
Soft, Formstable (Co)Polyester Blend Elastomers


Abstract
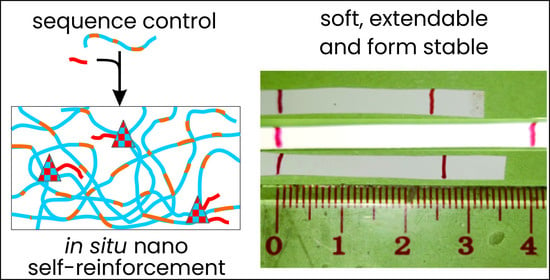
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. NMR
2.3. FT-IR
2.4. GPC
2.5. DSC
2.6. Tensile Tests
2.7. WAXS
2.8. Synthesis of Poly([(L-lactide)-co-(ε-caprolactone)] (PLC)
2.9. Synthesis of Poly(D-lactide) with an Mn of 16 kg·mol−1 (PDLA 16k)
2.10. Blending and Film Formation
3. Results and Discussion
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spontak, R.J.; Patel, N.P. Thermoplastic elastomers: Fundamentals and applications. Curr. Opin. Colloid Interface Sci. 2000, 5, 333–340. [Google Scholar] [CrossRef]
- Wisse, E.; Spiering, A.J.H.; van Leeuwen, E.N.M.; Renken, R.A.E.; Dankers, P.Y.W.; Brouwer, L.A.; van Luyn, M.J.A.; Harmsen, M.C.; Sommerdijk, N.A.J.M.; Meijer, E.W. Molecular Recognition in Poly(ε-caprolactone)-Based Thermoplastic Elastomers. Biomacromolecules 2006, 7, 3385–3395. [Google Scholar] [CrossRef]
- Leone, G.; Mauri, M.; Bertini, F.; Canetti, M.; Piovani, D.; Ricci, G. Ni(II) α-Diimine-Catalyzed α-Olefins Polymerization: Thermoplastic Elastomers of Block Copolymers. Macromolecules 2015, 48, 1304–1312. [Google Scholar] [CrossRef]
- Watts, A.; Kurokawa, N.; Hillmyer, M.A. Strong, Resilient, and Sustainable Aliphatic Polyester Thermoplastic Elastomers. Biomacromolecules 2017, 18, 1845–1854. [Google Scholar] [CrossRef]
- Shelke, N.B.; Nagarale, R.K.; Kumbar, S.G. Chapter 7—Polyurethanes. In Natural and Synthetic Biomedical Polymers; Kumbar, S.G., Laurencin, C.T., Deng, M., Eds.; Elsevier: Oxford, UK, 2014; pp. 123–144. [Google Scholar]
- Fabbri, M.; Soccio, M.; Gigli, M.; Guidotti, G.; Gamberini, R.; Gazzano, M.; Siracusa, V.; Rimini, B.; Lotti, N.; Munari, A. Design of fully aliphatic multiblock poly(ester urethane)s displaying thermoplastic elastomeric properties. Polymer 2016, 83, 154–161. [Google Scholar] [CrossRef]
- Ye, S.; Xiang, X.; Wang, S.; Han, D.; Xiao, M.; Meng, Y. Nonisocyanate CO2-Based Poly(ester-co-urethane)s with Tunable Performances: A Potential Alternative to Improve the Biodegradability of PBAT. ACS Sustain. Chem. Eng. 2020, 8, 1923–1932. [Google Scholar] [CrossRef]
- Malikmammadov, E.; Tanir, T.E.; Kiziltay, A.; Hasirci, V.; Hasirci, N. PCL and PCL-based materials in biomedical applications. J. Biomater. Sci., Polym. Ed. 2018, 29, 863–893. [Google Scholar] [CrossRef]
- Brzezinski, M.; Biela, T. Micro- and nanostructures of polylactide stereocomplexes and their biomedical applications. Polym. Int. 2015, 64, 1667–1675. [Google Scholar] [CrossRef]
