
Improved Water–Gas Shift Performance of Au/NiAl LDHs Nanostructured Catalysts via CeO2 Addition

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Sample Preparation
2.2.1. Adjusting the Procedure for NiAl-LDH Modification with CeO2
Synthesis of CeO2
Synthesis of NiAl-LDH
2.2.2. Synthesis of CeO2-Modified NiAl-LDH
2.2.3. Deposition of Gold on the Surface of NiAl and xCeNiAl LDHs
2.3. Sample Characterization Methods
2.4. WGS Activity Evaluation
3. Results and Discussions
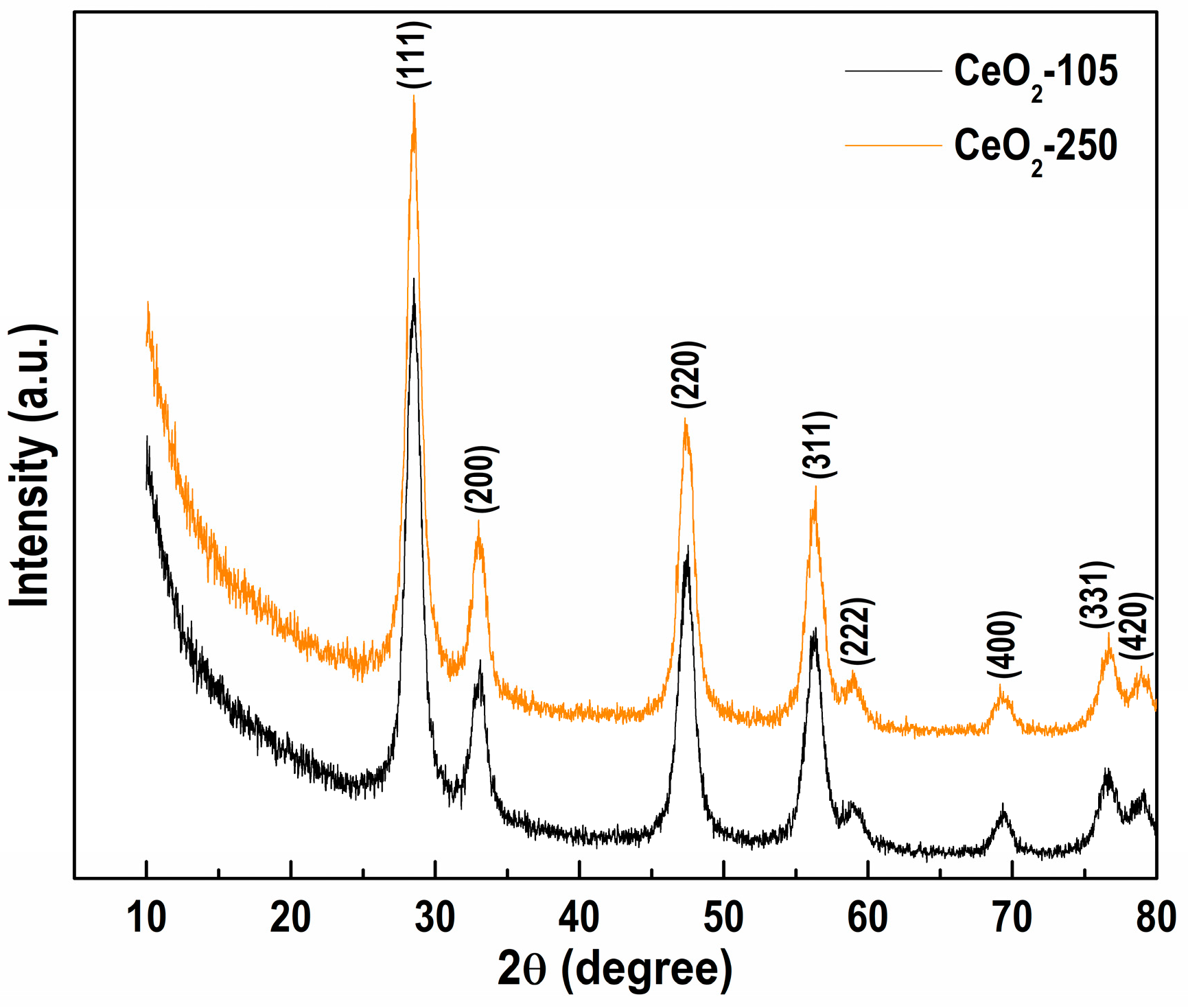
3.1. Tuning the Procedure for NiAl-LDH Modification with CeO2
3.2. Chemical Analysis
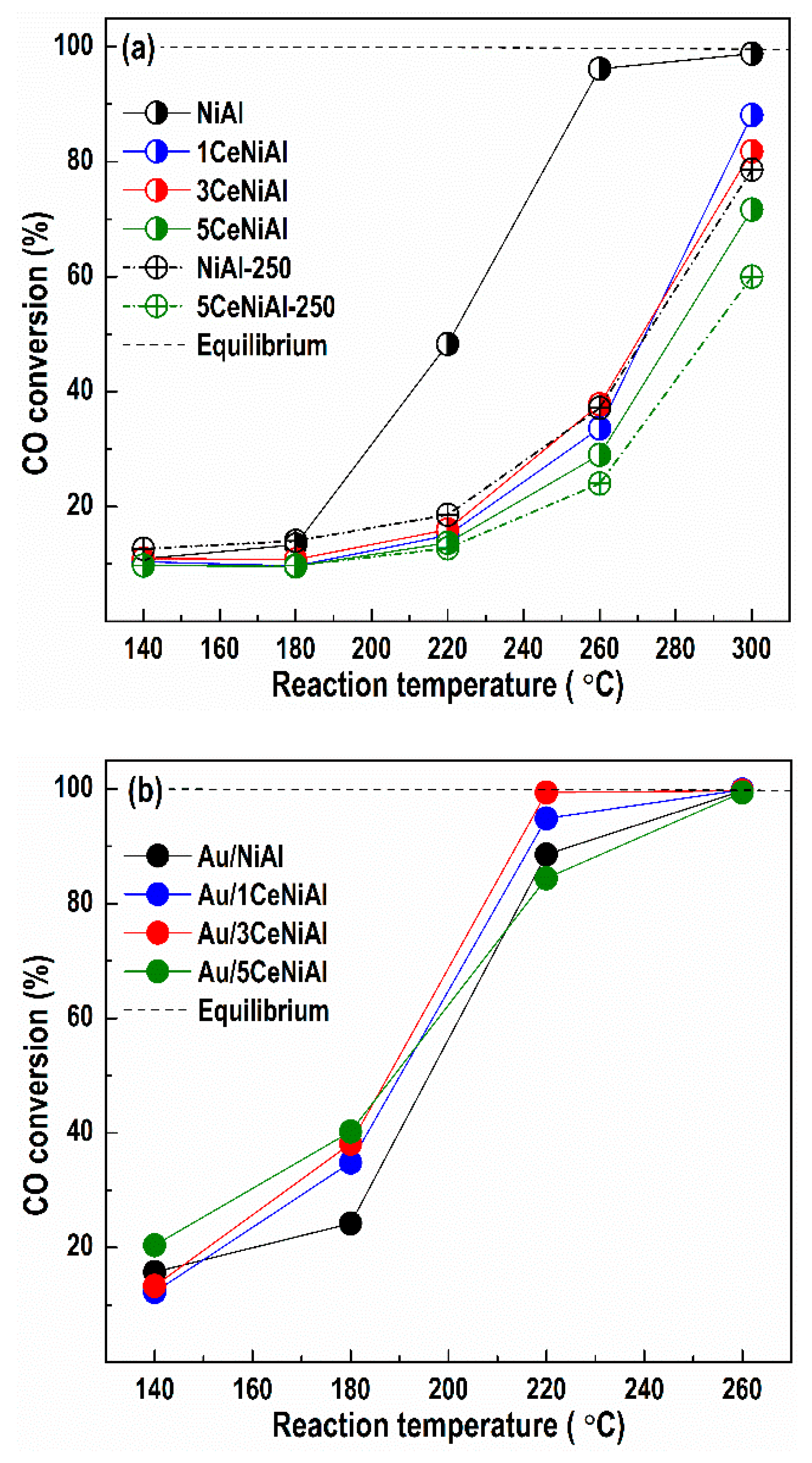
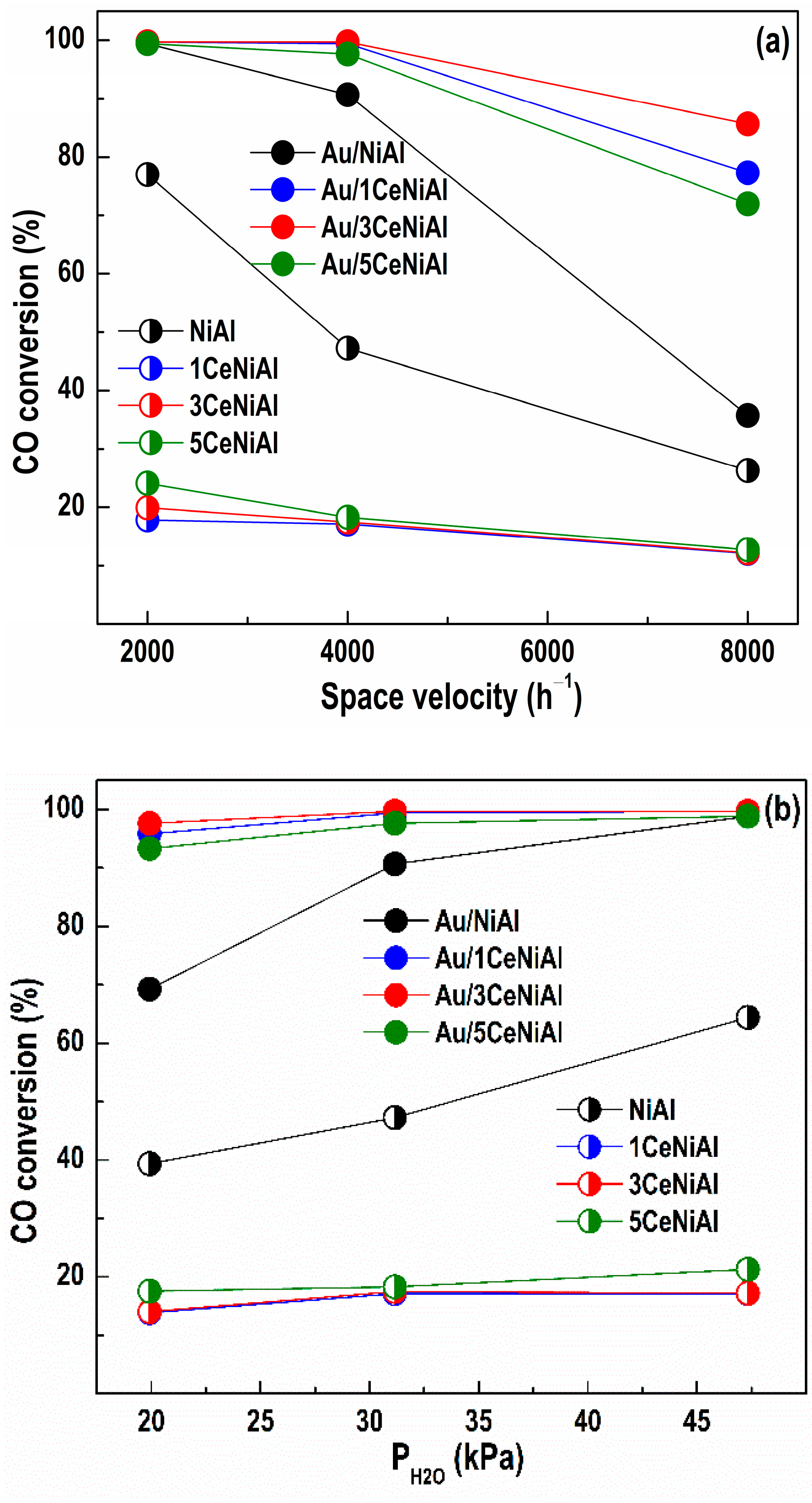
