.5. IVIS Imaging
2.6. Animal Studies
2.7. Statistics
3. Results and Discussion
3.1. p5RHH-Based siRNA Delivery Platform Delivers siRNA to the Tumor Site after Systemic Administration
3.2. Spontaneous Tumor Growth Is Suppressed by NF-κB Inhibitory Therapy during and after Treatment
3.3. NF-κB Inhibitory siRNA Nanoparticles Suppress Growth of Tumor Cell Transplants and Blood and Spleen Involvement in BNX Mice
3.4. mRNA and Protein Knokdown of p65 and p100 in BNX Mice
3.5. NF-κB Inhibitory siRNA Nanoparticles Sensitize ATLL Tumors to Adjunctive Chemotherapy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Hlela, C.; Shepperd, S.; Khumalo, N.P.; Taylor, G.P. The prevalence of human T-cell lymphotropic virus type 1 in the general population is unknown. Aids Rev. 2009, 11, 205–214. [Google Scholar] [PubMed]
- Chihara, D.; Ito, H.; Katanoda, K.; Shibata, A.; Matsuda, T.; Tajima, K.; Sobue, T.; Matsuo, K. Increase in incidence of adult T-cell leukemia/lymphoma in non-endemic areas of Japan and the United States. Cancer Sci. 2012, 103, 1857–1860. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, M.; Watanabe, T.; Yamaguchi, K. Adult T-cell leukemia: A review of epidemiological evidence. Front. Microbiol. 2012, 3, 322. [Google Scholar] [CrossRef] [PubMed]
- van Besien, K. Current status of allogeneic transplantation for aggressive non-Hodgkin lymphoma. Curr. Opin. Oncol. 2011, 23, 681–691. [Google Scholar] [CrossRef]
- Ishitsuka, K.; Tsukasaki, K.; Tamura, K. Interferon alfa and antiretroviral agents: A treatment option for adult T-cell leukemia/lymphoma. Drugs Today 2011, 47, 615–623. [Google Scholar] [CrossRef]
- Tanosaki, R.; Tobinai, K. Adult T-cell leukemia-lymphoma: Current treatment strategies and novel immunological approaches. Expert Rev. Hematol. 2010, 3, 743–753. [Google Scholar] [CrossRef]
- Ishitsuka, K.; Tamura, K. Treatment of adult T-cell leukemia/lymphoma: Past, present, and future. Eur. J. Haematol. 2008, 80, 185–196. [Google Scholar] [CrossRef]
- Ratner, L. Human T cell lymphotropic virus-associated leukemia/lymphoma. Curr. Opin. Oncol. 2005, 17, 469–473. [Google Scholar] [CrossRef]
- Munoz, E.; Israel, A. Activation of NF-kappa B by the Tax protein of HTLV. Immunobiology 1995, 193, 128–136. [Google Scholar] [CrossRef]
- Ratner, L. Adult T cell leukemia lymphoma. Front. Biosci. 2004, 9, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, L.; Lovly, C.; Ratner, L. The role of NF-κB-1 and NF-κB-2-mediated resistance to apoptosis in lymphomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9220–9225. [Google Scholar] [CrossRef]
- Horie, R. NF-kappaB in pathogenesis and treatment of adult T-cell leukemia/lymphoma. Int. Rev. Immunol. 2007, 26, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Rauch, D.A.; Ratner, L. Targeting HTLV-1 activation of NFkappaB in mouse models and ATLL patients. Viruses 2011, 3, 886–900. [Google Scholar] [CrossRef]
- Qu, Z.; Xiao, G. Human T-cell lymphotropic virus: A model of NF-kappaB-associated tumorigenesis. Viruses 2011, 3, 714–749. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Greene, W.C. Identification of HTLV-I tax trans-activator mutants exhibiting novel transcriptional phenotypes. Gene Dev. 1990, 4, 1875–1885. [Google Scholar] [CrossRef]
- Kimata, J.T.; Wong, F.-H.; Wang, J.J.; Ratner, L. Construction and Characterization of Infectious Human T-Cell Leukemia Virus Type 1 Molecular Clones. Virology 1994, 204, 656–664. [Google Scholar] [CrossRef]