- Park, Y.J.; Cha, J.H.; Bang, S.I.; Kim, S.Y. Clinical application of three-dimensionally printed biomaterial polycaprolactone (PCL) in augmentation rhinoplasty. Aesth. Plast. Surg. 2019, 43, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, M.A.; Zhang, J.; Sun, B.; El-Hamshary, H.; Al-Deyab, S.S.; Mo, X. Development of poly (L-lactide-co-caprolactone) multichannel nerve conduit with aligned electrospun nanofibers for Schwann cell proliferation. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 323–329. [Google Scholar] [CrossRef]
- Poh, P.S.P.; Hege, C.; Chhaya, M.P.; Balmayor, E.R.; Foehr, P.; Burgkart, R.H.; Schantz, J.-T.; Schiller, S.M.; Schilling, A.F.; Hutmacher, D.W. Evaluation of polycaprolactone-poly-D,L-lactide copolymer as biomaterial for breast tissue engineering. Polym. Int. 2017, 66, 77–84. [Google Scholar] [CrossRef]
- Sartoneva, R.; Nordback, P.H.; Haimi, S.; Grijpma, D.W.; Lehto, K.; Rooney, N.; Seppaenen-Kaijansinkko, R.; Miettinen, S.; Lahdes-Vasama, T. Comparison of Poly(L-lactide-co-ϵ-caprolactone) and Poly(trimethylene carbonate) Membranes for Urethral Regeneration: An In Vitro and In Vivo Study. Tissue Eng. Part A 2018, 24, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Armentano, I.; Dottori, M.; Fortunati, E.; Mattioli, S.; Kenny, J.M. Biodegradable polymer matrix nanocomposites for tissue engineering: A review. Polym. Degrad. Stab. 2010, 95, 2126–2146. [Google Scholar] [CrossRef]
- Sidle, D.M.; Stolovitzky, P.; Ow, R.A.; Silvers, S.; Matheny, K.; Bikhazi, N.; Wani, M.; Scurry, W.C.; Most, S.P. Twelve-month outcomes of a bioabsorbable implant for in-office treatment of dynamic nasal valve collapse. Laryngoscope 2020, 130, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- San Nicoló, M.; Stelter, K.; Sadick, H.; Bas, M.; Berghaus, A. Absorbable implant to treat nasal valve collapse. Facial Plast. Surg. 2017, 33, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Hu, J.-f.; Guo, W.-c.; Yu, L.; Zhao, S.-H. Research and application of absorbable screw in orthopedics: A clinical review comparing PDLLA screw with metal screw in patients with simple medial malleolus fracture. Chin. J. Traumatol. 2013, 16, 27–30. [Google Scholar] [PubMed]
- Tomihata, K.; Suzuki, M.; Tomita, N. Handling characteristics of poly (L-lactide-co-ε-caprolactone) monofilament suture. Bio-Med. Mater. Eng. 2005, 15, 381–391. [Google Scholar]
- Schmidt, S.C.; Hillmyer, M.A. Polylactide stereocomplex crystallites as nucleating agents for isotactic polylactide. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 300–313. [Google Scholar] [CrossRef]
- Ma, P.; Shen, T.; Xu, P.; Dong, W.; Lemstra, P.J.; Chen, M. Superior Performance of Fully Biobased Poly(lactide) via Stereocomplexation-Induced Phase Separation: Structure versus Property. ACS Sustain. Chem. Eng. 2015, 3, 1470–1478. [Google Scholar] [CrossRef]