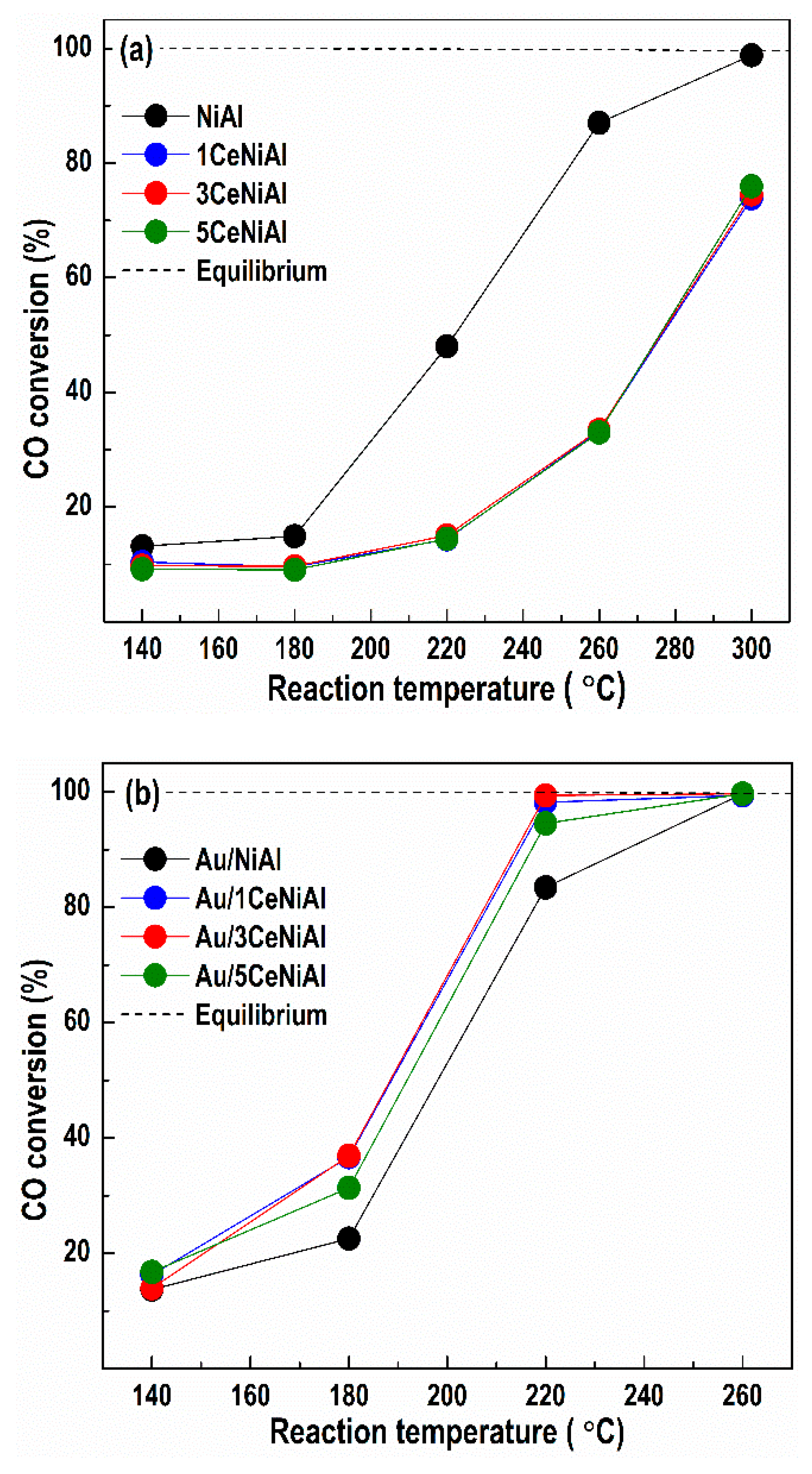
3.3. Water–Gas Shift Activity
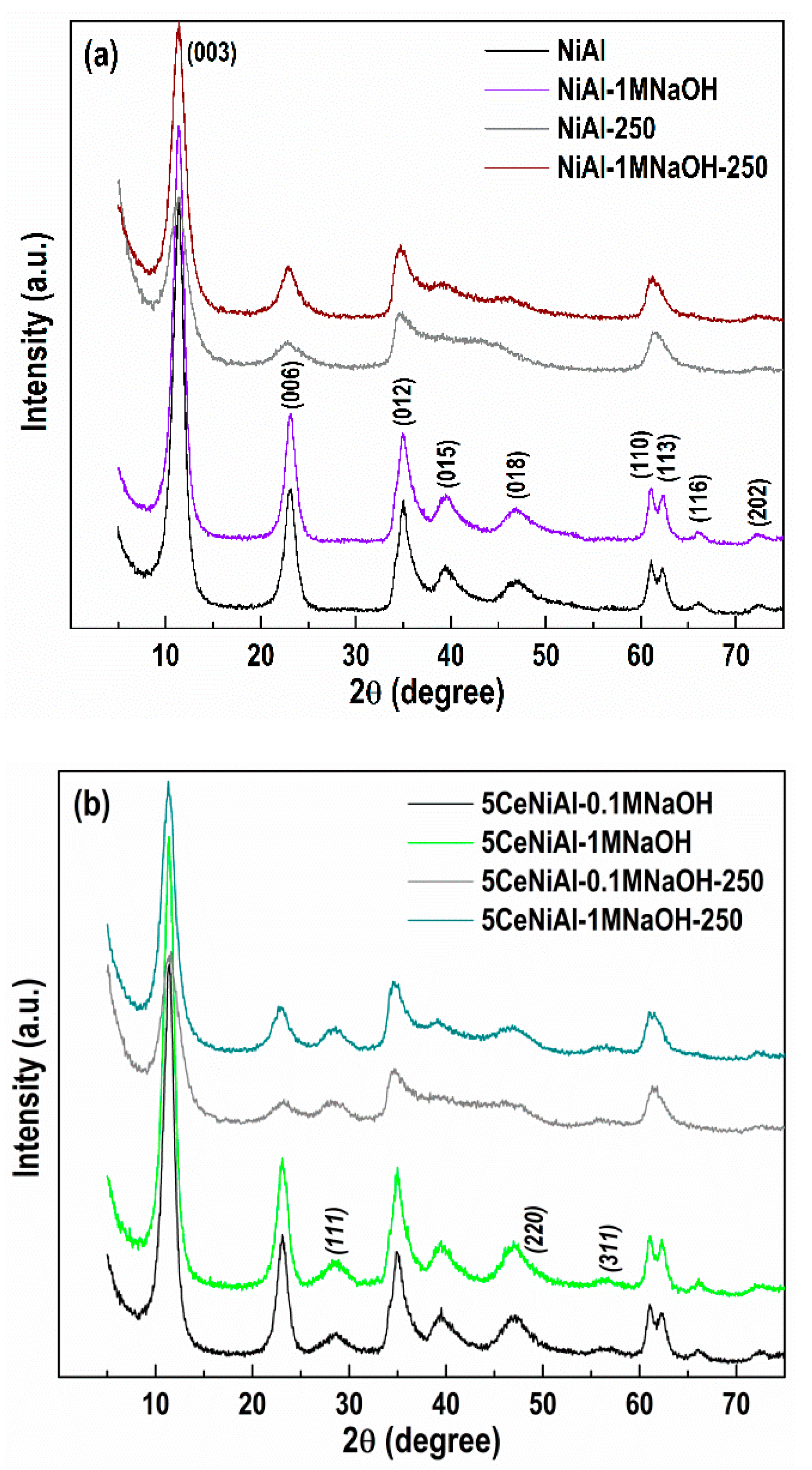
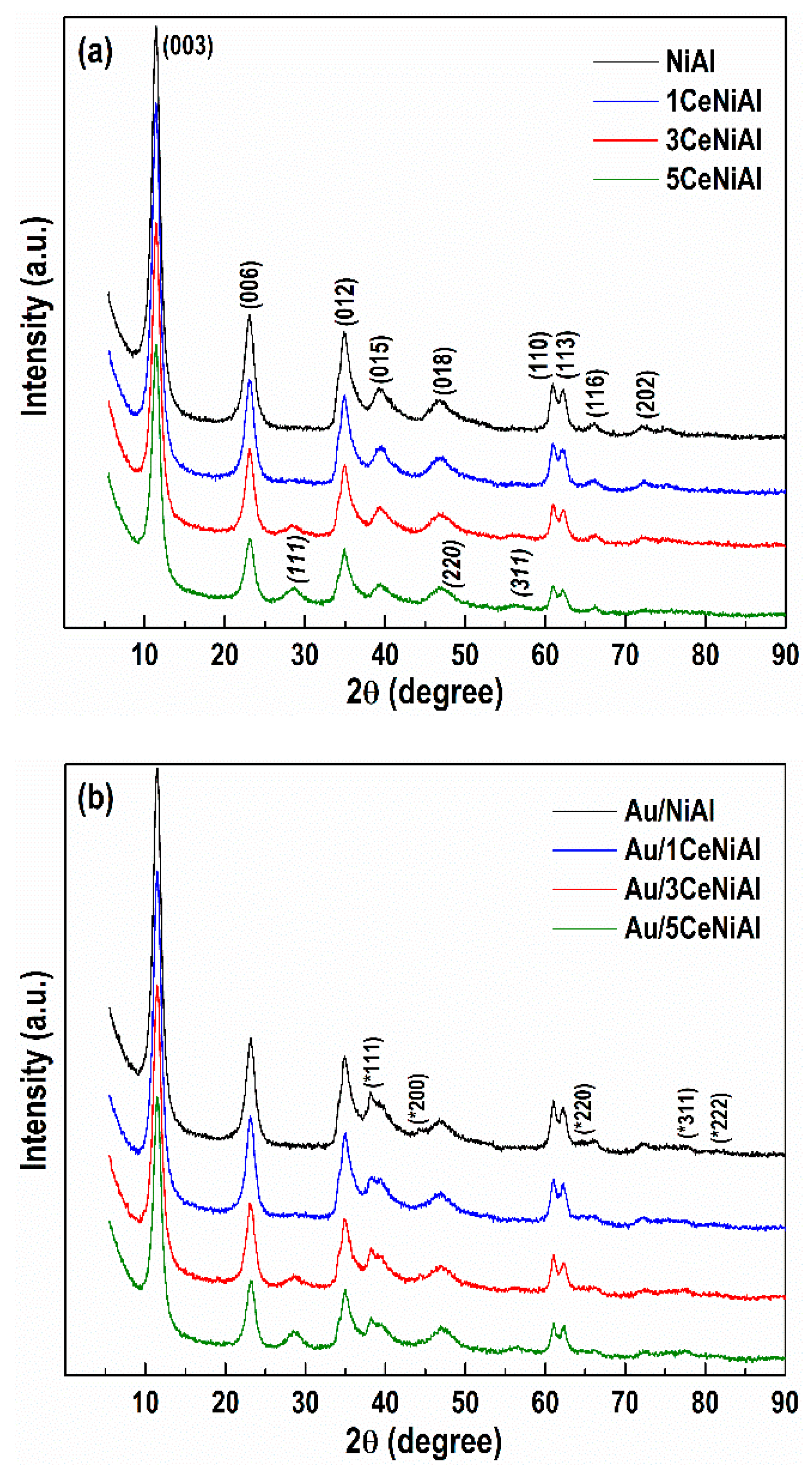
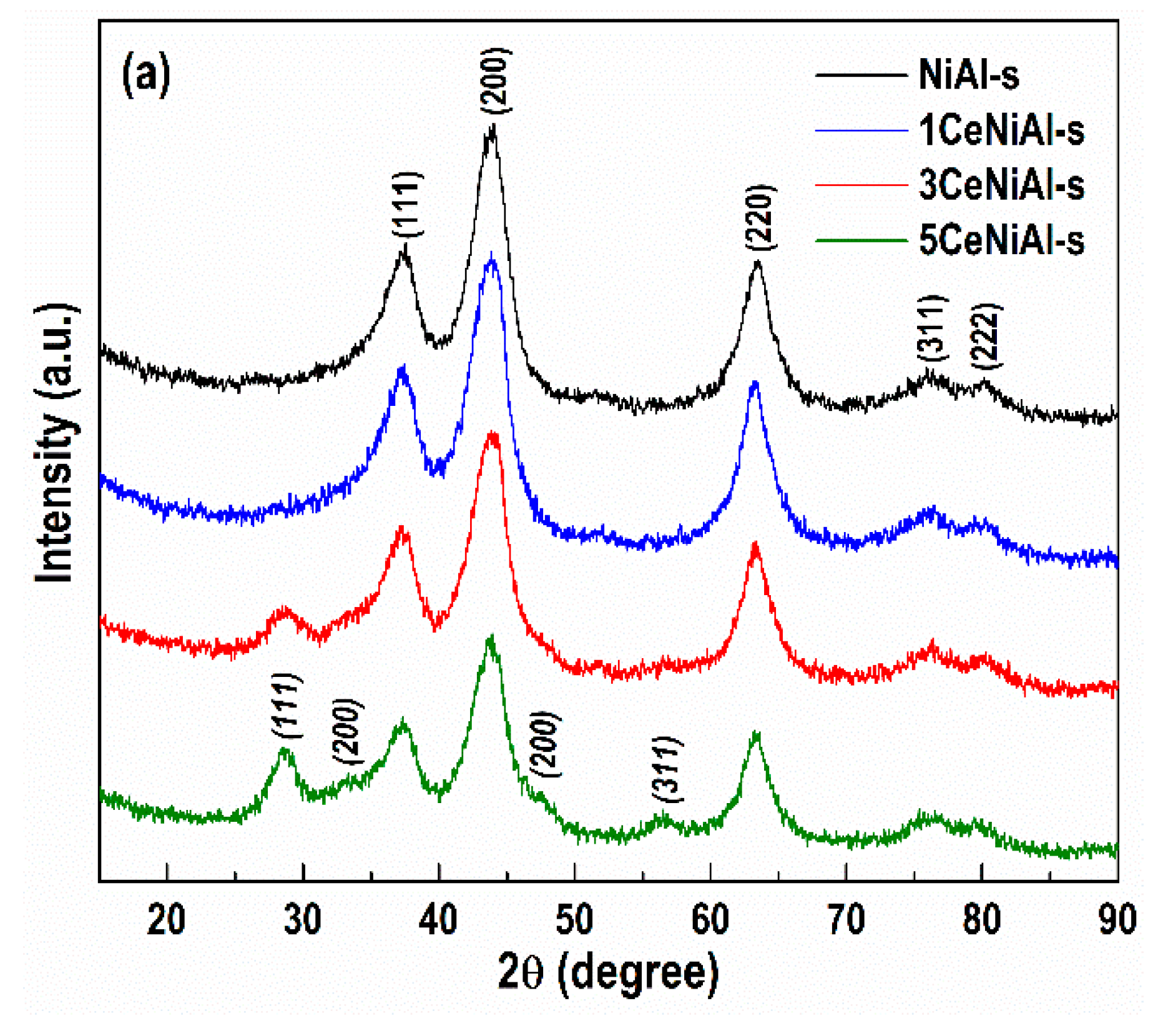
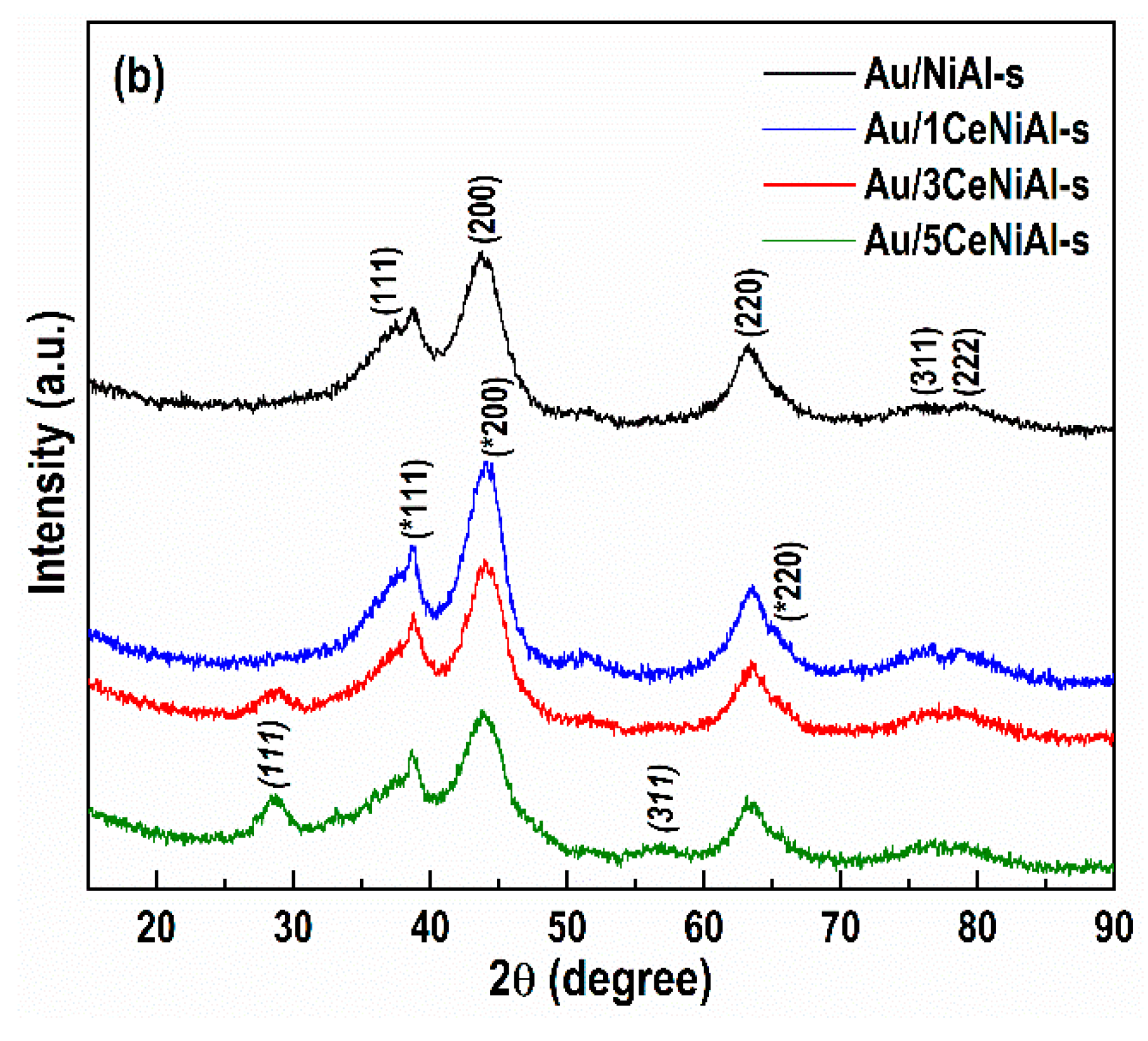
3.4. Structure and Phase Composition of As-Synthesized and Spent Catalysts
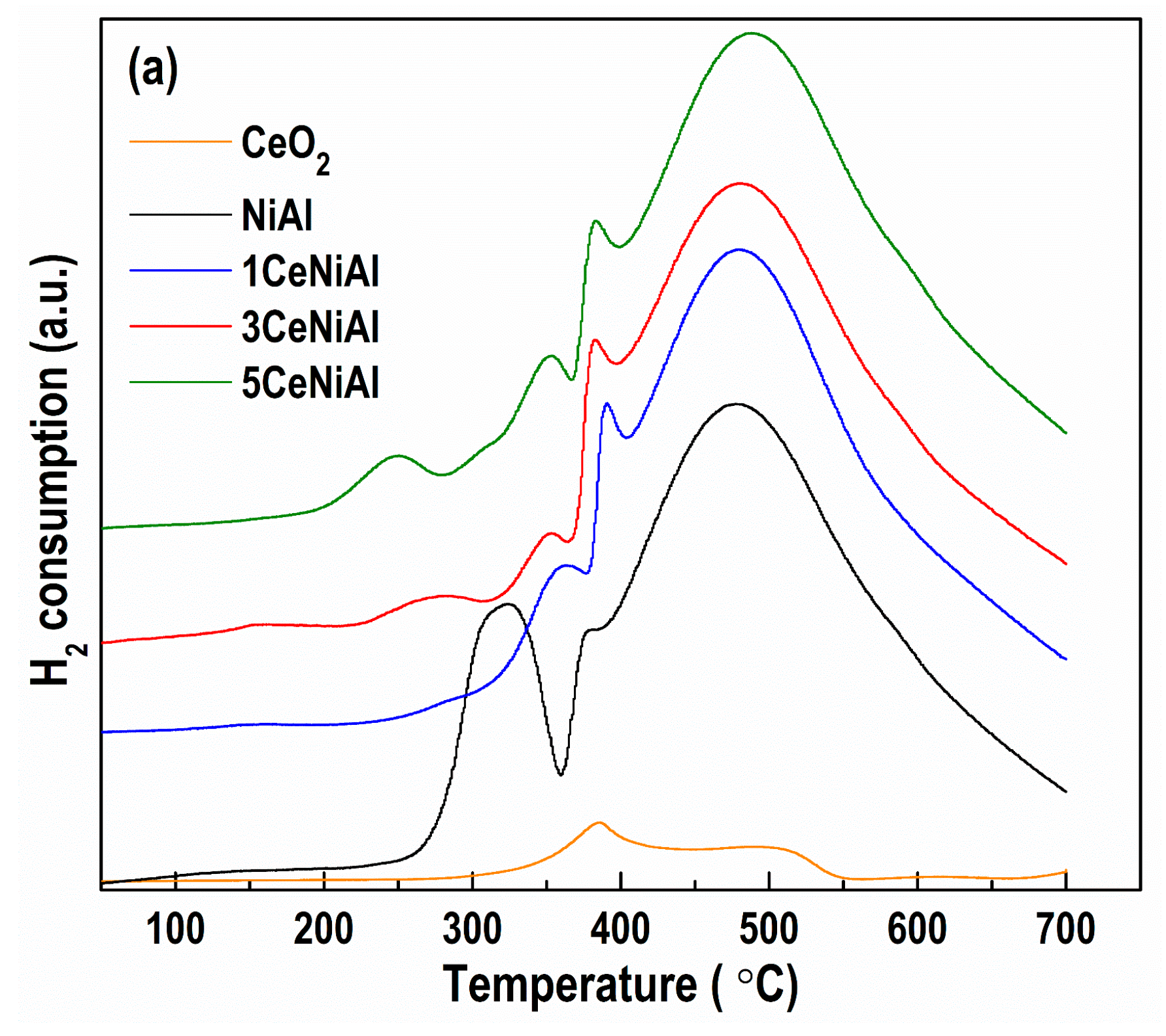