- Robek, M.D.; Ratner, L. Immortalization of CD4(+) and CD8(+) T lymphocytes by human T-cell leukemia virus type 1 Tax mutants expressed in a functional molecular clone. J. Virol. 1999, 73, 4856–4865. [Google Scholar] [CrossRef]
- Hiscott, J.; Kwon, H.; Genin, P. Hostile takeovers: Viral appropriation of the NF-kappaB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef]
- Fochi, S.; Mutascio, S.; Bertazzoni, U.; Zipeto, D.; Romanelli, M.G. HTLV Deregulation of the NF-kappaB Pathway: An Update on Tax and Antisense Proteins Role. Front. Microbiol. 2018, 9, 285. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Giam, C.Z. NF-kappaB signaling mechanisms in HTLV-1-induced adult T-cell leukemia/lymphoma. FEBS J. 2018, 285, 3324–3336. [Google Scholar] [CrossRef]
- Wong, R.W.J.; Tan, T.K.; Amanda, S.; Ngoc, P.C.T.; Leong, W.Z.; Tan, S.H.; Asamitsu, K.; Hibi, Y.; Ueda, R.; Okamoto, T.; et al. Feed-forward regulatory loop driven by IRF4 and NF-kappaB in adult T-cell leukemia/lymphoma. Blood 2020, 135, 934–947. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. NF-kappa B: Ten years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, J.L.; Baltimore, D. Two pathways to NF-kappaB. Mol. Cell 2002, 10, 693–695. [Google Scholar] [CrossRef]
- Scheidereit, C. IkappaB kinase complexes: Gateways to NF-kappaB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef] [PubMed]
- de Martin, R.; Schmid, J.A.; Hofer-Warbinek, R. The NF-kappaB/Rel family of transcription factors in oncogenic transformation and apoptosis. Mutat. Res. Rev. Mutat. Res. 1999, 437, 231–243. [Google Scholar] [CrossRef]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. Domain-specific interaction with the I kappa B kinase (IKK)regulatory subunit IKK gamma is an essential step in tax-mediated activation of IKK. J. Biol. Chem. 2000, 275, 34060–34067. [Google Scholar] [CrossRef]
- Xiao, G.; Cvijic, M.E.; Fong, A.; Harhaj, E.W.; Uhlik, M.T.; Waterfield, M.; Sun, S.C. Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: Evidence for the involvement of IKKalpha. EMBO J. 2001, 20, 6805–6815. [Google Scholar] [CrossRef]
- Qing, G.; Qu, Z.; Xiao, G. Regulation of NF-kappa B2 p100 processing by its cis-acting domain. J. Biol. Chem. 2005, 280, 18–27. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Jeang, K.T. Functional activities of the human T-cell leukemia virus type I Tax oncoprotein: Cellular signaling through NF-kappa B. Cytokine Growth Factor Rev. 2001, 12, 207–217. [Google Scholar] [CrossRef]
- Pontoriero, M.; Fiume, G.; Vecchio, E.; de Laurentiis, A.; Albano, F.; Iaccino, E.; Mimmi, S.; Pisano, A.; Agosti, V.; Giovannone, E.; et al. Activation of NF-kappaB in B cell receptor signaling through Bruton’s tyrosine kinase-dependent phosphorylation of IkappaB-alpha. J. Mol. Med. 2019, 97, 675–690. [Google Scholar] [CrossRef]
- Solan, N.J.; Miyoshi, H.; Carmona, E.M.; Bren, G.D.; Paya, C.V. RelB Cellular Regulation and Transcriptional Activity Are Regulated by p100. J. Biol. Chem. 2002, 277, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Dobrzanski, P.; Ryseck, R.P.; Bravo, R. Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100. Oncogene 1995, 10, 1003–1007. [Google Scholar] [PubMed]
