
Functionalizable Glyconanoparticles for a Versatile Redox Platform

 , ,
, , 

Abstract
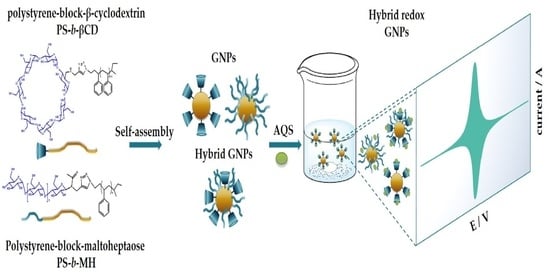
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Block Copolymers Synthesis (BCPs) Protocols
2.2.1. Synthesis of Hydroxyl-Terminated Polystyrene (PS-OH)
2.2.2. Synthesis of Azido-Functionalized Polystyrene (PS-N3)
2.2.3. Synthesis of Polystyrene-Block-Maltoheptaose (PS-b-MH) Block Copolymer
2.2.4. Synthesis of Polystyrene-Block-β-Cyclodextrin (PS-b-βCD) Block Copolymer
2.3. Preparation of Glyconanoparticles (GNPs) by Nanoprecipitation
2.4. Functionalization of GNPs with Sodium Anthraquinone-2-Sulfonate (AQS)
2.5. Characterization by 1H NMR and by Size Exclusion Chromatography
2.6. Characterization of the Glyconanoparticles by Scanning Electron Microscopy (SEM) and by Transmission Electron Microscopy (TEM)
2.7. Characterization of the Glyconanoparticles by Dynamic Light Scattering (DLS) and Nanoparticles Tracking Analysis (NTA)
2.8. Electrochemistry Measurements
3. Results and Discussions
3.1. Self-Assembly of Block-Copolymers
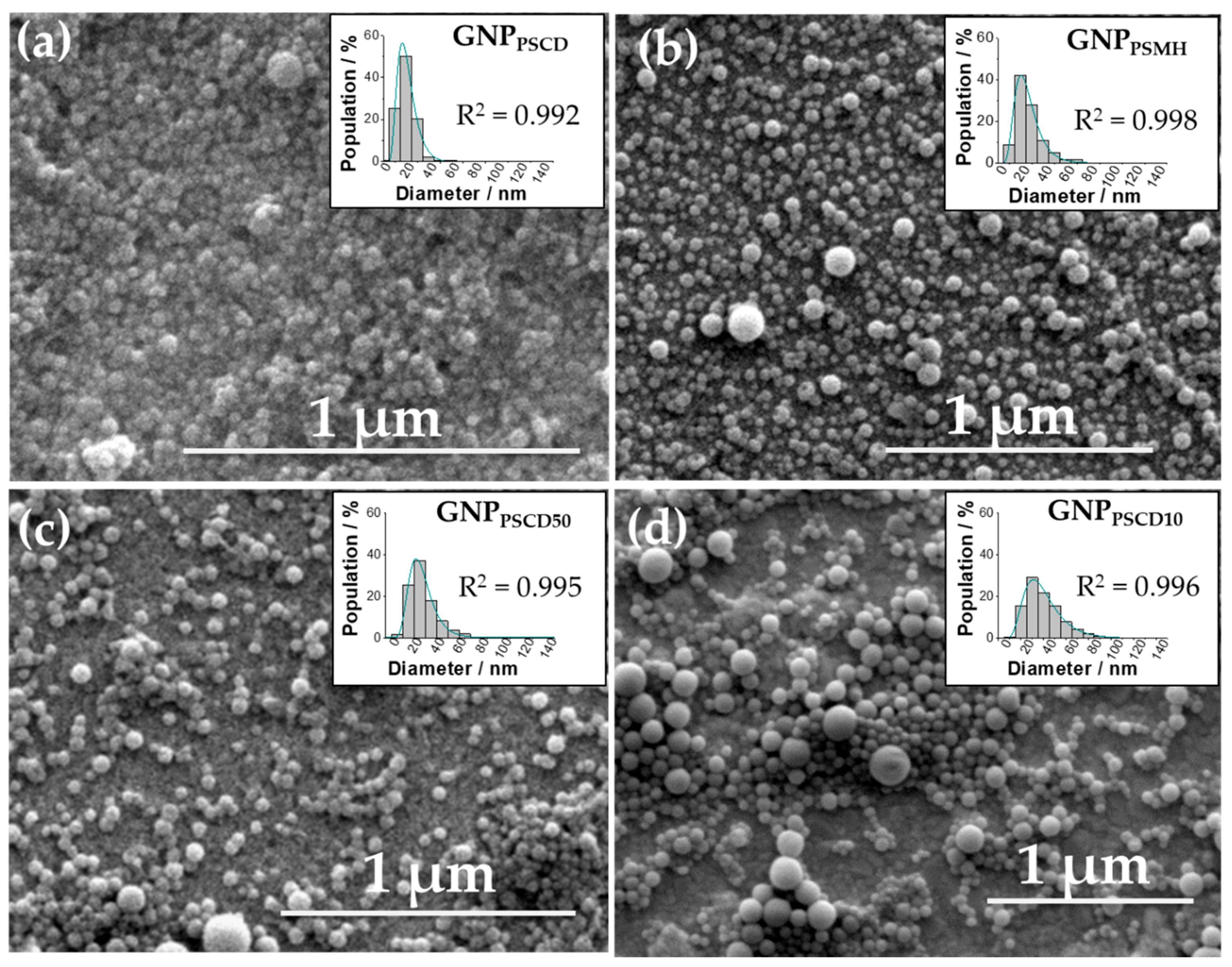
3.2. Morphology and Size Determination of GNPs by SEM and TEM
3.3. Hydrodynamic Diameter Determination of GNPs by Dynamic Light Scattering (DLS)
3.4. Characterization of the Host–Guest Properties of GNPs by Cyclic Voltammetry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giacomelli, C.; Schmidt, V.; Putaux, J.L.; Narumi, A.; Kakuchi, T.; Borsali, R. Aqueous self-assembly of polystyrene chains end-functionalized with β-cyclodextrin. Biomacromolecules 2009, 10, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Interfacing zwitterionic liposomes with inorganic nanomaterials: Surface forces, membrane integrity, and applications. Langmuir 2016, 32, 4393–4404. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.J.; Chen, X.; Giroud, F.; Travelet, C.; Borsali, R.; Cosnier, S. Redox-active glyconanoparticles as electron shuttles for mediated electron transfer with bilirubin oxidase in solution. J. Am. Chem. Soc. 2017, 139, 16076–16079. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.L.; Gross, A.J.; Giroud, F.; Travelet, C.; Borsali, R.; Cosnier, S. Solubilized enzymatic fuel cell (SEFC) for quasi-continuous operation exploiting carbohydrate block copolymer glyconanoparticle mediators. ACS Energy Lett. 2019, 4, 142–148. [Google Scholar] [CrossRef]