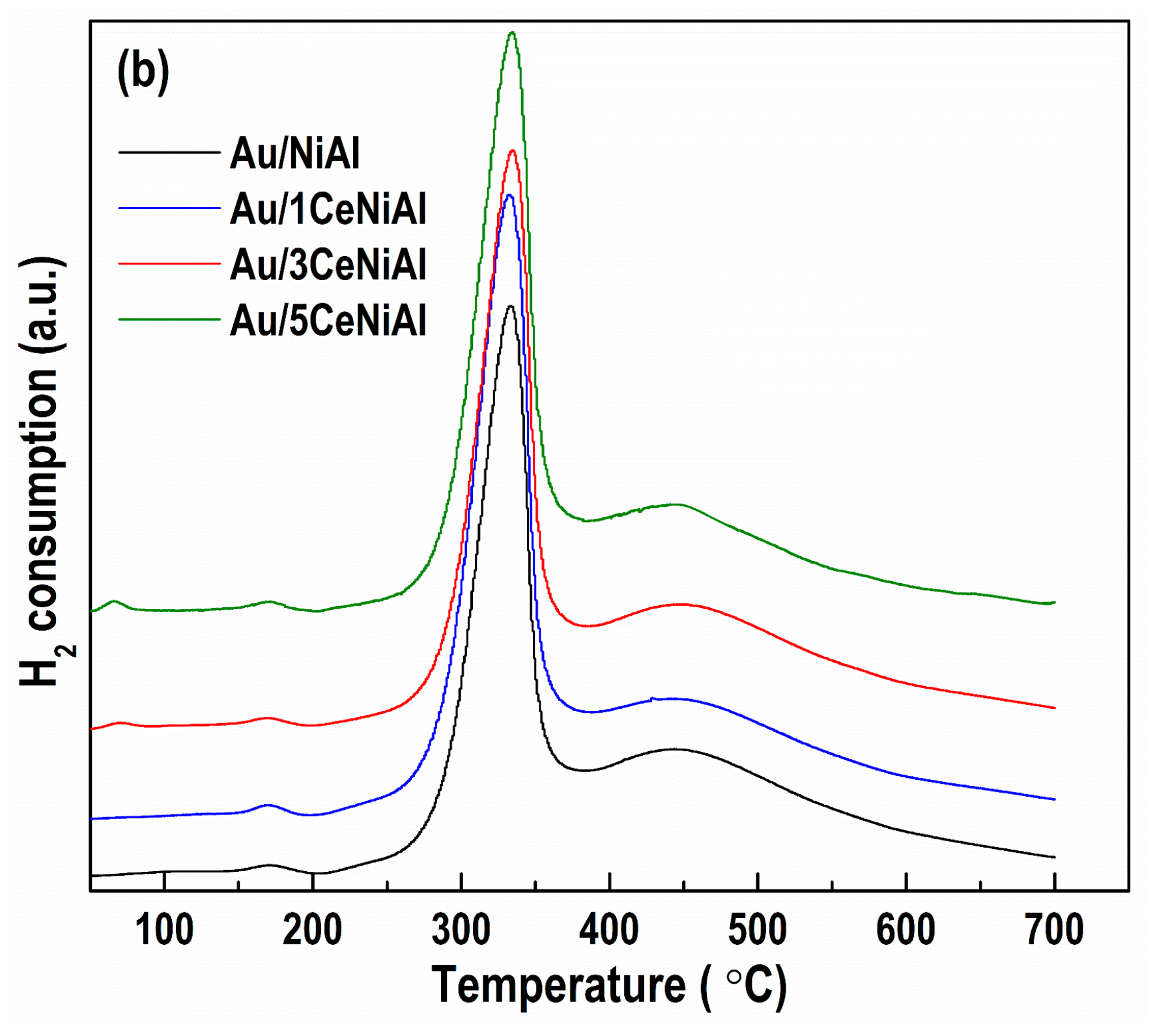
3.5. Catalyst Reduction Properties
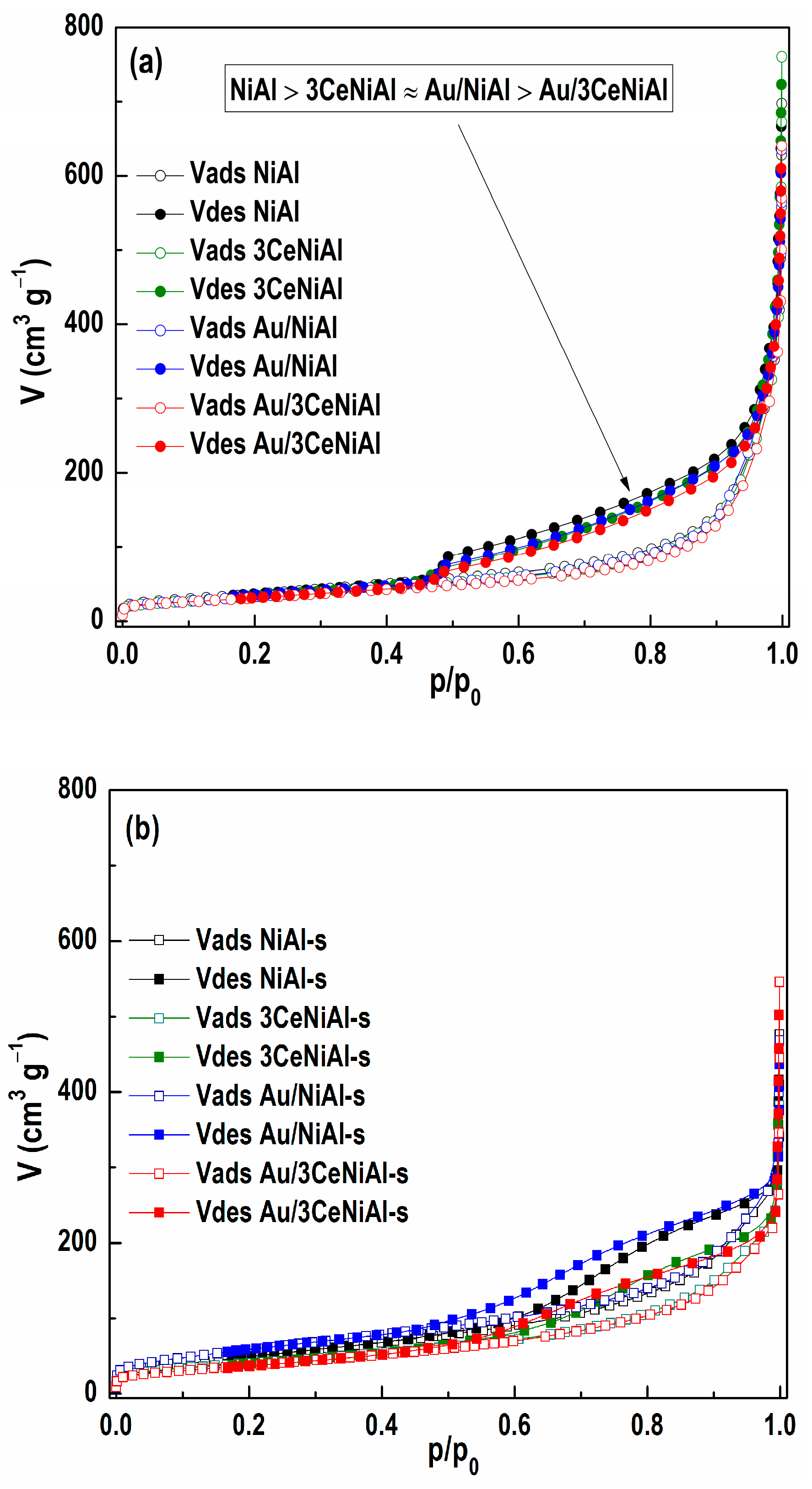
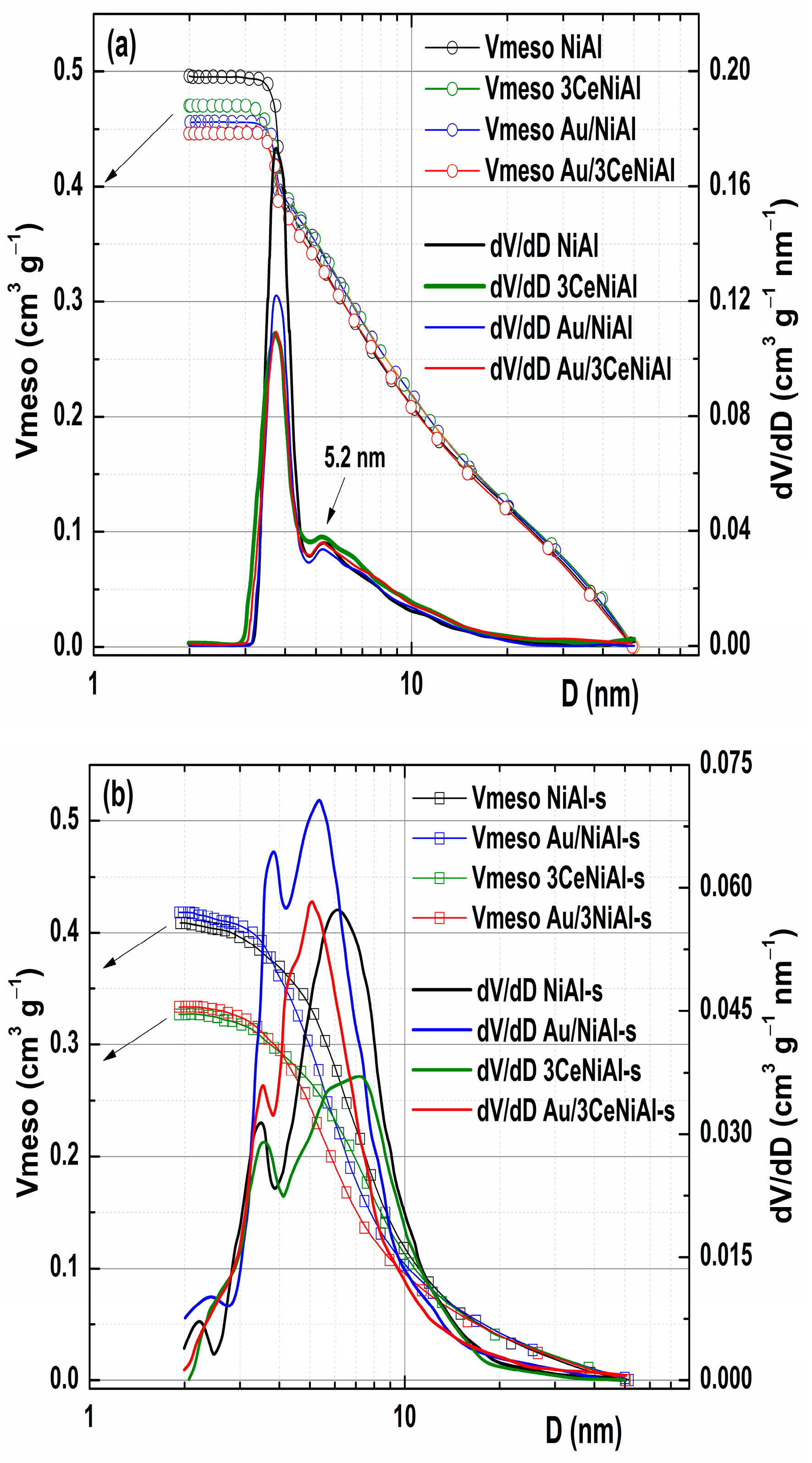
3.6. N2 Physisorption Analysis
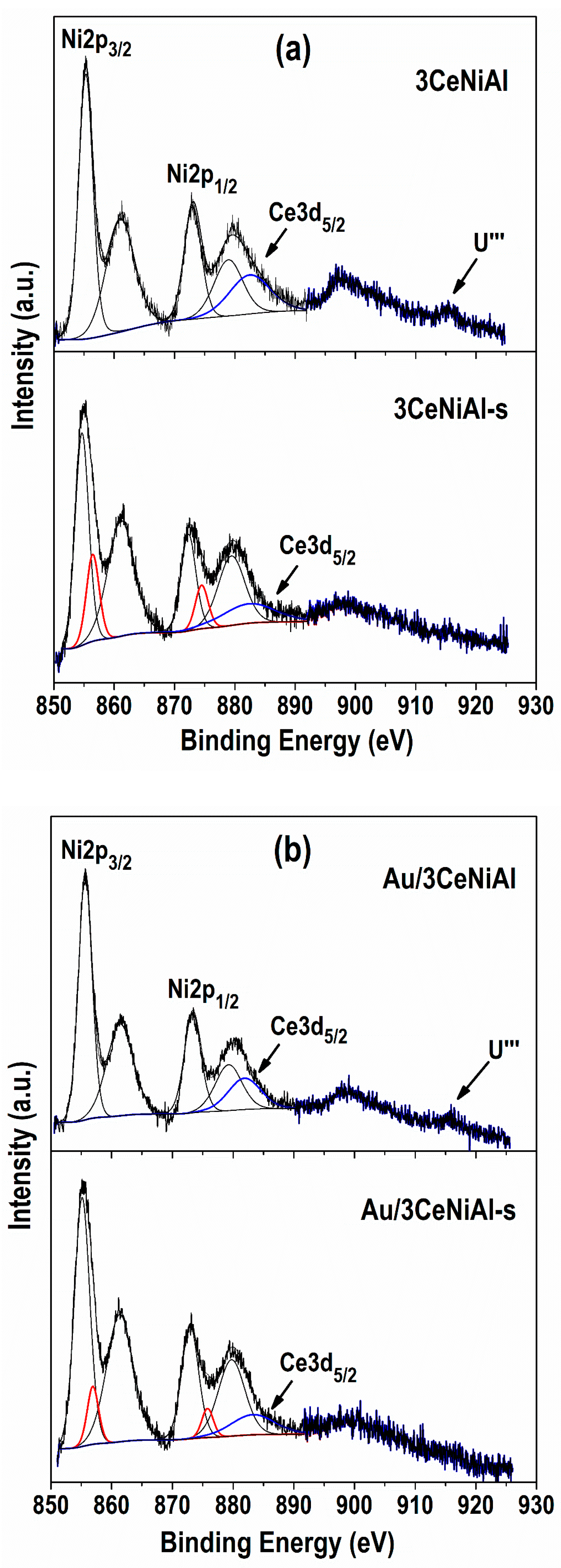
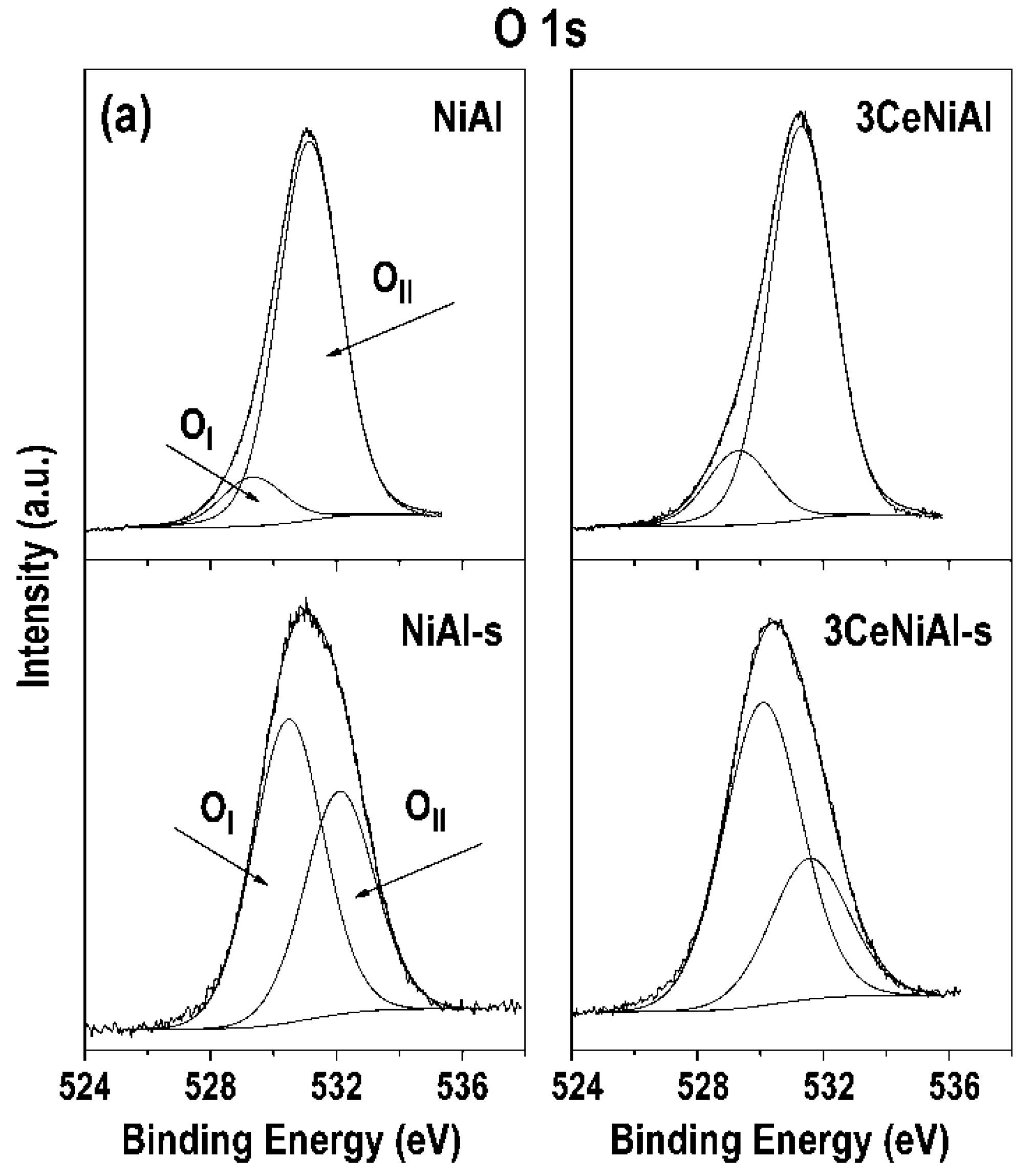
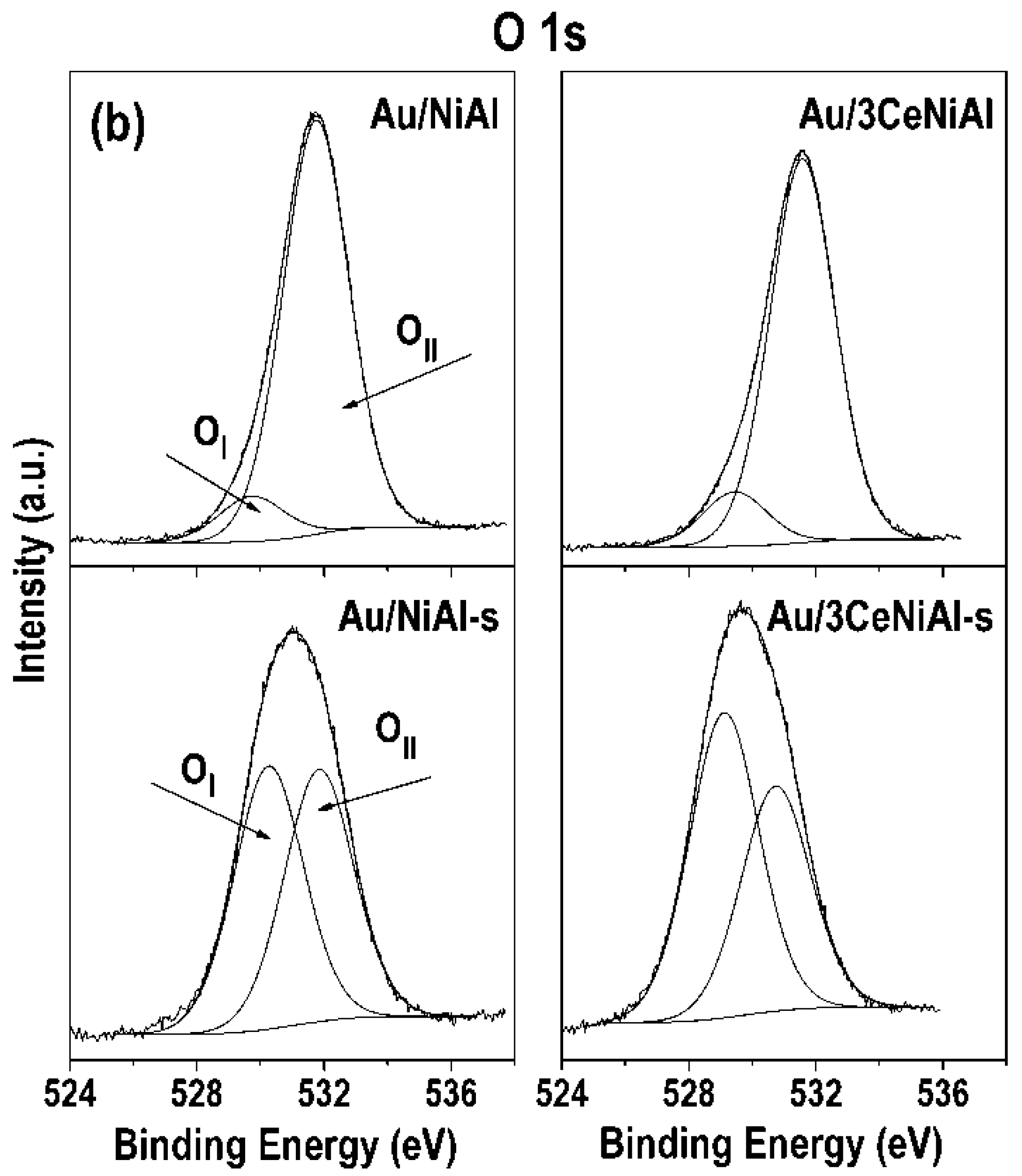
3.7. X-ray Photoelectron Spectroscopy (XPS)
4. Conclusions
- (i)
- The developed innovative approach for modification by ceria allows us to preserve the NiAl layered structure and to obtain a CeO2 phase with a good crystallinity in a relatively short time by a one-pot method, thus avoiding the calcination treatment, which simplifies the catalyst preparation procedure.