- Tsuji, H. Poly(lactic acid) stereocomplexes: A decade of progress. Adv. Drug Deliv. Rev. 2016, 107, 97–135. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; Sojoudiasli, H.; Heuzey, M.-C.; Huneault, M.A.; Wood-Adams, P. Rheological study of crystallization behavior of polylactide and its flax fiber composites. J. Polym. Res. 2017, 24, 46. [Google Scholar] [CrossRef]
- Izraylit, V.; Heuchel, M.; Gould, O.E.; Kratz, K.; Lendlein, A. Strain recovery and stress relaxation behaviour of multiblock copolymer blends physically cross-linked with PLA stereocomplexation. Polymer 2020, 209, 122984. [Google Scholar] [CrossRef]
- Su, X.; Feng, L.; Yu, D. Formation of Stereocomplex Crystal and Its Effect on the Morphology and Property of PDLA/PLLA Blends. Polymers 2020, 12, 2515. [Google Scholar] [CrossRef]
- Tsuji, H. Poly (lactide) stereocomplexes: Formation, structure, properties, degradation, and applications. Macromol. Biosci. 2005, 5, 569–597. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, N.; Hotta, A. Thermomechanical properties of highly transparent self-reinforced polylactide composites with electrospun stereocomplex polylactide nanofibers. Polymer 2018, 153, 214–222. [Google Scholar] [CrossRef]
- Peterlin, A. Molecular model of drawing polyethylene and polypropylene. J. Mater. Sci. 1971, 6, 490–508. [Google Scholar] [CrossRef]
- Wang, H.; Yu, J.; Fang, H.; Wei, H.; Wang, X.; Ding, Y. Largely improved mechanical properties of a biodegradable polyurethane elastomer via polylactide stereocomplexation. Polymer 2018, 137, 1–12. [Google Scholar] [CrossRef]
- Izraylit, V.; Hommes-Schattmann, P.J.; Neffe, A.T.; Gould, O.E.; Lendlein, A. Polyester urethane functionalizable through maleimide side-chains and cross-linkable by polylactide stereocomplexes. Eur. Polym. J. 2020, 137, 109916. [Google Scholar] [CrossRef]
- Nakayama, Y.; Aihara, K.; Yamanishi, H.; Fukuoka, H.; Tanaka, R.; Cai, Z.; Shiono, T. Synthesis of biodegradable thermoplastic elastomers from ε-caprolactone and lactide. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 489–495. [Google Scholar] [CrossRef]
- Huang, Y.; Chang, R.; Han, L.; Shan, G.; Bao, Y.; Pan, P. ABA-type thermoplastic elastomers composed of poly (ε-caprolactone-co-δ-valerolactone) soft midblock and polymorphic poly (lactic acid) hard end blocks. ACS Sustain. Chem. Eng. 2016, 4, 121–128. [Google Scholar] [CrossRef]
- García-Valle, F.M.; Cuenca, T.; Mosquera, M.E.G.; Milione, S.; Cano, J. Ring-Opening Polymerization (ROP) of cyclic esters by a versatile aluminum Diphenoxyimine Complex: From polylactide to random copolymers. Eur. Polym. J. 2020, 125, 109527. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, L.; Qu, C.; Qi, M. Microstructure analysis and thermal properties of l-lactide/ɛ-caprolactone copolymers obtained with magnesium octoate. Polymer 2009, 50, 1423–1429. [Google Scholar] [CrossRef]
