
First-Principle Investigations on the Electronic and Transport Properties of PbBi2Te2X2 (X = S/Se/Te) Monolayers


Abstract
:1. Introduction
2. Materials and Methods
3. Results
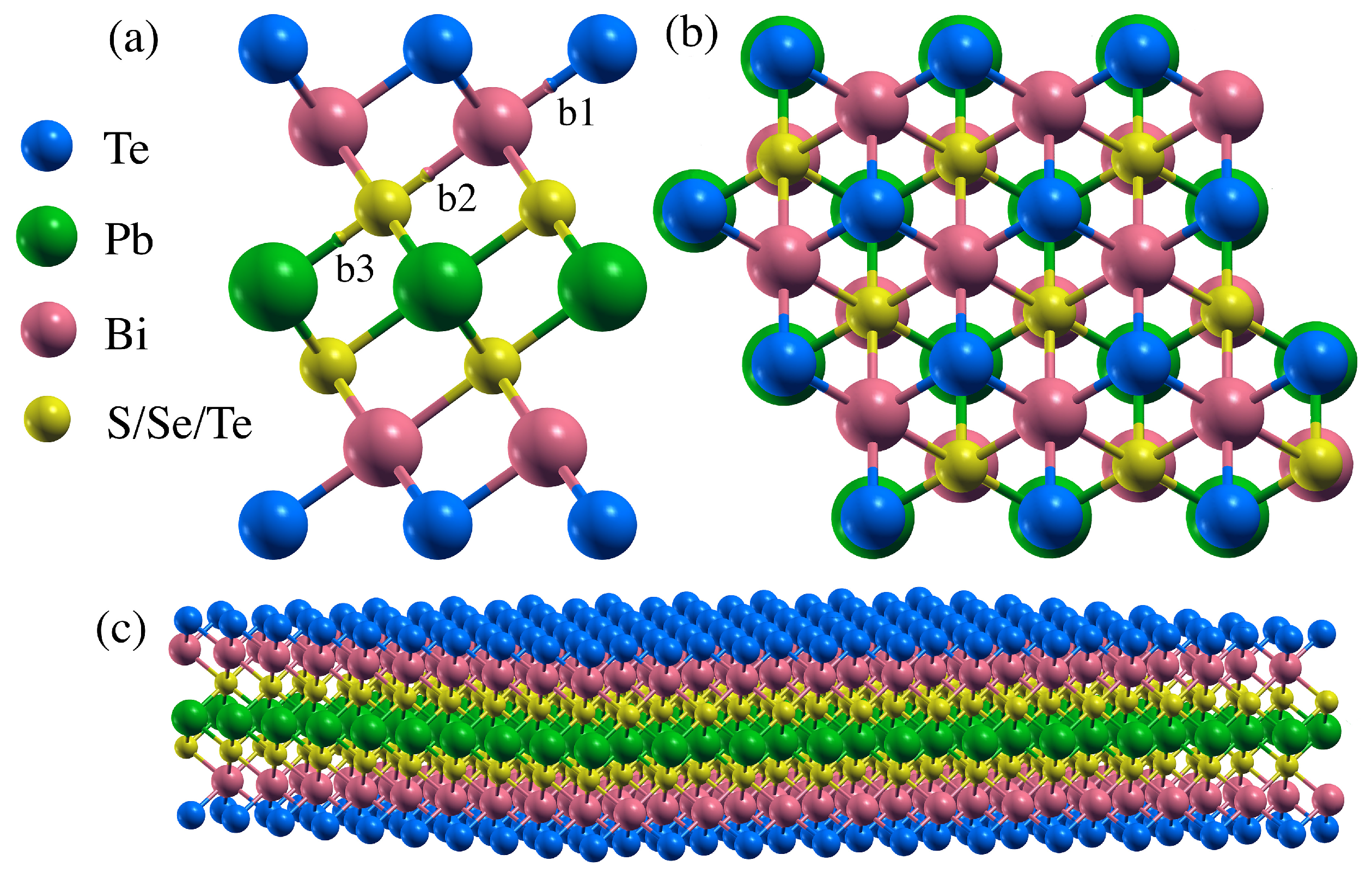
3.1. Structural Data
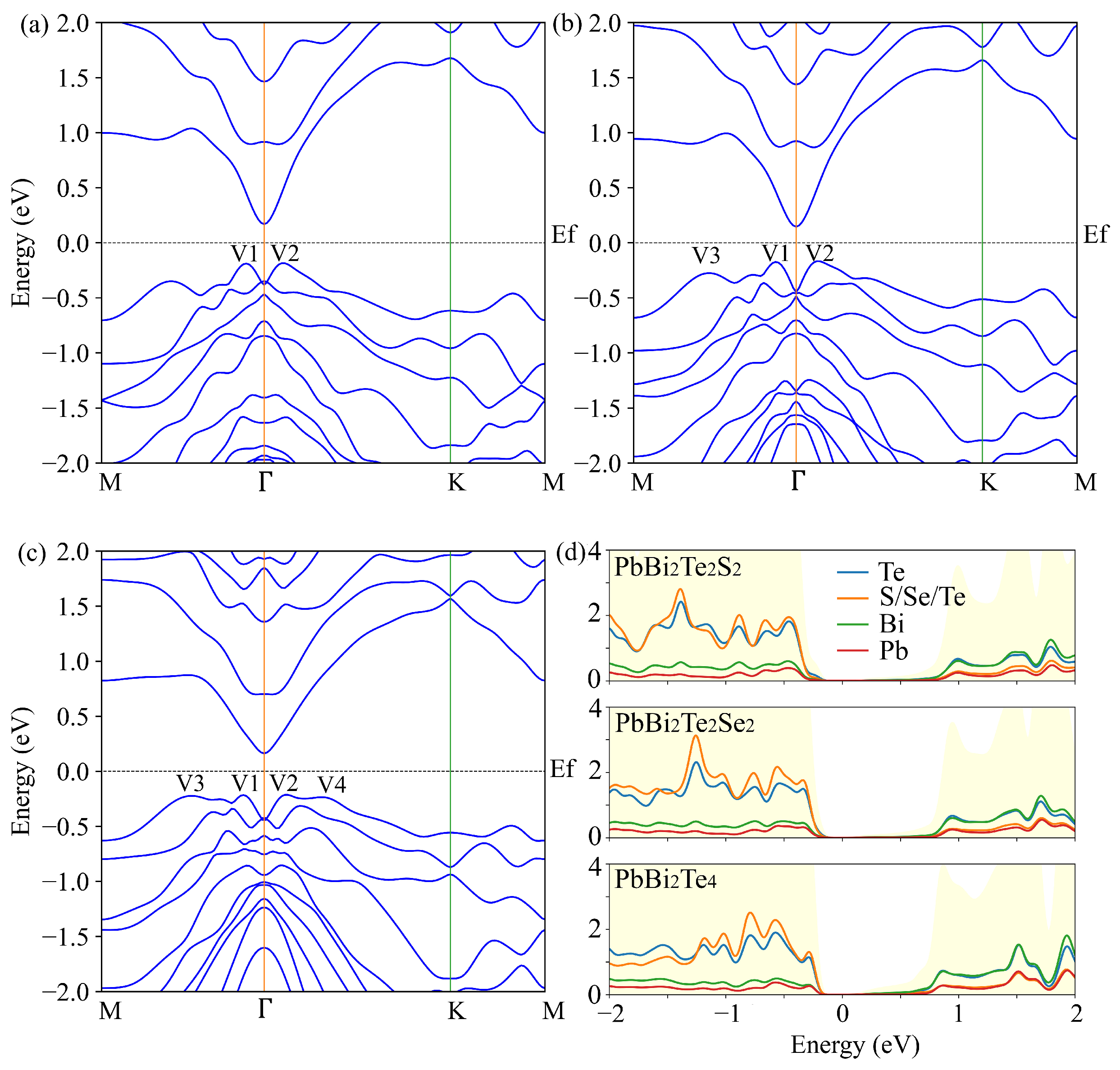
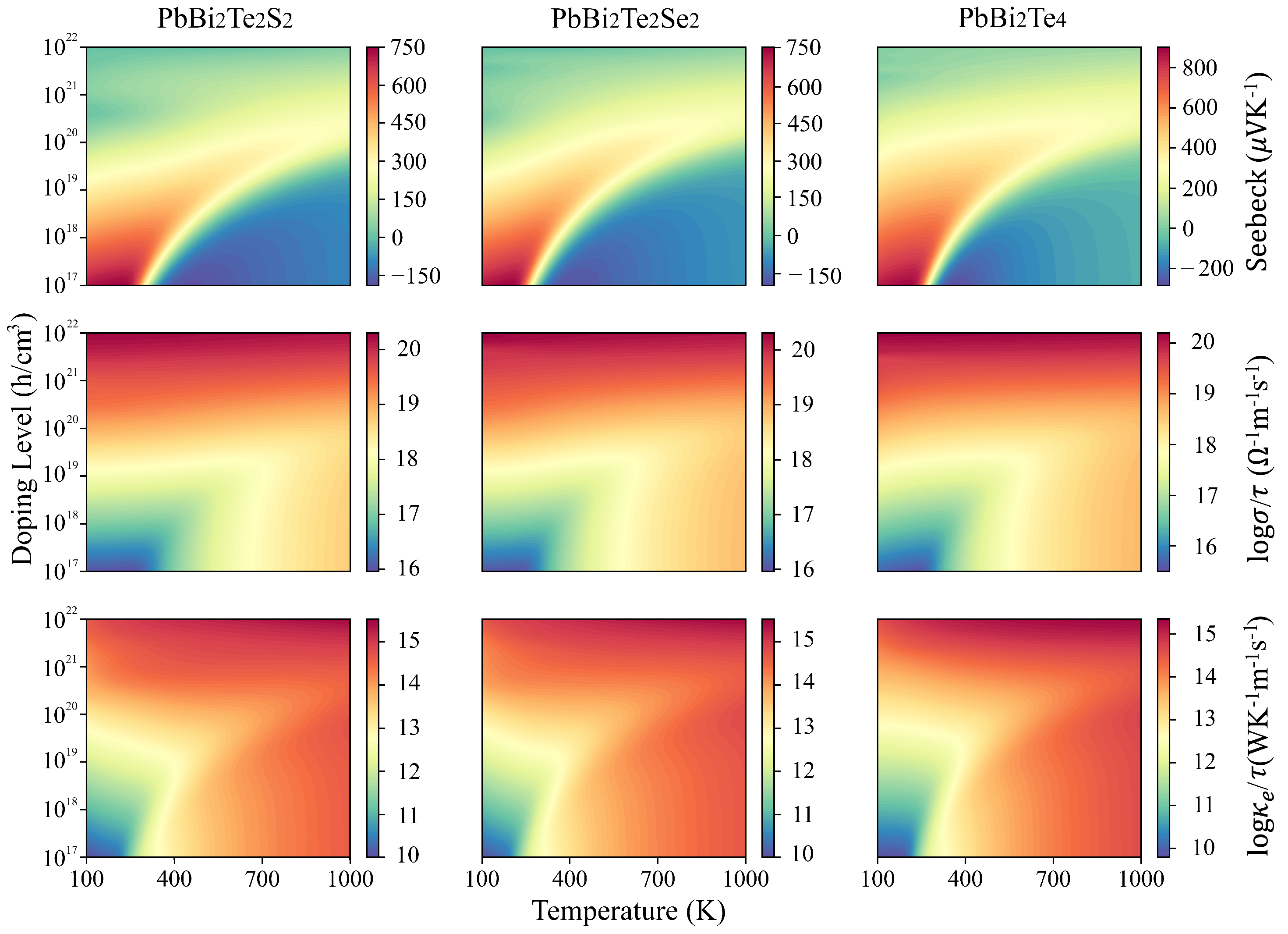
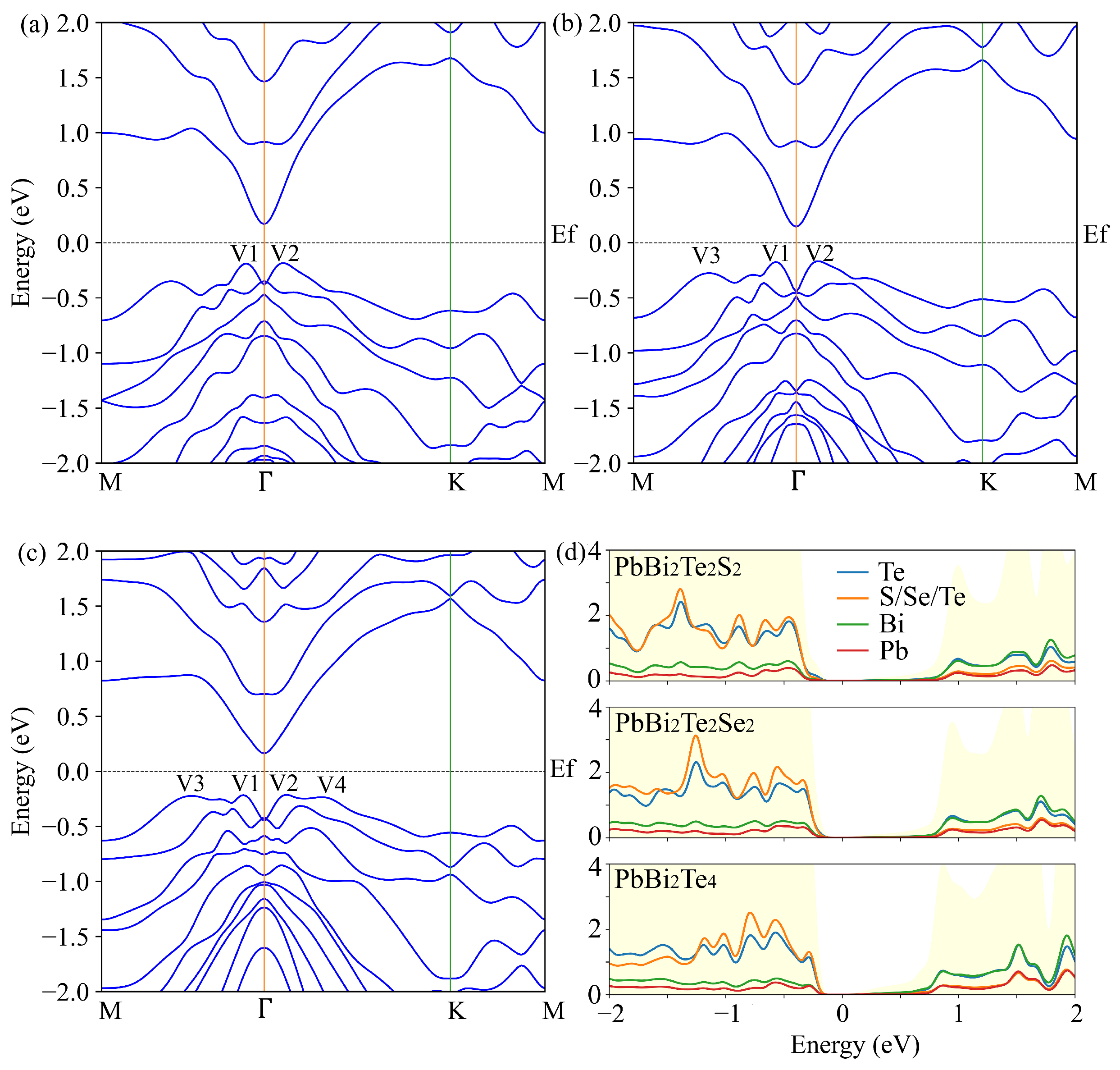
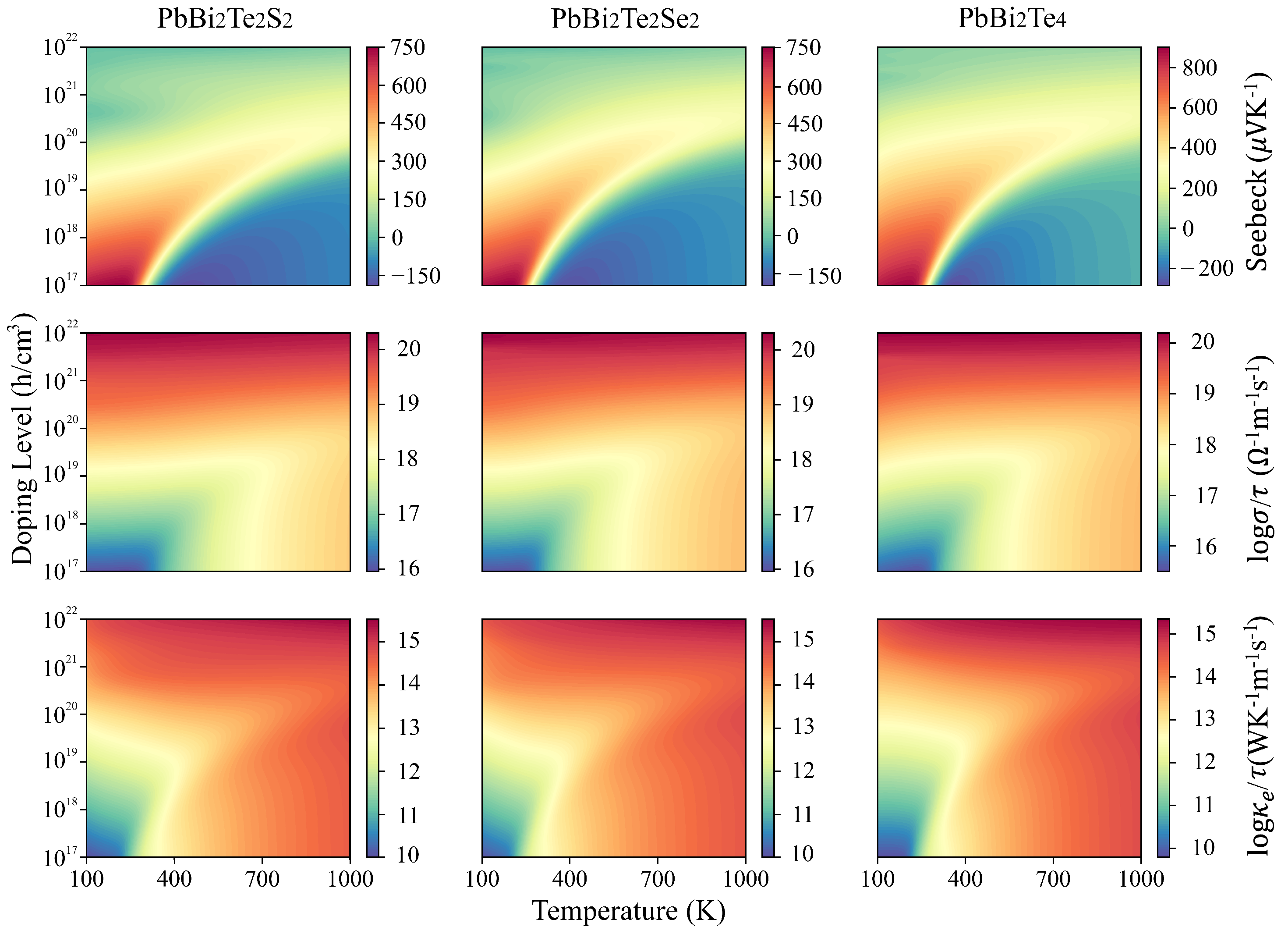
3.2. Electronic and Transport Properties
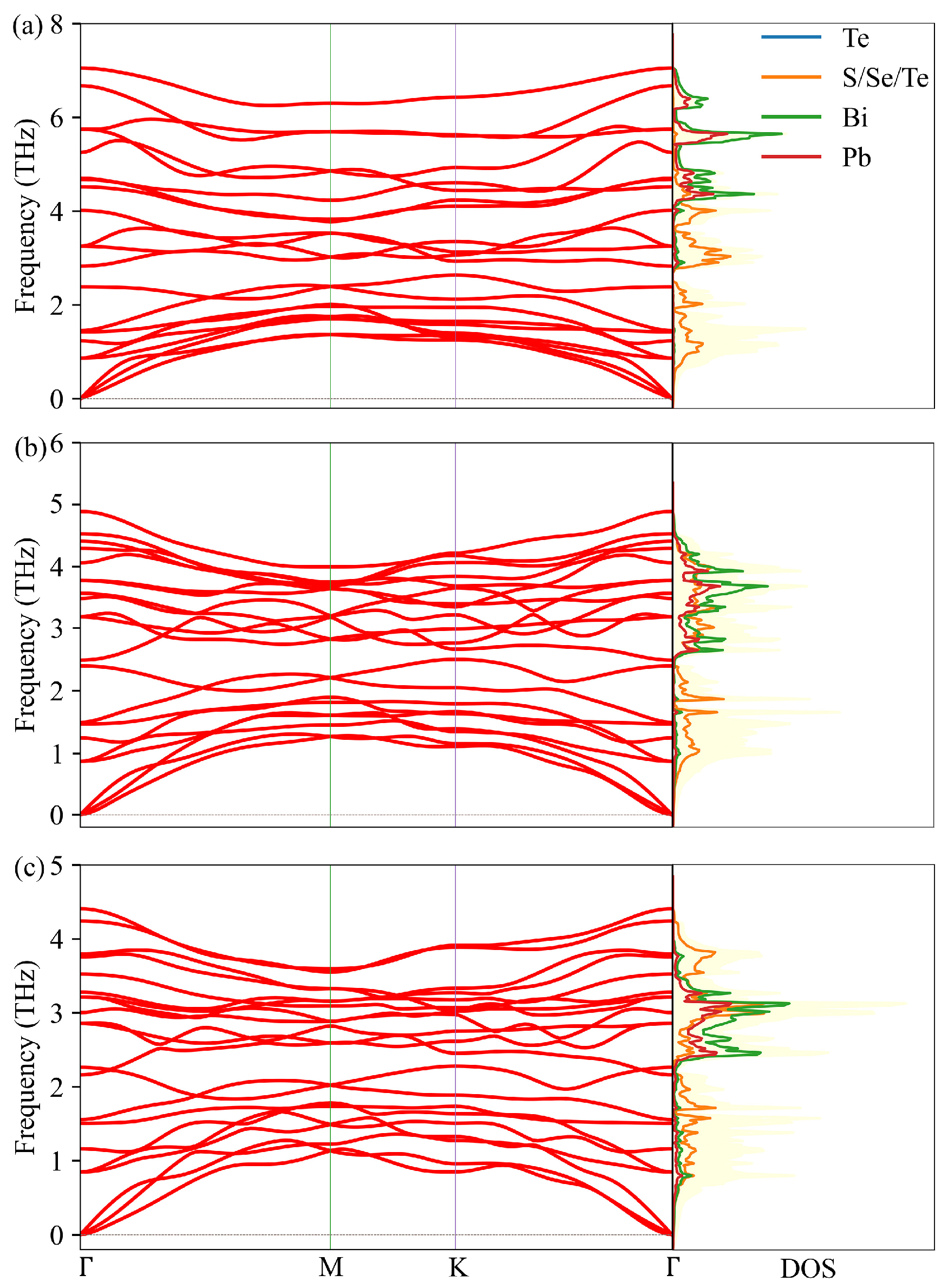
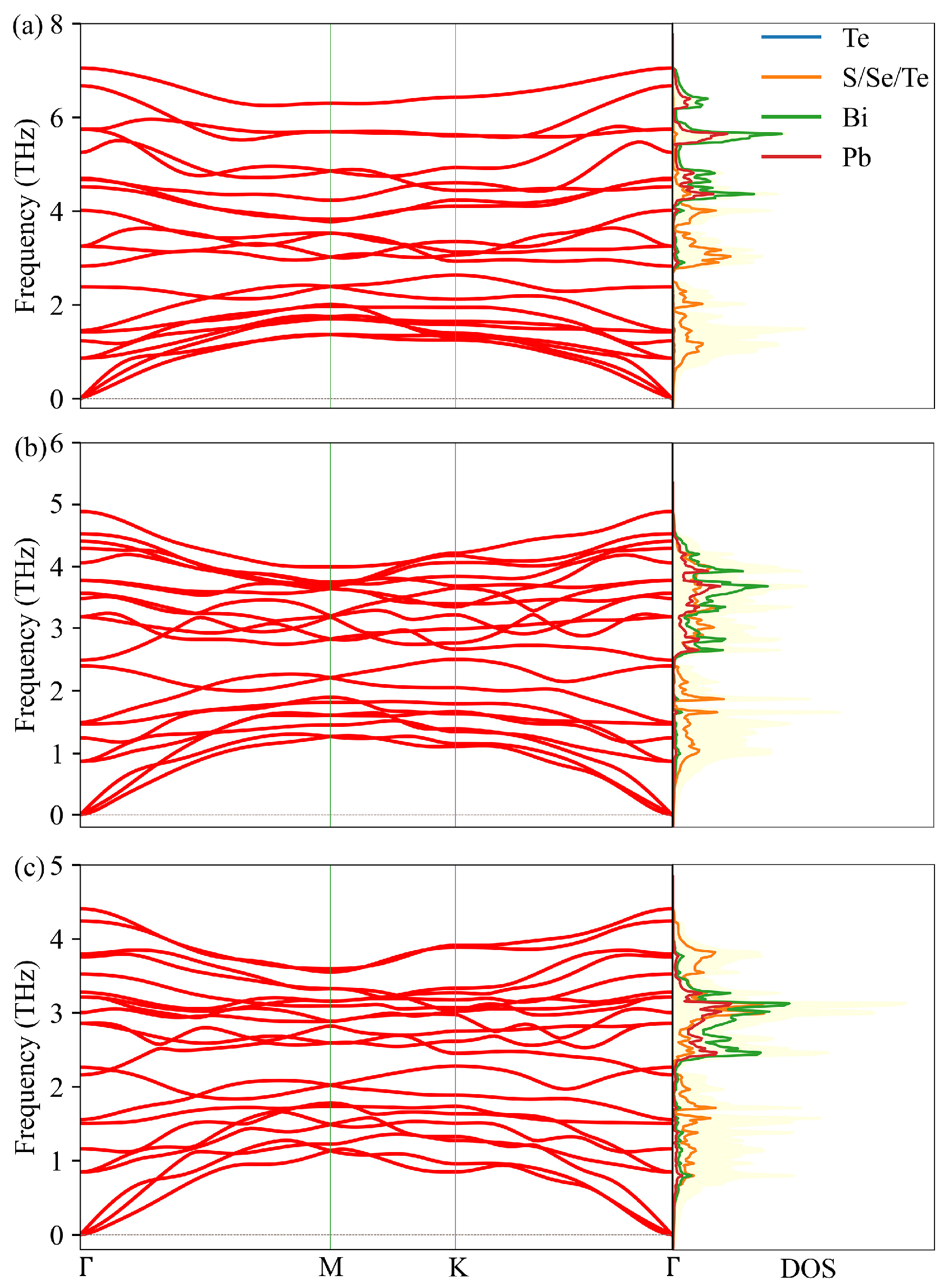
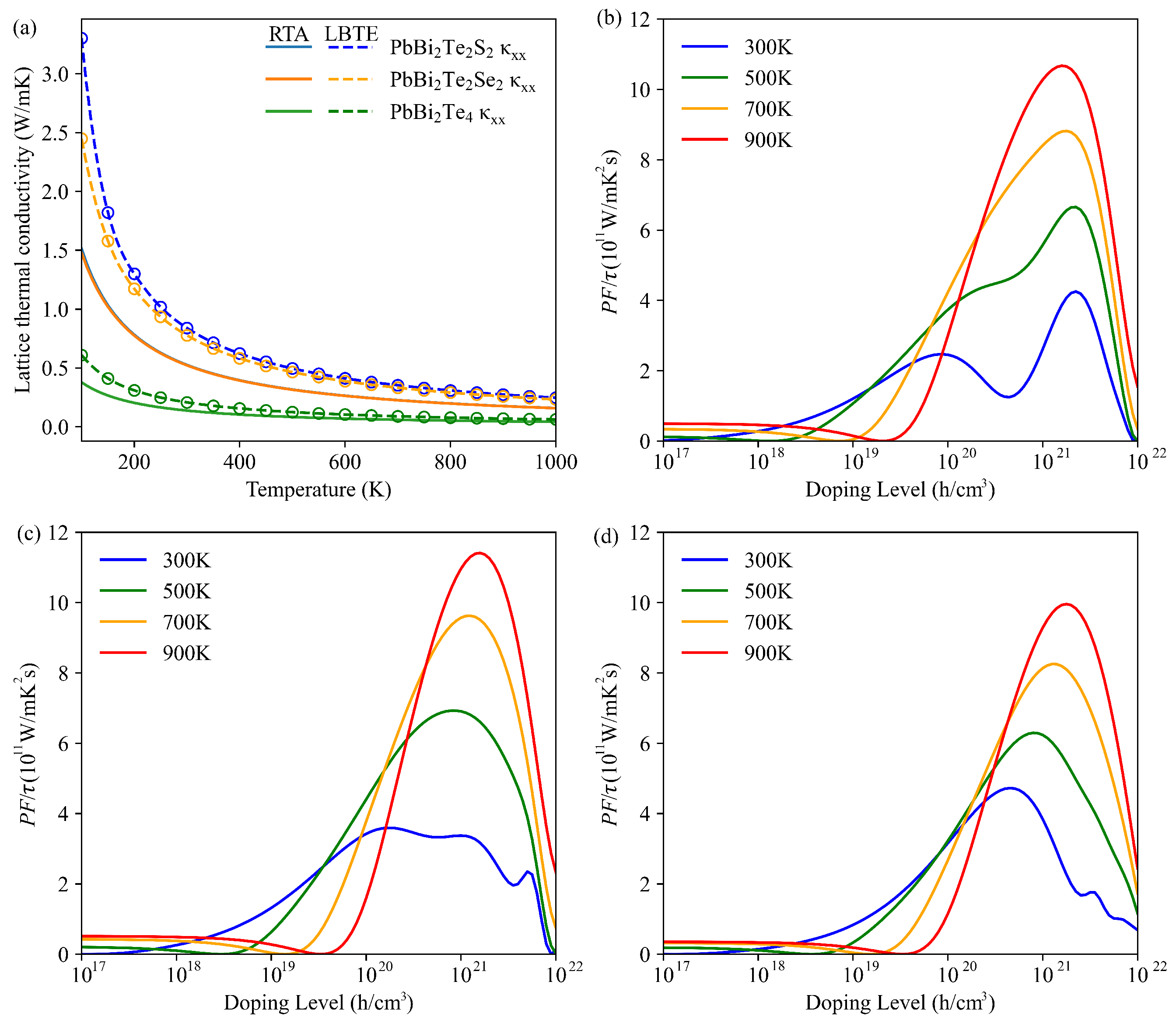
3.3. Lattice Thermal Conductivity
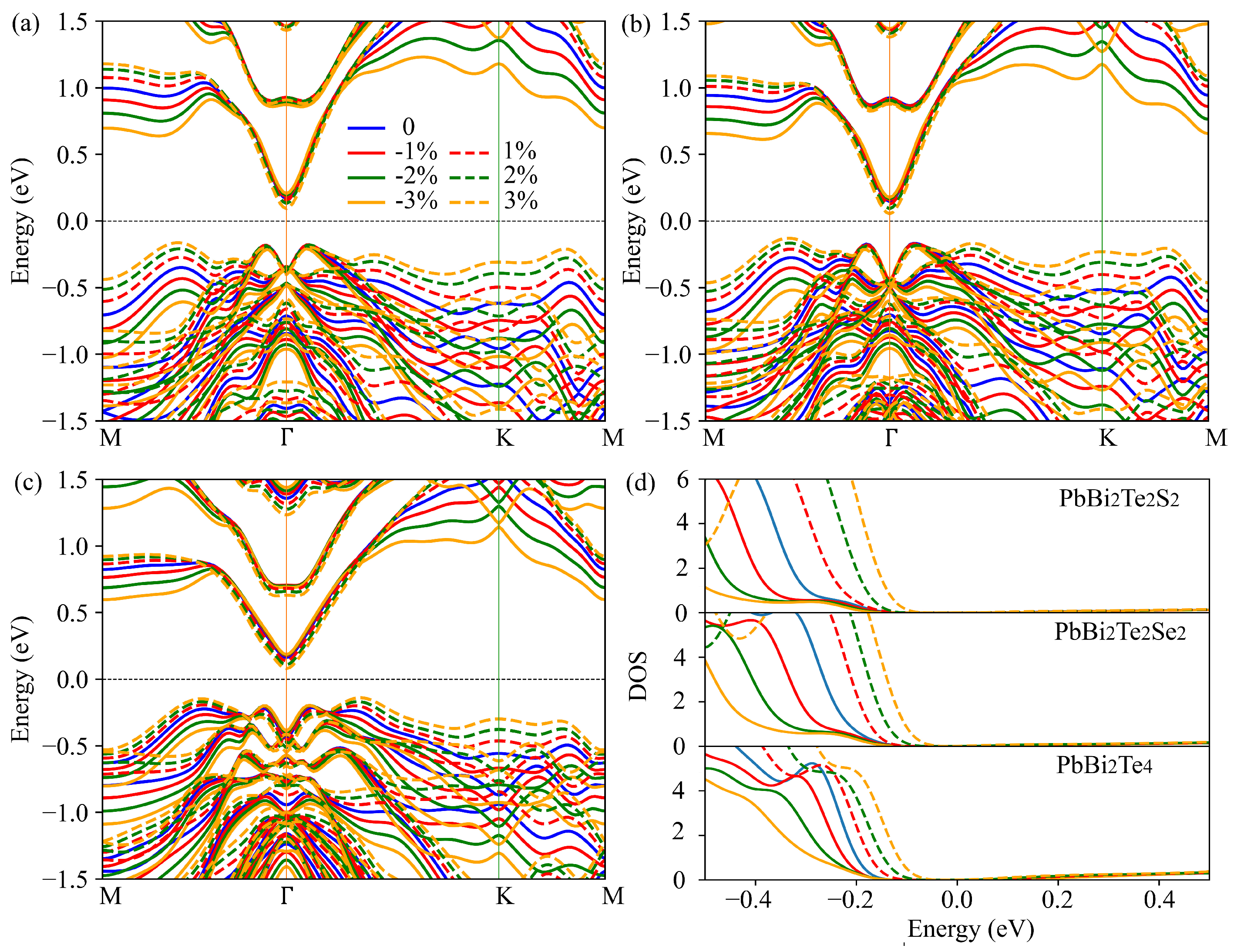
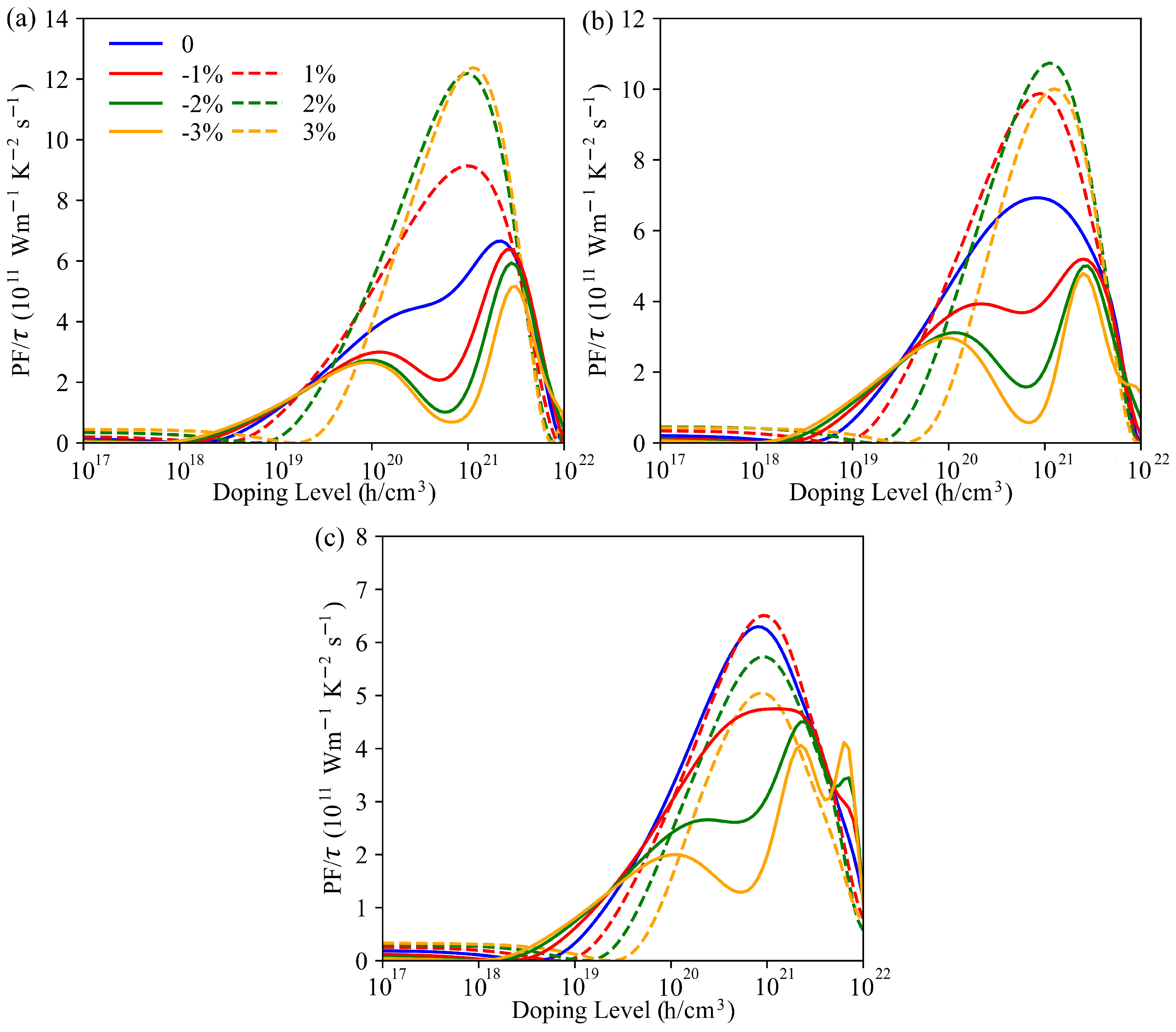
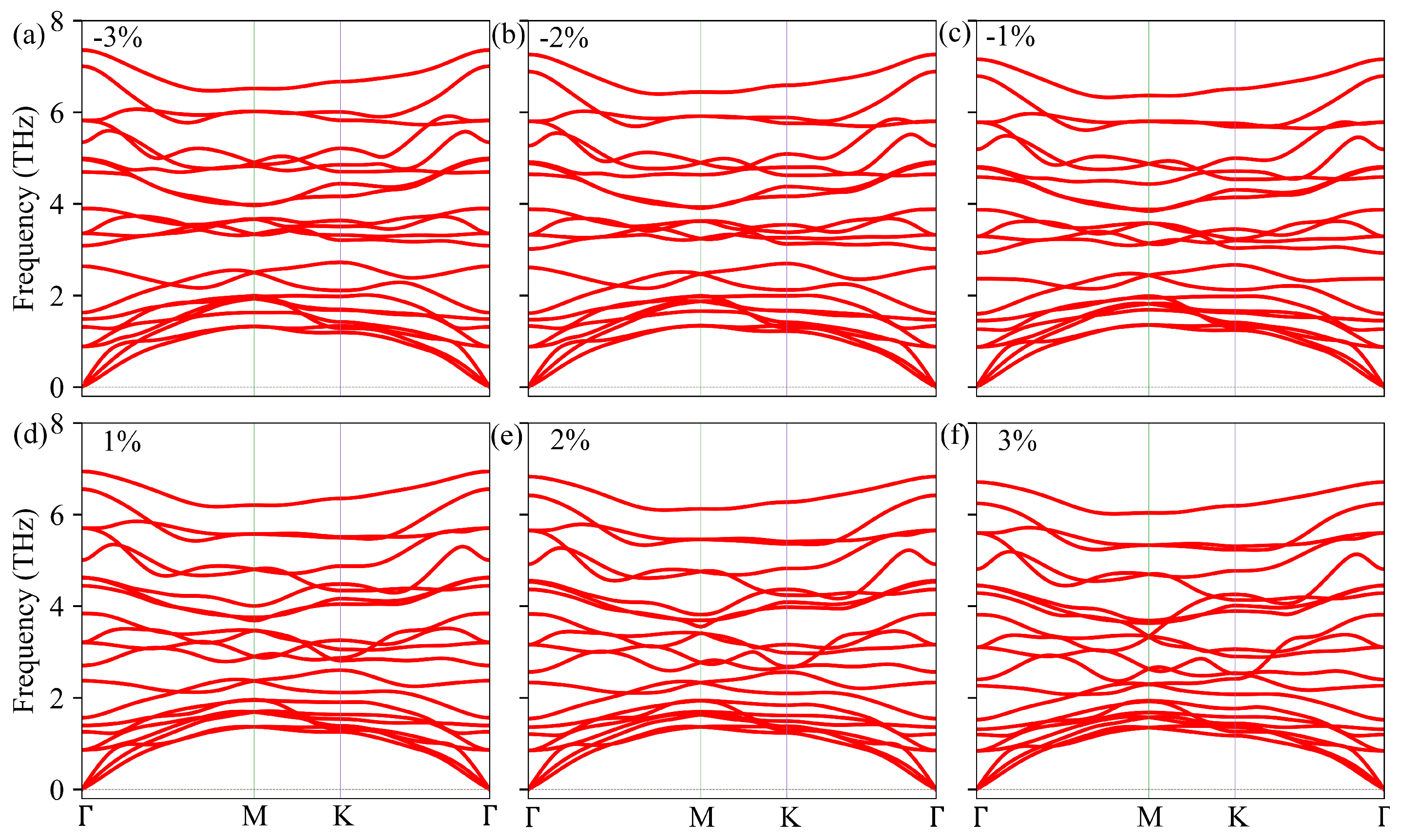
3.4. Strain Engineering of Electronic and Phonon Transport Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, X.; Zhang, Q.; Liao, J.; Chen, H.; Fan, Y.; Xing, J.; Gu, S.; Huang, J.; Ma, J.; Wang, J.; et al. High-Efficiency Thermoelectric Power Generation Enabled by Homogeneous Incorporation of MXene in (Bi,Sb)2Te3 Matrix. Adv. Energy Mater. 2020, 10, 1902986. [Google Scholar] [CrossRef]