- Ruben, S.M.; Klement, J.F.; Coleman, T.A.; Maher, M.; Chen, C.H.; Rosen, C.A. I-Rel: A novel rel-related protein that inhibits NF-kappa B transcriptional activity. Genes Dev. 1992, 6, 745–760. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fusco, A.J.; Savinova, O.V.; Talwar, R.; Kearns, J.D.; Hoffmann, A.; Ghosh, G. Stabilization of RelB Requires Multidomain Interactions with p100/p52. J. Biol. Chem. 2008, 283, 12324–12332. [Google Scholar] [CrossRef]
- Murakami, T.; Hirai, H.; Suzuki, T.; Fujisawa, J.; Yoshida, M. HTLV-1 Tax enhances NF-kappa B2 expression and binds to the products p52 and p100, but does not suppress the inhibitory function of p100. Virology 1995, 206, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Lanoix, J.; Lacoste, J.; Pepin, N.; Rice, N.; Hiscott, J. Overproduction of NFKB2 (lyt-10) and c-Rel: A mechanism for HTLV-I Tax-mediated trans-activation via the NF-kappa B signalling pathway. Oncogene 1994, 9, 841–852. [Google Scholar]
- Béraud, C.; Sun, S.C.; Ganchi, P.; Ballard, D.W.; Greene, W.C. Human T-cell leukemia virus type I Tax associates with and is negatively regulated by the NF-kappa B2 p100 gene product: Implications for viral latency. Mol. Cell. Biol. 1994, 14, 1374–1382. [Google Scholar] [CrossRef][Green Version]
- Cramer, P.; Larson, C.J.; Verdine, G.L.; Muller, C.W. Structure of the human NF-kappaB p52 homodimer-DNA complex at 2.1 A resolution. EMBO J. 1997, 16, 7078–7090. [Google Scholar] [CrossRef]
- Mills, K.A.; Quinn, J.M.; Roach, S.T.; Palisoul, M.; Nguyen, M.; Noia, H.; Guo, L.; Fazal, J.; Mutch, D.G.; Wickline, S.A.; et al. p5RHH nanoparticle-mediated delivery of AXL siRNA inhibits metastasis of ovarian and uterine cancer cells in mouse xenografts. Sci. Rep. 2019, 9, 4762. [Google Scholar] [CrossRef]
- Kabir, A.U.; Lee, T.-J.; Pan, H.; Berry, J.C.; Krchma, K.; Wu, J.; Liu, F.; Kang, H.-K.; Hinman, K.; Yang, L.; et al. Requisite endothelial reactivation and effective siRNA nanoparticle targeting of Etv2/Er71 in tumor angiogenesis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Mohankumar, K.; Namachivayam, K.; Song, T.; Cha, B.J.; Slate, A.; Hendrickson, J.E.; Pan, H.; Wickline, S.A.; Oh, J.-Y.; Patel, R.P.; et al. A murine neonatal model of necrotizing enterocolitis caused by anemia and red blood cell transfusions. Nat. Commun. 2019, 10, 3494. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Rohatgi, N.; Brestoff, J.R.; Moley, J.R.; Li, Y.; Williams, J.W.; Alippe, Y.; Pan, H.; Pietka, T.A.; Mbalaviele, G.; et al. Myeloid-specific Asxl2 deletion limits diet-induced obesity by regulating energy expenditure. J. Clin. Investig. 2020, 130, 2644–2656. [Google Scholar] [CrossRef] [PubMed]
- Lozhkin, A.; Vendrov, A.E.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J. Mol. Cell. Cardiol. 2017, 102, 10–21. [Google Scholar] [CrossRef]
- Pan, H.; Palekar, R.U.; Hou, K.K.; Bacon, J.; Yan, H.; Springer, L.E.; Akk, A.; Yang, L.; Miller, M.J.; Pham, C.T.; et al. Anti-JNK2 peptide-siRNA nanostructures improve plaque endothelium and reduce thrombotic risk in atherosclerotic mice. Int. J. Nanomed. 2018, 13, 5187–5205. [Google Scholar] [CrossRef]
- Strand, M.S.; Krasnick, B.A.; Pan, H.; Zhang, X.; Bi, Y.; Brooks, C.; Wetzel, C.; Sankpal, N.; Fleming, T.; Goedegebuure, S.P.; et al. Precision delivery of RAS-inhibiting siRNA to KRAS driven cancer via peptide-based nanoparticles. Oncotarget 2019, 10, 4761–4775. [Google Scholar] [CrossRef]