- Garlotta, D. A literature review of poly (lactic acid). J. Polym. Environ. 2001, 9, 63–84. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W. Synthesis of polycaprolactone: A review. Chem. Soc. Rev. 2009, 38, 3484–3504. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.; van Dijk-Wolthuis, W.; Kettenes-Van den Bosch, J.; Schuyl, P.; Hennink, W. Monodisperse enantiomeric lactic acid oligomers: Preparation, characterization, and stereocomplex formation. Macromolecules 1998, 31, 6397–6402. [Google Scholar] [CrossRef]
- Tsuji, H.; Bouapao, L. Stereocomplex formation between poly (L-lactic acid) and poly (D-lactic acid) with disproportionately low and high molecular weights from the melt. Polym. Int. 2012, 61, 442–450. [Google Scholar] [CrossRef]
- Grijpma, D.; Pennings, A. Polymerization temperature effects on the properties of l-lactide and ε-caprolactone copolymers. Polym. Bull. 1991, 25, 335–341. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Huang, B.-H.; Lin, C.-C. A Highly efficient initiator for the ring-opening polymerization of lactides and ε-caprolactone: A kinetic study. Macromolecules 2005, 38, 5400–5405. [Google Scholar] [CrossRef]
- Lendlein, A.; Neuenschwander, P.; Suter, U.W. Hydroxy-telechelic copolyesters with well defined sequence structure through ring-opening polymerization. Macromol. Chem. Phys. 2000, 201, 1067–1076. [Google Scholar] [CrossRef]
- Lipik, V.T.; Abadie, M.J. Synthesis of block copolymers of varying architecture through suppression of transesterification during coordinated anionic ring opening polymerization. Int. J. Biomater. 2012, 2012, 390947. [Google Scholar] [CrossRef]
- Kasperczyk, J.; Bero, M. Coordination polymerization of lactides, 4. The role of transesterification in the copolymerization of l, l-lactide and ε-caprolactone. Makromol. Chem. 1993, 194, 913–925. [Google Scholar] [CrossRef]
- Fernández, J.; Etxeberria, A.; Sarasua, J.-R. Synthesis, structure and properties of poly (L-lactide-co-ε-caprolactone) statistical copolymers. J. Mech. Behav. Biomed. Mater. 2012, 9, 100–112. [Google Scholar] [CrossRef]
- Herbert, I. Statistical analysis of copolymer sequence distribution. In NMR Spectroscopy of Polymers; Springer: Dordrecht, The Netherlands, 1993; pp. 50–79. [Google Scholar]
- Fernandez, J.; Etxeberria, A.; Sarasua, J.-R. Crystallization and melting behavior of poly(ε-caprolactone-co-δ-valerolactone) and poly(ε-caprolactone-co-L-lactide) copolymers with novel chain microstructures. J. Appl. Polym. Sci. 2015, 132, 42534. [Google Scholar] [CrossRef]