- (ii)
- The modification of NiAl LDHs with CeO2 neither improves reducibility nor enhances the WGS efficiency; however, the simultaneous presence of gold and ceria has a beneficial effect.
- (iii)
- It can be deduced that hydrogen production via WGS reaction is affected by the amount of ceria in the Au/NiAl catalyst.
- (iv)
- The addition of 3 wt.% CeO2 to the Au/NiAl catalyst provides the highest dispersion of gold particles in the spent catalyst (Au/3CeNiAl-s) and contributes to a good WGS performance—highest activity and significant stability.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baykara, S.Z. Hydrogen: A brief overview on its sources, production and environmental impact. Int. J. Hydrog. Energy 2018, 43, 10605–10614. [Google Scholar] [CrossRef]
- Sazali, N. Emerging technologies by hydrogen: A review. Int. J. Hydrog. Energy 2020, 45, 18753–18771. [Google Scholar] [CrossRef]
- Ratnasamy, C.; Wagner, J.P. Water gas shift catalysis. Catal. Rev. Sci. Eng. 2009, 51, 325–340. [Google Scholar] [CrossRef]
- LeValley, T.L.; Richard, A.R.; Fan, M. The progress in water gas shift and steam reforming hydrogen production technologies—A review. Int. J. Hydrog. Energy 2014, 39, 16983–17000. [Google Scholar] [CrossRef]
- Reddy, G.K.; Smirniotis, P.G. Water Gas Shift Reaction: Research Developments and Applications; Elsevier: Amsterdam, The Netherlands, 2015; p. 269. [Google Scholar]
- Chen, W.-H.; Chen, C.-Y. Water gas shift reaction for hydrogen production and carbon dioxide capture: A review. Appl. Energy 2020, 258, 114078. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A review of recent advances in water-gas shift catalysis for hydrogen production. Emergent Mater. 2020, 3, 881–917. [Google Scholar] [CrossRef]
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sust. Energy Rev. 2018, 93, 549–556. [Google Scholar] [CrossRef]
- Cavalcanti, F.; Schmal, M.; Giudici, R.; Alves, R. A catalyst selection method for hydrogen production through Water-Gas Shift Reaction using artificial neural networks. J. Environ. Manag. 2019, 237, 585–594. [Google Scholar] [CrossRef]
- Burch, R. Gold catalysts for pure hydrogen production in the water–gas shift reaction: Activity, structure and reaction mechanism. Phys. Chem. Chem. Phys. 2006, 8, 5483–5500. [Google Scholar] [CrossRef]
- Tao, F.; Ma, Z. Water–gas shift on gold catalysts: Catalyst systems and fundamental studies. Phys. Chem. Chem. Phys. 2013, 15, 15260–15270. [Google Scholar] [CrossRef]
- Reina, T.R.; González, M.C.; Palma, S.; Ivanova, S.; Odriozola, J.A. Twenty years of golden future in the water-gas shift reaction. In Heterogeneous Gold Catalysts and Catalysis; RSC Catalysis Series; Ma, Z., Dai, S., Eds.; RSC: Cambridge, UK, 2014; Volume 18, pp. 111–139, Chapter 5. [Google Scholar]
- Odabasi, C.; Günay, M.E.; Yildirim, R. Knowledge extraction for water gas shift reaction over noble metal catalysts from publications in the literature between 2002 and 2012. Int. J. Hydrog. Energy 2014, 39, 5733–5746. [Google Scholar] [CrossRef]
- Carter, J.H.; Hutchings, G.J. Recent advances in the gold-catalyzed low-temperature water-gas shift reaction. Catalysts 2018, 8, 627. [Google Scholar] [CrossRef]
- Tabakova, T. Recent advances in design of gold-based catalysts for H2 clean-up reactions. Review article. Front. Chem. 2019, 7, 517. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, A.; Gomez-Cortes, A.; Zanella, R.; Diaz, G.; Saniger, J.M. Gold nanoparticles: Support effects for the WGS reaction. J. Mol. Catal. Chem. 2007, 278, 200–208. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wang, A.; Zhang, T.; Mou, C.-Y. Catalysis by gold: New insights into the support effect. Review. Nano Today 2013, 8, 403–416. [Google Scholar] [CrossRef]
- Yang, M.; Flytzani-Stephanopoulos, M. Design of single-atom metal catalysts on various supports for the low temperature water-gas shift reaction. Catal. Today 2017, 298, 216–225. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Trovarelli, A. Structural properties and nonstoichiometric behavior of CeO2. In Catalysis by Ceria and Related Materials; Trovarelli, A., Ed.; Imperial College Press: London, UK, 2002; pp. 15–50. [Google Scholar]
- Polychronopoulou, K.; Kalamaras, C.M.; Efstathiou, A.M. Ceria-Based Materials for Hydrogen Production Via Hydrocarbon Steam Reforming and Water-Gas Shift Reactions. Recent Pat. Mater. Sci. 2011, 4, 122–145. [Google Scholar] [CrossRef]
- Sun, C.; Li, H.; Chen, L. Nanostructured ceria-based materials: Synthesis, properties, and applications. Energy Environ. Sci. 2012, 5, 8475–8505. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and catalytic applications of CeO2‑based materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Schmitt, R.; Nenning, A.; Kraynis, O.; Korobko, R.; Frenkel, A.I.; Lubomirsky, I.; Haile, S.M.; Rupp, J.L.M. A review of defect structure and chemistry in ceria and its solid solutions. Chem. Soc. Rev. 2020, 49, 554–592. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au-CeO2 catalysts for low-temperature water-gas shift. Catal. Lett. 2001, 77, 87–95. [Google Scholar] [CrossRef]
- Andreeva, D.; Idakiev, V.; Tabakova, T.; Ilieva, L.; Falaras, P.; Bourlinos, A.; Travlos, A. Low-temperature water–gas shift reaction over Au/CeO2 catalysts. Catal. Today 2002, 72, 51–57. [Google Scholar] [CrossRef]
- Luengnaruemitchai, A.; Osuwan, S.; Gulari, E. Comparative studies of low temperature water–gas shift reaction over Pt/CeO2, Au/CeO2, and Au/Fe2O3 catalysts. Catal. Commun. 2003, 4, 215–221. [Google Scholar] [CrossRef]
- Tabakova, T.; Boccuzzi, F.; Manzoli, M.; Sobczak, J.W.; Idakiev, V.; Andreeva, D. A comparative study of nanosized IB/ceria catalysts for low-temperature water-gas shift reaction. Appl. Catal. A Gen. 2006, 298, 127–143. [Google Scholar] [CrossRef]
- Karpenko, A.; Leppelt, R.; Plzak, V.; Cai, J.; Chuvilin, A.; Schumacher, B.; Kaiser, U.; Behm, R. Influence of the catalyst surface area on the activity and stability of Au/CeO2 catalysts for the low- temperature water gas shift reaction. Top. Catal. 2007, 44, 183–197. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Kucěrova, G.; Bansmann, J.; Behm, R.J. Active Au species during the low-temperature water gas shift reaction on Au/CeO2: A time-resolved operando XAS and DRIFTS study. ACS Catal. 2017, 7, 6471–6484. [Google Scholar] [CrossRef]
- Lenite, B.A.; Galletti, C.; Specchia, S. Studies on Au catalysts for water gas shift reaction. Int. J. Hydrog. Energy 2011, 36, 7750–7758. [Google Scholar] [CrossRef]
- Andreeva, D.; Tabakova, T.; Ilieva, L. Ceria-based gold catalysts: Synthesis, properties and catalytic performance for the WGS and PROX processes. In Catalysis by Ceria and Related Materials, 2nd ed.; Trovarelli, A., Fornasiero, P., Eds.; Imperial College Press: London, UK, 2013; p. 497. [Google Scholar]
- Yi, N.; Flytzani-Stephanopoulos, M. Gold/ceria: The making of a robust catalyst for fuel processing and hydrogen production. In Catalysis by Materials with Well-Defined Structures; Wu, Z., Overbury, S.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 133–158. [Google Scholar]
- Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. The role of Au, Cu & CeO2 and their interactions for an enhanced WGS performance. Appl. Catal. B Environ. 2016, 187, 98–107. [Google Scholar]
- Fu, X.-P.; Guo, L.-W.; Wang, W.-W.; Ma, C.; Jia, C.-J.; Wu, K.; Si, R.; Sun, L.-D.; Yan, C.-H. Direct identification of active surface species for the water–gas shift reaction on a gold–ceria catalyst. J. Am. Chem. Soc. 2019, 41, 4613–4623. [Google Scholar] [CrossRef]
- Tabakova, T.; Ivanov, I.; Zanella, R.; Karakirova, Y.; Sobczak, J.W.; Lisowski, W.; Kaszkur, Z.; Ilieva, L. Unraveling the effect of alumina-supported Y-doped ceria composition and method of preparation on the WGS activity of gold catalysts. Int. J. Hydrog. Energy 2020, 45, 26238–26253. [Google Scholar] [CrossRef]
- Santos, J.L.; Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Gold promoted Cu/ZnO/Al2O3 catalysts prepared from hydrotalcite precursors: Advanced materials for the WGS reaction. Appl. Catal. B Environ. 2017, 201, 310–317. [Google Scholar] [CrossRef]
- Santos, J.L.; Reina, T.R.; Ivanov, I.; Penkova, A.; Ivanova, S.; Tabakova, T.; Centeno, M.A.; Idakiev, V.; Odriozola, J.A. Multicomponent Au/Cu-ZnO-Al2O3 catalysts: Robust materials for clean hydrogen production. Appl. Catal. A Gen. 2018, 558, 91–98. [Google Scholar] [CrossRef]