- Liu, W.; Jie, Q.; Kim, H.S.; Ren, Z. Current Progress and Future Challenges in Thermoelectric Power Generation: From Materials to Devices. Acta Mater. 2015, 87, 357–376. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, I.; Prasher, R.; Lofgreen, K.; Chrysler, G.; Narasimhan, S.; Mahajan, R.; Koester, D.; Alley, R.; Venkatasubramanian, R. On-Chip Cooling by Superlattice-Based Thin-Film Thermoelectrics. Nat. Nanotechnol. 2009, 4, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, W.A.; Mecklenburg, M.; Lodico, J.J.; Chen, Y.; Ling, X.Y.; Patil, R.; Kessel, W.A.; Flatt, G.J.K.; Chan, H.L.; Vareskic, B.; et al. Electron-Transparent Thermoelectric Coolers Demonstrated with Nanoparticle and Condensation Thermometry. ACS Nano 2020, 14, 11510–11517. [Google Scholar] [CrossRef]
- Vining, C.B. An Inconvenient Truth about Thermoelectrics. Nat. Mater. 2009, 8, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Song, S.; Zhu, H.; Ren, Z. Realizing High Conversion Efficiency of Mg3Sb2-Based Thermoelectric Materials. J. Power Sour. 2019, 414, 393–400. [Google Scholar] [CrossRef]
- Balout, H.; Boulet, P.; Record, M.C. Strain-Induced Electronic Band Convergence: Effect on the Seebeck Coefficient of Mg2Si for Thermoelectric Applications. J. Mol. Model. 2017, 23, 130. [Google Scholar] [CrossRef] [Green Version]