- Tashiro, K.; Kouno, N.; Wang, H.; Tsuji, H. Crystal structure of poly (lactic acid) stereocomplex: Random packing model of PDLA and PLLA chains as studied by X-ray diffraction analysis. Macromolecules 2017, 50, 8048–8065. [Google Scholar] [CrossRef]
- Höhne, G.W.H. Another approach to the Gibbs–Thomson equation and the melting point of polymers and oligomers. Polymer 2002, 43, 4689–4698. [Google Scholar] [CrossRef]
- Shao, J.; Xiang, S.; Bian, X.; Sun, J.; Li, G.; Chen, X. Remarkable Melting Behavior of PLA Stereocomplex in Linear PLLA/PDLA Blends. Ind. Eng. Chem. Res. 2015, 54, 2246–2253. [Google Scholar] [CrossRef]
- Izraylit, V.; Gould, O.E.; Rudolph, T.; Kratz, K.; Lendlein, A. Controlling Actuation Performance in Physically Cross-Linked Polylactone Blends Using Polylactide Stereocomplexation. Biomacromolecules 2019, 21, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shan, G.; Bao, Y.; Pan, P. Exclusive stereocomplex crystallization of linear and multiarm star-shaped high-molecular-weight stereo diblock poly (lactic acid)s. J. Phys. Chem. B 2015, 119, 14270–14279. [Google Scholar] [CrossRef] [PubMed]
- Purnama, P.; Jung, Y.; Kim, S.H. Stereocomplexation of Poly(l-lactide) and Random Copolymer Poly(d-lactide-co-ε-caprolactone) To Enhance Melt Stability. Macromolecules 2012, 45, 4012–4014. [Google Scholar] [CrossRef]
- Sarasua, J.R.; Arraiza, A.L.; Balerdi, P.; Maiza, I. Crystallinity and mechanical properties of optically pure polylactides and their blends. Polym. Eng. Sci. 2005, 45, 745–753. [Google Scholar] [CrossRef]
- Kállay-Menyhárd, A.; Suba, P.; László, Z.; Fekete, H.; Mester, A.; Horváth, Z.; Varga, J.; Móczó, J. Direct correlation between modulus and the crystalline structure in isotactic polypropylene. eXPRESS Polym. Lett. 2015, 9, 308–320. [Google Scholar] [CrossRef]
- Hong, K.; Rastogi, A.; Strobl, G. A model treating tensile deformation of semicrystalline polymers: Quasi-static stress−strain relationship and viscous stress determined for a sample of polyethylene. Macromolecules 2004, 37, 10165–10173. [Google Scholar] [CrossRef]
- Behl, M.; Ridder, U.; Feng, Y.; Kelch, S.; Lendlein, A. Shape-memory capability of binary multiblock copolymer blends with hard and switching domains provided by different components. Soft Matter. 2009, 5, 676–684. [Google Scholar] [CrossRef]
- Abdallah-Elhirtsi, S.; Fitoussi, J.; Rashmi, B.-J.; Prashantha, K.; Farzaneh, S.; Lacrampe, M.-F.; Krawczak, P.; Tcharkhtchi, A. Study of Partial Shape Memory Effect of Polymers by Multicycle Tests. Polym. Compos. 2015, 36, 1145–1151. [Google Scholar] [CrossRef]