- Yan, H.; Duan, X.; Pan, H.; Akk, A.; Sandell, L.J.; Wickline, S.A.; Rai, M.F.; Pham, C.T.N. Development of a peptide-siRNA nanocomplex targeting NF-kappaB for efficient cartilage delivery. Sci. Rep. 2019, 9, 442. [Google Scholar] [CrossRef]
- Stansel, T.; Wickline, S.A.; Pan, H. NF-kappaB Inhibition Suppresses Experimental Melanoma Lung Metastasis. J. Cancer Sci. Clin. Ther. 2020, 4, 256–265. [Google Scholar] [CrossRef]
- Zhou, H.F.; Yan, H.; Pan, H.; Hou, K.K.; Akk, A.; Springer, L.E.; Hu, Y.; Allen, J.S.; Wickline, S.A.; Pham, C.T. Peptide-siRNA nanocomplexes targeting NF-kappaB subunit p65 suppress nascent experimental arthritis. J. Clin. Investig. 2014, 124, 4363–4374. [Google Scholar] [CrossRef]
- Hou, K.K.; Pan, H.; Ratner, L.; Schlesinger, P.H.; Wickline, S.A. Mechanisms of Nanoparticle-Mediated siRNA Transfection by Melittin-Derived Peptides. ACS Nano 2013, 7, 8605–8615. [Google Scholar] [CrossRef]
- Hou, K.K.; Pan, H.; Lanza, G.M.; Wickline, S.A. Melittin derived peptides for nanoparticle based siRNA transfection. Biomaterials 2013, 34, 3110–3119. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.K.; Pan, H.; Schlesinger, P.H.; Wickline, S.A. A role for peptides in overcoming endosomal entrapment in siRNA delivery—A focus on melittin. Biotechnol. Adv. 2015, 33, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Rauch, D.; Gross, S.; Harding, J.; Niewiesk, S.; Lairmore, M.; Piwnica-Worms, D.; Ratner, L. Imaging spontaneous tumorigenesis: Inflammation precedes development of peripheral NK tumors. Blood 2009, 113, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Grossman, W.J.; Ratner, L. Cytokine expression and tumorigenicity of large granular lymphocytic leukemia cells from mice transgenic for the tax gene of human T-cell leukemia virus type I. Blood 1997, 90, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, H.; Sonobe, H.; Yamato, K.; Takeuchi, T.; Ookawa, K.; Kodama, H.; Ohtsuki, Y.; Miyoshi, I. Central nervous system involvement of adult T-cell leukemia–lymphoma with multinucleated giant cells in the brain, skin, and kidney. Cancer 1993, 71, 133–137. [Google Scholar] [CrossRef]
- Ma, W.-L.; Li, C.-C.; Yu, S.-C.; Tien, H.-F. Adult T-Cell Lymphoma/Leukemia Presenting as Isolated Central Nervous System T-Cell Lymphoma. Case Rep. Hematol. 2014, 2014, 917369. [Google Scholar] [CrossRef][Green Version]
- Kitajima, M.; Korogi, Y.; Shigematsu, Y.; Liang, L.; Matsuoka, M.; Yamamoto, T.; Jhono, M.; Eto, K.; Takahashi, M. Central nervous system lesions in adult T-cell leukaemia: MRI and pathology. Neuroradiology 2002, 44, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, C.; Ikeda, H.; Fukumoto, T. Cerebral mass lesions associated with adult T-cell leukemia/lymphoma. Int. J. Hematol. 1995, 61, 97–102. [Google Scholar] [CrossRef]
- Lotan, I.; Khlebtovsky, A.; Inbar, E.; Strenov, J.; Djaldetti, R.; Steiner, I. Primary brain T-cell lymphoma in an HTLV-1 serologically positive male. J. Neurol. Sci. 2012, 314, 163–165. [Google Scholar] [CrossRef]
- Portis, T.; Harding, J.C.; Ratner, L. The contribution of NF-kappa B activity to spontaneous proliferation and resistance to apoptosis in human T-cell leukemia virus type 1 Tax-induced tumors. Blood 2001, 98, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Eladl, E.; Tremblay-LeMay, R.; Rastgoo, N.; Musani, R.; Chen, W.; Liu, A.; Chang, H. Role of CD47 in Hematological Malignancies. J. Hematol. Oncol. 2020, 13, 96. [Google Scholar] [CrossRef]