- Diznab, M.R.; Maleki, I.; Vaez Allaei, S.M.; Xia, Y.; Naghavi, S.S. Achieving an Ultrahigh Power Factor in Sb2Te2Se Monolayers via Valence Band Convergence. ACS Appl. Mater. Interfaces 2019, 11, 46688–46695. [Google Scholar] [CrossRef]
- Thébaud, S.; Adessi, C.; Pailhès, S.; Bouzerar, G. Boosting the Power Factor with Resonant States: A Model Study. Phys. Rev. B 2017, 96, 075201. [Google Scholar] [CrossRef] [Green Version]
- Heremans, J.P.; Wiendlocha, B.; Chamoire, A.M. Resonant Levels in Bulk Thermoelectric Semiconductors. Energy Environ. Sci. 2012, 5, 5510–5530. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.; Zhang, Q.; Liu, W.; Yu, B.; Wang, H.; Wang, D.; Ni, G.; Chen, G.; Ren, Z. Effect of Silicon and Sodium on Thermoelectric Properties of Thallium-Doped Lead Telluride-Based Materials. Nano Lett. 2012, 12, 2324–2330. [Google Scholar] [CrossRef]
- Zhang, G.; Kirk, B.; Jauregui, L.A.; Yang, H.; Xu, X.; Chen, Y.P.; Wu, Y. Rational Synthesis of Ultrathin N-Type Bi2Te3 Nanowires with Enhanced Thermoelectric Properties. Nano Lett. 2012, 12, 56–60. [Google Scholar] [CrossRef]