- Mohr, R.; Kratz, K.; Weigel, T.; Lucka-Gabor, M.; Moneke, M.; Lendlein, A. Initiation of shape-memory effect by inductive heating of magnetic nanoparticles in thermoplastic polymers. Proc. Natl. Acad. Sci. USA 2006, 103, 3540–3545. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, J.; Wang, J.; Liu, X.; Zhang, R.; Hu, G.; Na, H.; Zhu, J. Soft segment free thermoplastic polyester elastomers with high performance. J. Mater. Chem. A 2015, 3, 13637–13641. [Google Scholar] [CrossRef]
- Aoyama, T.; Carlos, A.J.; Saito, H.; Inoue, T.; Niitsu, Y. Strain recovery mechanism of PBT/rubber thermoplastic elastomer. Polymer 1999, 40, 3657–3663. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Feed | Reaction Time | Conversion | Comp. | Sequence Structure | Molar Masses and Distribution | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LA:CL | LA | CL | LA:CL | lLA | lCL | R | Mn (kg·mol−1) | Mw (kg·mol−1) | Đ | ||
| (mol%) | (h) | (%) | (%) | (mol%) | |||||||
| PLC-84-24.0-49 a | 75:25 | 48 | 99 | 93 | 89:11 | 24.0 | 3.0 | 0.38 | 26 | 49 | 1.88 |
| PLC-82-21.1-51 b,d | 75:25 | 44 | 99 | 96 | 88:12 | 21.1 | 2.9 | 0.39 | 35 | 51 | 1.46 |
| PLC-57-5.9-56 a | 70:30 | 48 | 99 | 99 | 68:32 | 5.9 | 2.8 | 0.53 | 30 | 56 | 1.86 |
| PLC-65-10.8-80 b,e | 70:30 | 44 | 99 | 96 | 75:25 | 10.8 | 3.6 | 0.37 | 62 | 80 | 1.29 |
| PLC-68-10.6-71 b,f | 70:30 | 5 | 98 | 90 | 77:23 | 10.6 | 3.2 | 0.41 | 37 | 71 | 1.92 |
| PLC-57-4.9-103 b,f | 70:30 | 21 | 98 | 99 | 68:32 | 4.9 | 2.3 | 0.64 | 59 | 103 | 1.75 |
| PLC-56-5.4-145 c,e | 70:30 | 53 | 98 | 98 | 67:33 | 5.4 | 2.6 | 0.57 | 70 | 145 | 2.1 |
| PLC-58-4.8-156 c,e | 69:31 | 26 | 95 | 98 | 69:31 | 4.8 | 2.2 | 0.67 | 67 | 156 | 2.3 |
| PLC-62-7.2-180 c | 67:33 | 23 | nd | nd | 72:28 | 7.2 | 2.8 | 0.50 | 76 | 180 | 2.4 |
| PLC-56-5.5-182 c | 67:33 | 48 | nd | nd | 67:33 | 5.5 | 2.7 | 0.55 | 80 | 182 | 2.3 |
| PLC-53-4.4-103 b,e | 66:34 | 24 | 98 | 99 | 64:36 | 4.4 | 2.5 | 0.64 | 66 | 103 | 1.56 |
| PLC-50-3.9-101 b,e | 66:34 | 27 | 98 | 99 | 61:39 | 3.9 | 2.5 | 0.66 | 64 | 101 | 1.58 |
| a54-PLC-62-6.6-159 e | 70:30 | 53 | nd | nd | 72 ± 1 | 6.6 | 2.6 | 0.54 | 61 ± 17 | 159 ± 68 | 2.5 ± 0.5 |
| a9-PLC-56-6.4-149 e | 67:33 | 53 | nd | nd | 69 ± 2 | 6.4 | 2.9 | 0.51 | 64 ± 17 | 149 ± 64 | 2.3 ± 0.6 |
| Sample ID | DSC | Tensile Properties | ||||||
|---|---|---|---|---|---|---|---|---|
| Tg (°C) | Tm (°C) | ΔHm (J·g−1) | χc,LA (%) | ϕc,LA (%) | E (MPa) | σmax (MPa) | εb (%) | |
| PLC-65-10.8-80 | 24 a | 153 | 20.7 | 34 | 22.3 | 560 ± 35 | 12.4 ± 0.6 | 134 ± 76 |
| PLC-56-5.4-145 b | −10 | 60–150 | 6.3 | 10 | 6.8 | 36 ± 3 | 30 ± 5 | 905 ± 51 |
| PLC-58-4.8-156 b | −7 | 70–140 | 4.3 | 7 | 4.7 | 19 ± 3 | 6.5 ± 4.4 | 920 ±327 |
| PLC-57-7.4-91 c | −12 | 156 | 11.0 | 17 | 11.8 | 51 ± 5 | 2.6 ± 0.1 | 505 ± 37 |
| PLC-62-7.2-180 b | −19 | 65–145 | 13.9 | 21 | 14.9 | 68 ± 4 | 40 ± 6 | 620 ± 40 |
| PLC-56-5.5-182 b | −9 | 65–130 | 7.137 | 14 | 7.7 | 16 ± 2 | 19 ± 7 | 692 ± 74 |