- Betancur, P.A.; Abraham, B.; Yiu, Y.Y.; Willingham, S.B.; Khameneh, F.; Zarnegar, M.; Kuo, A.H.; McKenna, K.; Kojima, Y.; Leeper, N.J.; et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat. Commun. 2017, 8, 14802. [Google Scholar] [CrossRef]
- Boukhari, A.; Alhosin, M.; Bronner, C.; Sagini, K.; Truchot, C.; Sick, E.; Schini-Kerth, V.B.; André, P.; Mély, Y.; Mousli, M.; et al. CD47 activation-induced UHRF1 over-expression is associated with silencing of tumor suppressor gene p16INK4A in glioblastoma cells. Anticancer Res. 2015, 35, 149–157. [Google Scholar] [PubMed]
- Lo, J.; Lau, E.Y.T.; Ching, R.H.H.; Cheng, B.Y.L.; Ma, M.K.F.; Ng, I.O.L.; Lee, T.K.W. Nuclear factor kappa B-mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology 2015, 62, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Rauch, D.A.; Conlon, K.C.; Janakiram, M.; Brammer, J.E.; Harding, J.C.; Ye, B.H.; Zang, X.; Ren, X.; Olson, S.; Cheng, X.; et al. Rapid progression of adult T-cell leukemia/lymphoma as tumor-infiltrating Tregs after PD-1 blockade. Blood 2019, 134, 1406–1414. [Google Scholar] [CrossRef]
- Rauch, D.A.; Olson, S.L.; Harding, J.C.; Sundaramoorthi, H.; Kim, Y.; Zhou, T.; MacLeod, A.R.; Challen, G.; Ratner, L. Interferon regulatory factor 4 as a therapeutic target in adult T-cell leukemia lymphoma. Retrovirology 2020, 17, 27. [Google Scholar] [CrossRef]
- Shen, H.; Sun, T.; Ferrari, M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012, 19, 367–373. [Google Scholar] [CrossRef]
- Miele, E.; Spinelli, G.P.; Miele, E.; Di Fabrizio, E.; Ferretti, E.; Tomao, S.; Gulino, A. Nanoparticle-based delivery of small interfering RNA: Challenges for cancer therapy. Int. J. Nanomed. 2012, 7, 3637–3657. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nat. Cell Biol. 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Lancellotti, P.; Oury, C. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, e38. [Google Scholar] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L.-S. Delivery of siRNA Therapeutics: Barriers and Carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef]
- Aigner, A. Cellular delivery in vivo of siRNA-based therapeutics. Curr. Pharm. Des. 2008, 14, 3603–3619. [Google Scholar] [CrossRef] [PubMed]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Kwon, Y.J. Before and after Endosomal Escape: Roles of Stimuli-Converting siRNA/Polymer Interactions in Determining Gene Silencing Efficiency. Acc. Chem. Res. 2012, 45, 1077–1088. [Google Scholar] [CrossRef]
- Nimesh, S.; Gupta, N.; Chandra, R. Strategies and advances in nanomedicine for targeted siRNA delivery. Nanomedicine 2011, 6, 729–746. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Villanueva, D.; El-Sherbiny, I.M.; Herrera-Ruiz, D.; Vlassov, A.V.; Smyth, H.D.C. Formulation Approaches to Short Interfering RNA and MicroRNA: Challenges and Implications. J. Pharm. Sci. 2012, 101, 4046–4066. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, I.R.; Fox, S.P.; Hollins, A.J.; Akhtar, S. Delivery strategies for siRNA-mediated gene silencing. Curr. Drug Deliv. 2006, 3, 147–155. [Google Scholar] [CrossRef]