- Wu, H.J.; Zhao, L.D.; Zheng, F.S.; Wu, D.; Pei, Y.L.; Tong, X.; Kanatzidis, M.G.; He, J.Q. Broad Temperature Plateau for Thermoelectric Figure of Merit ZT>2 in Phase-Separated PbTe0.7S0.3. Nat. Commun. 2014, 5, 4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Kim, S.I.; Lee, K.H.; Kim, S.W.; Snyder, G.J. Phonon Scattering by Dislocations at Grain Boundaries in Polycrystalline Bi0.5Sb1.5Te3. Phys. Status Solidi (b) 2017, 254, 1600103. [Google Scholar] [CrossRef]
- Wu, H.; Lu, X.; Wang, G.; Peng, K.; Chi, H.; Zhang, B.; Chen, Y.; Li, C.; Yan, Y.; Guo, L.; et al. Sodium-Doped Tin Sulfide Single Crystal: A Nontoxic Earth-Abundant Material with High Thermoelectric Performance. Adv. Energy Mater. 2018, 8, 1800087. [Google Scholar] [CrossRef]
- Lu, Z.; Wu, Y.; Xu, Y.; Ma, C.; Chen, Y.; Xu, K.; Zhang, H.; Zhu, H.; Fang, Z. Ultrahigh Electron Mobility Induced by Strain Engineering in Direct Semiconductor Monolayer Bi2TeSe2. Nanoscale 2019, 11, 20620–20629. [Google Scholar] [CrossRef]
- Mishra, P.; Singh, D.; Sonvane, Y.; Ahuja, R. Two-Dimensional Boron Monochalcogenide Monolayer for Thermoelectric Material. Sustain. Energy Fuels 2020, 4, 2363–2369. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cheng, H.; Gao, S.; Liu, Q.; Sun, Z.; Xiao, C.; Wu, C.; Wei, S.; Xie, Y. Atomically Thick Bismuth Selenide Freestanding Single Layers Achieving Enhanced Thermoelectric Energy Harvesting. J. Am. Chem. Soc. 2012, 134, 20294–20297. [Google Scholar] [CrossRef]
- Mounet, N.; Gibertini, M.; Schwaller, P.; Campi, D.; Merkys, A.; Marrazzo, A.; Sohier, T.; Castelli, I.E.; Cepellotti, A.; Pizzi, G.; et al. Two-Dimensional Materials from High-Throughput Computational Exfoliation of Experimentally Known Compounds. Nat. Nanotechnol. 2018, 13, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, A.; Pumera, M. Exfoliation of Layered Materials Using Electrochemistry. Chem. Soc. Rev. 2018, 47, 7213–7224. [Google Scholar] [CrossRef]
- Chatterjee, A.; Biswas, K. Solution-Based Synthesis of Layered Intergrowth Compounds of the Homologous PbmBi2nTe3n+m Series as Nanosheets. Angew. Chem. Int. Ed. 2015, 54, 5623–5627. [Google Scholar] [CrossRef]
- Park, K.; Heremans, J.J.; Scarola, V.W.; Minic, D. Robustness of Topologically Protected Surface States in Layering of Bi2Te3 Thin Films. Phys. Rev. Lett. 2010, 105, 186801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shvets, I.A.; Klimovskikh, I.I.; Aliev, Z.S.; Babanly, M.B.; Sánchez-Barriga, J.; Krivenkov, M.; Shikin, A.M.; Chulkov, E.V. Impact