| a54-PLC-62-6.6-159 b | −3 | 65–140 | 13.7 ± 5.1 | 24 | 14.7 | 60 ± 36 | 32 ± 12 | 739 ± 143 |
| a9-PLC-56-6.4-149 b | −5 | 65–140 | 13.8 ± 4.8 | 26 | 14.8 | 65 ± 40 | 27 ± 8 | 652 ± 78 |
| Blend | LA (wt%) | Tg (°C) | Tm,HC (°C) | χc,LAHC
(%) | Tm,SC (°C) | χc,LASC
(%) | ϕc,LA (%) | E (MPa) | σmax (MPa) | εb (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| B80-PLC-65-10.8-80-147k a,b | 72.4 | 17 | 112–152,177 | 38 | 207 | 11 | 30 | 106 ± 4 | 5.2 ± 1 | 338 ± 16 |
| B80-PLC-65-10.8-80 | 72.4 | 10 c | 70–150 | 13.5 | 211 | 48 | 19.4 | 91 ± 3 | 6.9 ± 0.2 | 135 ± 9 |
| B90-PLC-65-10.8-80 | 68.9 | 3 c | 120–155 | 21 | 210 | 50 | 22.2 | 270 ± 30 | 20.5 ± 3 | 472 ± 40 |
| B95-PLC-65-10.8-80 | 67.2 | 0 | 110–155 | 20 | 210 | 48 | 17.2 | 79 ± 30 | 4.2 ± 1 | 435 ± 25 |
| B80-PLC-50-3.9-101 | 59.8 | −8 c | 169 | 36 | 170 | - | 14.5 | 5 ± 6 | 1.0 ± 0.1 | 244 ± 102 |
| B90-PLC-57-7.4-91 | 61.6 | −3 | 82,135 | 9 | 208 | 53 | 14.6 | 90 ± 7 | 10.6 ± 0.9 | 576 ± 60 |
| B90-PLC-56-5.4-145 b | 60.6 | −13 | 81,171 | 6 | 198 | 27 | 8.2 | 27 ± 1 | 27 ± 4 | 1020 ± 32 |
| B95-PLC-56-5.4-145 b | 58.4 | −16 | 65–145 | 8 | 200 | 22 | 8.7 | 26 ± 2 | 18 ± 10 | 889 ± 83 |
| B90-PLC-58-4.8-156 | 62.6 | −2 | 84,171 | 8 | 191 | 21 | 8.3 | 20 ± 2 | 9.7 ± 1.6 | 689 ± 88 |
| B95-PLC-58-4.8-156 | 60.5 | −2 | 84,172 | 2 | 191 | 40 | 4.9 | 12 ± 3 | 8.4 ± 4.1 | 845 ± 267 |
| B95-PLC-62-7.2-180 b | 63.8 | −10 | 70–145 | 15 | 199 | 54 | 14 | 73 ± 3 | 42 ± 11 | 564 ± 78 |
| B90-PLC-56-5.5-182 b | 60.6 | −14 | 67–130,171 | 17 | 189 | 16 | 12.7 | 26 ± 2 | 22 ± 7 | 657 ± 90 |
| B95-PLC-56-5.5-182 b | 58.4 | −15 | 70–110,172 | 20 | 187 | 44 | 15.1 | 16 ± 3 | 18 ± 4 | 607 ± 33 |
| B95-a54-PLC-62-6.6-159 b | 63.8 | −14 ± 5 | 70–150 | 19 | 199 | 52 | 16.6 | 68 ± 43 | 35 ± 12 | 676 ± 111 |
| B95-a9-PLC-56-6.4-149 b | 58.4 | −17 ± 6 | 70–150 | 16 | 198 | 49 | 14.6 | 74 ± 43 | 29 ± 11 | 601 ± 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neffe, A.T.; Izraylit, V.; Hommes-Schattmann, P.J.; Lendlein, A. Soft, Formstable (Co)Polyester Blend Elastomers. Nanomaterials 2021, 11, 1472. https://doi.org/10.3390/nano11061472
Neffe AT, Izraylit V, Hommes-Schattmann PJ, Lendlein A. Soft, Formstable (Co)Polyester Blend Elastomers. Nanomaterials. 2021; 11(6):1472. https://doi.org/10.3390/nano11061472
Chicago/Turabian StyleNeffe, Axel T., Victor Izraylit, Paul J. Hommes-Schattmann, and Andreas Lendlein. 2021. "Soft, Formstable (Co)Polyester Blend Elastomers" Nanomaterials 11, no. 6: 1472. https://doi.org/10.3390/nano11061472
APA StyleNeffe, A. T., Izraylit, V., Hommes-Schattmann, P. J., & Lendlein, A. (2021). Soft, Formstable (Co)Polyester Blend Elastomers. Nanomaterials, 11(6), 1472. https://doi.org/10.3390/nano11061472