- Filion, M.C.; Phillips, N.C. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta 1997, 1329, 345–356. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Soenen, S.; Brisson, A.R.; De Cuyper, M. Addressing the problem of cationic lipid-mediated toxicity: The magnetoliposome model. Biomaterials 2009, 30, 3691–3701. [Google Scholar] [CrossRef]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Int. Med. 2009, 267, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Love, K.T.; Chen, Y.; Eltoukhy, A.A.; Kastrup, C.; Sahay, G.; Jeon, A.; Dong, Y.; Whitehead, K.A.; Anderson, D.G. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J. Am. Chem. Soc. 2012, 134, 6948–6951. [Google Scholar] [CrossRef]
- Plank, C.; Mechtler, K.; Szoka, F.C.; Wagner, E. Activation of the Complement System by Synthetic DNA Complexes: A Potential Barrier for Intravenous Gene Delivery. Hum. Gene Ther. 1996, 7, 1437–1446. [Google Scholar] [CrossRef]
- Abes, R.; Arzumanov, A.A.; Moulton, H.M.; Abes, S.; Ivanova, G.D.; Iversen, P.L.; Gait, M.J.; Lebleu, B. Cell-penetrating-peptide-based delivery of oligonucleotides: An overview. Biochem. Soc. Trans. 2007, 35, 775–779. [Google Scholar] [CrossRef]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: From molecular mechanisms to therapeutics. Biol. Cell 2008, 100, 201–217. [Google Scholar] [CrossRef]
- Meade, B.R.; Dowdy, S.F. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv. Drug Deliv. Rev. 2008, 60, 530–536. [Google Scholar] [CrossRef]
- Endoh, T.; Ohtsuki, T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv. Drug Deliv. Rev. 2009, 61, 704–709. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, N.Y.; Bin Choi, Y.; Park, S.H.; Yang, J.M.; Shin, S. RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system. J. Control. Release 2010, 143, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.H.; Choe, J.; Park, T.G. Intracellular small interfering RNA delivery using genetically engineered double-stranded RNA binding protein domain. J. Gene Med. 2009, 11, 804–812. [Google Scholar] [CrossRef]
- Ezzat, K.; Andaloussi, S.E.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef] [PubMed]
- Arthanari, Y.; Pluen, A.; Rajendran, R.; Aojula, H.; Demonacos, C. Delivery of therapeutic shRNA and siRNA by Tat fusion peptide targeting bcr–abl fusion gene in Chronic Myeloid Leukemia cells. J. Control. Release 2010, 145, 272–280. [Google Scholar] [CrossRef]
- Mae, M.; Andaloussi, S.E.; Lehto, T.; Langel, U. Chemically modified cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2009, 6, 1195–1205. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rauch, D.A.; Harding, J.C.; Ratner, L.; Wickline, S.A.; Pan, H. Targeting NF-κB with Nanotherapy in a Mouse Model of Adult T-Cell Leukemia/Lymphoma. Nanomaterials 2021, 11, 1582. https://doi.org/10.3390/nano11061582
Rauch DA, Harding JC, Ratner L, Wickline SA, Pan H. Targeting NF-κB with Nanotherapy in a Mouse Model of Adult T-Cell Leukemia/Lymphoma. Nanomaterials. 2021; 11(6):1582. https://doi.org/10.3390/nano11061582
Chicago/Turabian StyleRauch, Daniel A., John C. Harding, Lee Ratner, Samuel A. Wickline, and Hua Pan. 2021. "Targeting NF-κB with Nanotherapy in a Mouse Model of Adult T-Cell Leukemia/Lymphoma" Nanomaterials 11, no. 6: 1582. https://doi.org/10.3390/nano11061582
APA StyleRauch, D. A., Harding, J. C., Ratner, L., Wickline, S. A., & Pan, H. (2021). Targeting NF-κB with Nanotherapy in a Mouse Model of Adult T-Cell Leukemia/Lymphoma. Nanomaterials, 11(6), 1582. https://doi.org/10.3390/nano11061582