of Stoichiometry and Disorder on the Electronic Structure of the PbBi2Te4−xSex Topological Insulator. Phys. Rev. B 2017, 96, 235124. [Google Scholar] [CrossRef] [Green Version]
- Peng, R.; Ma, Y.; Wang, H.; Huang, B.; Dai, Y. Stacking-Dependent Topological Phase in Bilayer MBi2Te4 (M = Ge, Sn, Pb). Phys. Rev. B 2020, 101, 115427. [Google Scholar] [CrossRef]
- Hung, N.T.; Nugraha, A.R.; Saito, R. Designing High-Performance Thermoelectrics in Two-Dimensional Tetradymites. Nano Energy 2019, 58, 743–749. [Google Scholar] [CrossRef]
- Sharma, S.; Schwingenschlögl, U. Thermoelectric Response in Single Quintuple Layer Bi2Te3. ACS Energy Lett. 2016, 1, 875–879. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APW+lo Program for Calculating the Properties of Solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef]
- Madsen, G.K.; Carrete, J.; Verstraete, M.J. BoltzTraP2, a Program for Interpolating Band Structures and Calculating Semi-Classical Transport Coefficients. Comput. Phys. Commun. 2018, 231, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tanaka, I. Distribution of Phonon Lifetime in Brillouin Zone. Phys. Rev. B 2015, 91, 094306. [Google Scholar] [CrossRef] [Green Version]