- Gross, A.J.; Haddad, R.; Travelet, C.; Reynaud, E.; Audebert, P.; Borsali, R.; Cosnier, S. Redox-active carbohydrate-coated nanoparticles: Self-assembly of a cyclodextrin-polystyrene glycopolymer with tetrazine-naphthalimide. Langmuir 2016, 32, 11939–11945. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, R.; Tachibana, S.; Cao, R.; Ortiz, P.D.; Gomez, L.; Asano, Y. Supramolecular-mediated immobilisation of L-phenylalanine dehydrogenase on β-cyclodextrin-modified gold nanospheres. J. Exp. Nanosci. 2006, 1, 249–260. [Google Scholar] [CrossRef]
- Villalonga, R.; Fragoso, A.; Cao, R.; Ortiz, P.D.; Villalonga, M.L.; Damiao, A.E. Supramolecular-mediated immobilization of trypsin on cyclodextrin-modified gold nanospheres. Supramol. Chem. 2005, 17, 387–391. [Google Scholar] [CrossRef]
- Otsuka, I.; Osaka, M.; Sakai, Y.; Travelet, C.; Putaux, J.L.; Borsali, R. Self-assembly of maltoheptaose-block-polystyrene into micellar nanoparticles and encapsulation of gold nanoparticles. Langmuir 2013, 29, 15224–15230. [Google Scholar] [CrossRef] [PubMed]
- Djedaïni, F.; Perly, B. Anthraquinone sulphonate as a general-purpose shift reagent for the NMR analysis of cyclodextrins. Magn. Reson. Chem. 1990, 28, 372–374. [Google Scholar] [CrossRef]
- Shen, A.; Guo, Z.; Cai, X.; Xue, X.; Liang, X. Preparation and chromatographic evaluation of a cysteine-bonded zwitterionic hydrophilic interaction liquid chromatography stationary phase. J. Chromatogr. A 2012, 1228, 175–182. [Google Scholar] [CrossRef]
- Otsuka, I.; Fuchise, K.; Halila, S.; Fort, S.; Aissou, K.; Pignot-Paintrand, I.; Chen, Y.; Narumi, A.; Kakuchi, T.; Borsali, R. Thermoresponsive vesicular morphologies obtained by self-assemblies of hybrid oligosaccharide-block-poly(N-isopropylacrylamide) copolymer systems. Langmuir 2010, 26, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Loos, K.; Böker, A.; Zettl, H.; Zhang, M.; Krausch, G.; Müller, A.H.E. Micellar aggregates of amylose-block-polystyrene rod-coil block copolymers in water and THF. Macromolecules 2005, 38, 873–879. [Google Scholar] [CrossRef]
- Houga, C.; Le Meins, J.F.; Borsali, R.; Taton, D.; Gnanou, Y. Synthesis of ATRP-induced dextran-b-polystyrene diblock copolymers and preliminary investigation of their self-assembly in water. Chem. Commun. 2007, 3063–3065. [Google Scholar] [CrossRef]