- Meng, Y.; Chen, Y.; Zhou, X.; Pan, G.; Xia, S. Experimental and theoretical investigations into the activity and mechanism of the water-gas shift reaction catalyzed by Au nanoparticles supported on Zn-Al/Cr/Fe layered double hydroxides. Int. J. Hydrog. Energy 2020, 45, 464–476. [Google Scholar] [CrossRef]
- Xia, S.; Fang, L.; Meng, Y.; Zhang, X.; Zhang, L.; Yang, C.; Ni, Z. Water-gas shift reaction catalyzed by layered double hydroxides supported Au-Ni/Cu/Pt bimetallic alloys. Appl. Catal. B Environ. 2020, 272, 118949. [Google Scholar] [CrossRef]
- Fuentes, E.M.; Faro, A.C., Jr.; Silva, T.F.; Assaf, J.M.; Rangel, M.C. A comparison between copper and nickel-based catalysts obtained from hydrotalcite-like precursors for WGSR. Catal. Today 2011, 171, 290–296. [Google Scholar] [CrossRef]
- Gabrovska, M.; Idakiev, V.; Tenchev, K.; Nikolova, D.; Edreva-Kardjieva, R.; Crisan, D. Catalytic performance of Ni-Al layered double hydroxides in CO purification processes. Russ. J. Phys. Chem. A 2013, 87, 2152–2159. [Google Scholar] [CrossRef]
- Fuentes, E.M.; Aires, F.J.C.S.; Prakash, S.; Faro, A.C., Jr.; Silva, T.F.; Assaf, J.M.; Rangel, M.C. The effect of metal content on nickel-based catalysts obtained from hydrotalcites for WGSR in one step. Int. J. Hydrog. Energy 2014, 39, 815–828. [Google Scholar] [CrossRef]
- Gabrovska, M.; Tabakova, T.; Ivanov, I.; Kovacheva, D. Water-gas shift reaction over gold deposited on NiAl layered double hydroxides. React. Kinet. Mech. Cat. 2019, 127, 187–203. [Google Scholar] [CrossRef]
- Meza-Fuentes, E.; Rodriguez-Ruiz, J.; Solano-Polo, C.; Rangel, M.C.; Faro, A. Monitoring the structural and textural changes of Ni-Zn-Al hydrotalcites under heating. Thermochim. Acta 2020, 687, 178594. [Google Scholar] [CrossRef]
- Xia, S.; Dai, T.; Meng, Y.; Zhou, X.; Pan, G.; Zhang, X.; Ni, Z. Low temperature water-gas shift reaction catalyzed by hybrid NiO@NiCr elayered double hydroxides: Catalytic property, kinetics and mechanism investigation. Phys. Chem. Chem. Phys. 2020, 22, 12630–12643. [Google Scholar] [CrossRef] [PubMed]
- Gabrovska, M.; Ivanov, I.; Nikolova, D.; Kovacheva, D.; Tabakova, T. Hydrogen production via water-gas shift reaction over gold supported on Ni-based layered double hydroxides. Int. J. Hydrog. Energy 2021, 46, 458–473. [Google Scholar] [CrossRef]
- Bish, D.; Brindley, G. A reinvestigation of takovite, a nickelaluminium hydroxycarbonate of the pyroaurite group. Am. Mineral 1977, 62, 458–464. [Google Scholar]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Clause, O.; Goncalves Coelho, M.; Gazzano, M.; Matteuzzi, D.; Trifirò, F.; Vaccari, A. Synthesis and thermal reactivity of nickel-containing anionic clays. Appl. Clay Sci. 1993, 8, 169–186. [Google Scholar] [CrossRef]
- Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chaleogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Sanati, S.; Rezvani, Z. Co-intercalation of acid Red-27/sodium dodecyl sulfate in a Ce-containing Ni–Al-layered double hydroxide matrix and characterization of its luminescent properties. J. Mol. Liq. 2018, 249, 318–325. [Google Scholar] [CrossRef]
- Golovin, S.N.; Yapryntsev, M.N.; Ryltsova, I.G.; Veligzhanin, A.A.; Lebedeva, O.E. Novel cerium‑containing layered double hydroxide. Chem. Pap. 2020, 74, 367–370. [Google Scholar] [CrossRef]
- Reni, M.L.; Nesaraj, A.S. Preparative methods and recent technological applications of ceria -based nanostructured catalyst materials in chemical and other fields-a review. Mater. Res. Innov. 2020. [Google Scholar] [CrossRef]
- Matijević, E.; Hsu, W.P. Preparation and properties of monodispersed colloidal particles of lanthanide compounds. I. Gadolinium, Europium, Terbium, Samarium, and Cerium (lll). J. Colloid Interf. Sci. 1987, 118, 506–523. [Google Scholar] [CrossRef]
- Chen, P.L.; Chen, I.W. Reactive cerium (IV) oxide powders by the homogeneous precipitation method. J. Am. Ceram. Soc. 1993, 76, 1577–1583. [Google Scholar] [CrossRef]
- Zhou, X.D.; Huebner, W.; Anderson, H.U. Room-temperature homogeneous nucleation synthesis and thermal stability of nanometer single crystal CeO2. Appl. Phys. Lett. 2002, 80, 3814–3816. [Google Scholar] [CrossRef]
- Li, J.-G.; Ikegami, T.; Wang, Y.; Mori, T. Reactive Ceria Nanopowders via Carbonate Precipitation. J. Am. Ceram. Soc. 2002, 85, 2376–2378. [Google Scholar] [CrossRef]
- Yamashita, M.; Kameyama, K.; Yabe, S.; Yoshida, S.; Fujishiro, Y.; Kawai, T.; Sato, T. Synthesis and microstructure of ceria doped ceria as UV filters. J. Mater. Sci. 2002, 37, 683–687. [Google Scholar] [CrossRef]
- Uekawa, N.; Ueta, M.; Wu, Y.J.; Kakegawa, K. Synthesis of CeO2 spherical fine particles by homogeneous precipitation method with polyethylene glycol. Chem. Lett. 2002, 8, 854–855. [Google Scholar] [CrossRef]
- Zhou, X.D.; Huebner, W.; Anderson, H.U. Processing of nanometer-CeO2 particles. Chem. Mater. 2003, 15, 378–382. [Google Scholar] [CrossRef]
- Djuričić, B.; Pickering, S. Nanostructured cerium oxide: Preparation and properties of weakly agglomerated powders. J. Eur. Ceram. Soc. 1999, 19, 1925–1934. [Google Scholar] [CrossRef]
- Hirano, M.; Kato, E. Hydrothermal synthesis of nanocrystalline cerium (IV) oxide powders. J. Am. Ceram. Soc. 1999, 82, 768–788. [Google Scholar] [CrossRef]
- Chen, H.-I.; Chang, H.-Y. Synthesis of nanocrystalline cerium oxide particles by the precipitation method. Ceram. Int. 2005, 31, 795–802. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Niemark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Dubinin, M.M. Physical Adsorption of Gases and Vapors in Micropores. Prog. Surf. Membr. Sci. 1975, 9, l-70. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Lecloux, A.; Pirard, J.P. The importance of standard isotherms in the analysis of adsorption isotherms for determining the porous texture of solids. J. Colloid Interface Sci. 1979, 70, 265–281. [Google Scholar] [CrossRef]
- Monti, D.A.M.; Baiker, A. Temperature-programmed reduction. Parametric sensitivity and estimation of kinetic parameters. J. Catal. 1983, 83, 323–335. [Google Scholar] [CrossRef]
- Russo, M.; La Parola, V.; Testa, M.L.; Pantaleo, G.; Venezia, A.M.; Gupta, R.K.; Bordoloi, A.; Bordoloi, B. Structural insight in TiO2 supported CoFe catalysts for Fisher Tropsch synthesis at ambient pressure. Appl. Catal. A Gen. 2020, 600, 117621. [Google Scholar] [CrossRef]
- Benito, P.; Labajos, F.; Rives, V. Microwave-treated layered double hydroxides containing Ni2+ and Al3+: The effect of added Zn2+. J. Solid State Chem. 2006, 179, 3784–3797. [Google Scholar] [CrossRef]
- Tichit, D.; Medina, F.; Coq, B.; Dutartre, R. Activation under oxidizing and reducing atmospheres of Ni-containing layered double hydroxides. Appl. Catal. A Gen. 1997, 159, 241–258. [Google Scholar] [CrossRef]
- Trifirò, F.; Vaccari, A.; Clause, O. Nature and properties of nickel-containing mixed oxides obtained from hydrotalcite-type anionic clays. Catal. Today 1994, 21, 185–195. [Google Scholar] [CrossRef]
- Świrk, K.; Rønning, M.; Motak, M.; Beaunier, P.; Da Costa, P.; Grzybek, T. Ce-and Y-Modified Double-Layered Hydroxides as Catalysts for Dry Reforming of Methane: On the Effect of Yttrium Promotion. Catalysts 2019, 9, 56. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K.S.W.; Llewellyn, P.; Maurin, G. Adsorption by Powders and Porous Solids Principles, Methodology and Applications, 2nd ed.; Academic Press: Cambridge, MA, USA; Elsevier Ltd.: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Groen, J.C.; Peffer, L.A.A.; Pérez-Ramırez, J. Pore size determination in modified micro- and mesoporous materials. Pitfalls and limitations in gas adsorption data analysis. Micropor. Mesopor. Mat. 2003, 60, 1–17. [Google Scholar] [CrossRef]
- Kim, K.S.; Winograd, N. X-ray photoelectron spectroscopic studies of nickel-oxygen surfaces using oxygen and argon ion-bombardment. Surf. Sci. 1974, 43, 625–643. [Google Scholar] [CrossRef]
- Löchel, B.P.; Strehblow, H.H. Breakdown of passivity of nickel by fluoride: II. Surface analytical studies. J. Electrochem. Soc. 1984, 131, 713–723. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.S.C.; McIntyre, N.S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Zhang, X.; Yu, B.; Lv, C.; Zeng, N.; Luo, J.; Zhang, Z.; Song, J.; Liu, Y. Ni-Based Catalyst Derived from NiAl layered double hydroxide for vapor phase catalytic exchange between hydrogen and water. Nanomaterials 2019, 9, 1688. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Yi, H.; Tang, X.; Gao, F.; Yu, Q.; Zhou, Y.; Wang, J.; Huang, Y.; Yang, Z. Enhancement effects of ultrasound assisted in the synthesis of NiAl hydrotalcite for carbonyl sulfide removal. Ultrason. Sonochem. 2016, 32, 336–342. [Google Scholar] [CrossRef]