- Chaput, L. Direct Solution to the Linearized Phonon Boltzmann Equation. Phys. Rev. Lett. 2013, 110, 265506. [Google Scholar] [CrossRef]
- Ma, W.; Record, M.C.; Tian, J.; Boulet, P. Strain Effects on the Electronic and Thermoelectric Properties of n(PbTe)-m(Bi2Te3) System Compounds. Materials 2021, 14, 4086. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cohen, R.E. A More Accurate Generalized Gradient Approximation for Solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef] [Green Version]
- Shelimova, L.E.; Karpinskii, O.G.; Konstantinov, P.P.; Avilov, E.S.; Kretova, M.A.; Zemskov, V.S. Crystal Structures and Thermoelectric Properties of Layered Compounds in the ATe–Bi2Te3 (A = Ge, Sn, Pb) Systems. Inorg. Mater. 2004, 40, 451–460. [Google Scholar] [CrossRef]
- Heyd, J.; Peralta, J.E.; Scuseria, G.E.; Martin, R.L. Energy Band Gaps and Lattice Parameters Evaluated with the Heyd-Scuseria-Ernzerhof Screened Hybrid Functional. J. Chem. Phys. 2005, 123, 174101. [Google Scholar] [CrossRef]
- Park, S.; Ryu, B. Hybrid-Density Functional Theory Study on Band Structures of Tetradymite-Bi2Te3, Sb2Te3, Bi2Se3, and Sb2Se3 Thermoelectric Materials. J. Korean Phys. Soc. 2016, 69, 1683–1687. [Google Scholar] [CrossRef] [Green Version]