- Houga, C.; Giermanska, J.; Lecommandoux, S.; Borsali, R.; Taton, D.; Gnanou, Y.; Meins, J.F. Le Micelles and polymersomes obtained by self-assembly of dextran and polystyrene based block copolymers. Biomacromolecules 2009, 10, 32–40. [Google Scholar] [CrossRef]
- Batchelor-McAuley, C.; Li, Q.; Dapin, S.M.; Compton, R.G. Voltammetric characterization of DNA intercalators across the full pH range: Anthraquinone-2,6-disulfonate and anthraquinone-2-sulfonate. J. Phys. Chem. B 2010, 114, 4094–4100. [Google Scholar] [CrossRef]
- Jacq, J. Etablissement et Discussion de L’Equation Generale de la Coubre Intensite-Potentiel en Regime Stationnaire et Diffusion Convective. J. Electroanal. Chem. Interfacial Electrochem. 1971, 29, 149–180. [Google Scholar] [CrossRef]
- Bourourou, M.; Elouarzaki, K.; Lalaoui, N.; Agnès, C.; Le Goff, A.; Holzinger, M.; Maaref, A.; Cosnier, S. Supramolecular immobilization of laccase on carbon nanotube electrodes functionalized with (methylpyrenylaminomethyl)anthraquinone for direct electron reduction of oxygen. Chem. A Eur. J. 2013, 19, 9371–9375. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticles | GNPPSCD/AQS | GNPPSMH/AQS | GNPPSCD50/AQS | GNPPSCD10/AQS |
|---|---|---|---|---|
| Current (%) | 100 ± 7 | 30 ± 5 | 67 ± 16 | 32 ± 12 |
| E1/2 (V vs. SCE) | −0.426 | −0.460 | −0.419 | −0.414 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrière, M.; Buzzetti, P.H.M.; Gorgy, K.; Mumtaz, M.; Travelet, C.; Borsali, R.; Cosnier, S. Functionalizable Glyconanoparticles for a Versatile Redox Platform. Nanomaterials 2021, 11, 1162. https://doi.org/10.3390/nano11051162
Carrière M, Buzzetti PHM, Gorgy K, Mumtaz M, Travelet C, Borsali R, Cosnier S. Functionalizable Glyconanoparticles for a Versatile Redox Platform. Nanomaterials. 2021; 11(5):1162. https://doi.org/10.3390/nano11051162
Chicago/Turabian StyleCarrière, Marie, Paulo Henrique M. Buzzetti, Karine Gorgy, Muhammad Mumtaz, Christophe Travelet, Redouane Borsali, and Serge Cosnier. 2021. "Functionalizable Glyconanoparticles for a Versatile Redox Platform" Nanomaterials 11, no. 5: 1162. https://doi.org/10.3390/nano11051162
APA StyleCarrière, M., Buzzetti, P. H. M., Gorgy, K., Mumtaz, M., Travelet, C., Borsali, R., & Cosnier, S. (2021). Functionalizable Glyconanoparticles for a Versatile Redox Platform. Nanomaterials, 11(5), 1162. https://doi.org/10.3390/nano11051162