- Shyu, J.Z.; Otto, K.; Watkins, W.L.H.; Graham, G.W. Characterization of Pd/γ-alumina catalysts containing ceria. J. Catal. 1988, 114, 23–33. [Google Scholar] [CrossRef]
- Wang, L.; Meng, F. Oxygen vacancy and Ce3+ ion dependent magnetism of monocrystal CeO2 nanopoles synthesized by a facile hydrothermal method. Mater. Res. Bull. 2013, 48, 3492–3498. [Google Scholar] [CrossRef]
- Pan, Y.; Nilius, N.; Freund, H.-J.; Paier, J.; Penschke, C.; Sauer, J. Titration of Ce3+ Ions in the CeO2(111) Surface by Au Adatoms. Phys. Rev. Lett. 2013, 111, 206101. [Google Scholar] [CrossRef]
- Lu, B.; Zhuang, J.; Du, J.; Gu, F.; Xu, G.; Zhong, Z.; Liu, Q.; Su, F. Highly dispersed Ni nanocatalysts derived from NiMnAl-hydrotalcites as high-performing catalyst for low-temperature syngas methanation. Catalysts 2019, 9, 282. [Google Scholar] [CrossRef]
- Praline, G.; Koel, B.E.; Hance, R.L.; Lee, H.-I.; White, J.M. X-Ray photoelectron study of the reaction of oxygen with cerium. J. Electron Spectros. Relat. Phenom. 1980, 21, 17–30. [Google Scholar] [CrossRef]
- Laachir, A.; Perrichon, V.; Badri, A.; Lamotte, J.; Catherine, E.; Lavalley, J.C.; El Fallah, J.; Hilaire, L.; Le Normand, F.; Quéméré, E.; et al. Reduction of CeO2 by hydrogen. Magnetic susceptibility and Fourier-transform infrared, ultraviolet and X-ray photoelectron spectroscopy measurements. J. Chem. Soc. Faraday Trans. 1991, 87, 1601–1609. [Google Scholar] [CrossRef]
- Pereira, A.; Blouin, M.; Pillonnet, A.; Guay, D. Structure and valence properties of ceria films synthesized by laser ablation under reducing atmosphere. Mater. Res. Express 2014, 1, 015704. [Google Scholar] [CrossRef]
- Genty, E.; Brunet, J.; Poupin, C.; Casale, S.; Capelle, S.; Massiani, P.; Siffert, S.; Cousin, R. Co-Al mixed oxides prepared via LDH route using microwaves or ultrasound: Application for catalytic toluene total oxidation. Catalysts 2015, 5, 851–867. [Google Scholar] [CrossRef]
- Ingo, G.M.; Paparazzo, E.; Bagnarelli, O.; Zacchetti, N. XPS studies on cerium, zirconium and yttrium valence states in plasma-sprayed coatings. Surf. Interface Anal. 1990, 16, 515–519. [Google Scholar] [CrossRef]
- Mansour, A.N.; Melendres, C.A. Characterization of Electrochemically Prepared γ-NiOOH by XPS. Surf. Sci. Spectra 1994, 3, 271–278. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Di, Q.; Zhao, H.; Liang, B.; Yang, J. Mutual effects of fluorine dopant and oxygen vacancies on structural and luminescence characteristics of F doped SnO2 nanoparticles. Materials 2017, 10, 1398. [Google Scholar] [CrossRef]
- Tu, Y.; Chen, S.; Li, X.; Gorbaciova, J.; Gillin, W.P.; Krause, S.; Briscoe, J. Control of oxygen vacancies in ZnO nanorods by annealing and their influence on ZnO/PEDOT: PSS diode behavior. J. Mater. Chem. C 2018, 6, 1815–1821. [Google Scholar] [CrossRef]
- Jain, S.; Shah, J.; Negi, N.S.; Sharma, C.; Kotnala, R.K. Nickel substituted oxygen deficient nanoporous lithium ferrite based green energy device hydroelectric cell. J. Alloys Compd. 2020, 827, 154334. [Google Scholar]
- Rodriguez, J.A.; Wang, X.; Liu, P.; Wen, W.; Hanson, J.C.; Hrbek, J.; Perez, M.; Evans, J. Gold nanoparticles on ceria: Importance of O vacancies in the activation of gold. Top. Catal. 2007, 44, 73–81. [Google Scholar] [CrossRef]
- Vindigni, F.; Manzoli, M.; Damin, A.; Tabakova, T.; Zecchina, A. Surface and inner defects in Au/CeO2 WGS catalysts: Relation between Raman properties, reactivity and morphology. Chem. Eur. J. 2011, 17, 4356–4361. [Google Scholar] [CrossRef]
- Laguna, O.H.; Dominguez, M.I.; Romero-Sarria, F.; Odriozola, J.A.; Centeno, M.A. Role of oxygen vacancies in gold oxidation catalysis. In Heterogeneous Gold Catalysts and Catalysis; Ma, Z., Dai, S., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2014; pp. 489–511. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a (Å) | V (Å3) | L (nm) |
|---|---|---|---|
| CeO2-105 | 5.4211(10) | 159.32(9) | 7.27(6) |
| CeO2-250 | 5.4185(9) | 159.09(8) | 7.55(6) |
| CeO2-standard | 5.4113 | 158.46 | - |
| Sample | Chemical Composition (wt.%) | Ni2+/Al3+ Molar Ratio | |||
|---|---|---|---|---|---|
| NiO | CeO2 | Al2O3 | Au | ||
| NiAl | 78.55 | - | 21.45 | - | 2.5 |
| 1CeNiAl | 77.76 | 1.0 | 21.24 | - | 2.5 |
| 3CeNiAl | 76.19 | 3.0 | 20.81 | - | 2.5 |
| 5CeNiAl | 74.62 | 5.0 | 20.38 | - | 2.5 |
| Au/NiAl | 76.30 | - | 20.70 | 3.0 | 2.5 |
| Au/1CeNiAl | 75.43 | 0.97 | 20.60 | 3.0 | 2.5 |
| Au/3CeNiAl | 73.90 | 2.91 | 20.19 | 3.0 | 2.5 |
| Au/5CeNiAl | 72.38 | 4.85 | 19.77 | 3.0 | 2.5 |
| Sample | aTK (Å) | cTK (Å) | VTK (Å3) | LTK (nm) | LCeO2 (nm) | LAu (nm) |
|---|---|---|---|---|---|---|
| NiAl | 3.0333(6) | 22.964(13) | 182.98(13) | 9.90 | - | - |
| 1CeNiAl | 3.0334(6) | 22.958(13) | 182.98(13) | 9.80 | - | - |
| 3CeNiAl | 3.0336(6) | 22.974(12) | 183.07(12) | 10.10 | - | - |
| 5CeNiAl | 3.0338(6) | 22.972(13) | 183.11(13) | 10.20 | 2.16 | - |
| Au/NiAl | 3.0343(7) | 22.994(12) | 183.34(12) | 8.05 | - | 14.0 |
| Au/1CeNiAl | 3.0355(6) | 23.026(11) | 183.75(12) | 9.10 | 1.90 | 11.0 |
| Au/3CeNiAl | 3.0339(6) | 23.020(12) | 183.49(13) | 9.30 | 1.82 | 17.0 |
| Au/5CeNiAl | 3.0353(6) | 23.017(13) | 183.65(13) | 10.10 | 2.18 | 16.0 |
| * Takovite | 3.0250 | 22.5950 | 179.06 | - | - | - |
| Sample | LNiO (nm) | LCeO2 (nm) | LAu (nm) |
|---|---|---|---|
| NiAl-s | 3.09 | - | - |
| 1CeNiAl-s | 3.08 | n.d.* | - |
| 3CeNiAl-s | 2.98 | 3.10 | - |
| 5CeNiAl-s | 2.78 | 3.50 | - |
| Au/NiAl-s | 2.65 | - | 8.5 |
| Au/1CeNiAl-s | 2.63 | n.d. | 11.8 |
| Au/3CeNiAl-s | 2.41 | 4.95 | 5.8 |
| Au/5CeNiAl-s | 2.24 | 3.65 | 9.2 |
| Sample | SSA (m2 g−1) | Vmic (m3 g−1) | Vmeso (m3 g−1) | Vtot (m3 g−1) |
|---|---|---|---|---|
| NiAl | 138 | 0.048 | 0.496 | 0.575 |
| NiAl-s | 191 | 0.058 | 0.409 | 0.417 |
| 3CeNiAl | 126 | 0.042 | 0.474 | 0.569 |
| 3CeNiAl-s | 147 | 0.053 | 0.327 | 0.367 |
| Au/NiAl | 129 | 0.043 | 0.456 | 0.596 |
| Au/NiAl-s | 221 | 0.072 | 0.418 | 0.466 |
| Au/3CeNiAl | 118 | 0.041 | 0.446 | 0.534 |
| Au/3CeNiAl-s | 143 | 0.047 | 0.333 | 0.350 |
| Sample | Ni 2p3/2 | Al 2p | O 1s Position (eV) and Contribution (%) * | Au 4f7/2 | Ce 3d5/2 | |
|---|---|---|---|---|---|---|
| OI | OII | |||||
| NiAl | 855.4 | 74.2 | 529.3(11) | 531.1(89) | ||
| Au/NiAl | 855.7 | 74.5 | 529.7(10) | 531.7(90) | 84.7 | |
| 3CeNiAl | 855.3 | 74.2 | 529.3(16) | 531.3(84) | 882.2 | |
| Au/3CeNiAl | 855.7 | 74.5 | 529.4(12) | 531.6(88) | 84.5 | 881.8 |
| Catalyst | Ni 2p3/2 Position (eV) and Contribution (%) | Ni3+/Ni2+ Ratio | O 1s Position (eV) and Contribution (%) | OII/(OII+OI) | Au 4f7/2 | Ce3d5/2 | ||
|---|---|---|---|---|---|---|---|---|
| Ni2+ | Ni3+ | OI | OII | |||||
| NiAl-s | 855.2(90) | 856.8(10) | 0.11 | 530.5(58) | 532.1(42) | 0.42 | ||
| Au/NiAl-s | 855.2(93) | 856.9(7) | 0.08 | 530.3(51) | 531.8(49) | 0.49 | 84.4 | |
| 3CeNiAl-s | 854.7(84) | 856.4(16) | 0.19 | 530.1(68) | 531.6(32) | 0.32 | 881.9 | |
| Au/3CeNiAl-s | 855.1(92) | 856.9(8) | 0.09 | 529.1(58) | 531.7(42) | 0.42 | 84.4 | 881.9 |
| Catalyst | Au/(Ni + Al) | Au/(Ce + Ni + Al) | Ce/(Ni + Al) | Ni/Al |
|---|---|---|---|---|
| NiAl | 0.49 | |||
| NiAl-s | 0.51 | |||
| Au/NiAl | 0.0088 | 0.47 | ||
| Au/NiAl-s | 0.0040 | 0.54 | ||
| 3CeNiAl | 0.0140 | 0.47 | ||
| 3CeNiAl-s | 0.0077 | 0.49 | ||
| Au/3CeNiAl | 0.0089 | 0.0113 | 0.45 | |
| Au/3CeNiAl-s | 0.0041 | 0.0103 | 0.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabrovska, M.; Ivanov, I.; Nikolova, D.; Krstić, J.; Venezia, A.M.; Crişan, D.; Crişan, M.; Tenchev, K.; Idakiev, V.; Tabakova, T. Improved Water–Gas Shift Performance of Au/NiAl LDHs Nanostructured Catalysts via CeO2 Addition. Nanomaterials 2021, 11, 366. https://doi.org/10.3390/nano11020366
Gabrovska M, Ivanov I, Nikolova D, Krstić J, Venezia AM, Crişan D, Crişan M, Tenchev K, Idakiev V, Tabakova T. Improved Water–Gas Shift Performance of Au/NiAl LDHs Nanostructured Catalysts via CeO2 Addition. Nanomaterials. 2021; 11(2):366. https://doi.org/10.3390/nano11020366
Chicago/Turabian StyleGabrovska, Margarita, Ivan Ivanov, Dimitrinka Nikolova, Jugoslav Krstić, Anna Maria Venezia, Dorel Crişan, Maria Crişan, Krassimir Tenchev, Vasko Idakiev, and Tatyana Tabakova. 2021. "Improved Water–Gas Shift Performance of Au/NiAl LDHs Nanostructured Catalysts via CeO2 Addition" Nanomaterials 11, no. 2: 366. https://doi.org/10.3390/nano11020366
APA StyleGabrovska, M., Ivanov, I., Nikolova, D., Krstić, J., Venezia, A. M., Crişan, D., Crişan, M., Tenchev, K., Idakiev, V., & Tabakova, T. (2021). Improved Water–Gas Shift Performance of Au/NiAl LDHs Nanostructured Catalysts via CeO2 Addition. Nanomaterials, 11(2), 366. https://doi.org/10.3390/nano11020366