- Mouhat, F.; Coudert, F.X. Necessary and Sufficient Elastic Stability Conditions in Various Crystal Systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Campi, D.; Paulatto, L.; Fugallo, G.; Mauri, F.; Bernasconi, M. First-Principles Calculation of Lattice Thermal Conductivity in Crystalline Phase Change Materials: GeTe, Sb2Te3, and Ge2Sb2Te5. Phys. Rev. B 2017, 95, 024311. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566. [Google Scholar]
- Hellman, O.; Broido, D.A. Phonon Thermal Transport in Bi2Te3 from First Principles. Phys. Rev. B 2014, 90, 134309. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ke, X.; Chen, C.; Yang, J.; Kent, P.R.C. Thermodynamic Properties of PbTe, PbSe, and PbS: A First-Principles Study. Phys. Rev. B 2009, 80, 024304. [Google Scholar]
- Ma, W.; Record, M.C.; Tian, J.; Boulet, P. Influence of the Stacking Sequence on Layered-Chalcogenides Properties: First Principle Investigation of Pb2Bi2Te5. Phys. Chem. Chem. Phys. 2021, 23, 11300–11313. [Google Scholar] [CrossRef] [PubMed]
- King-Smith, R.; Vanderbilt, D. Theory of Polarization of Crystalline Solids. Phys. Rev. B 1993, 47, 1651. [Google Scholar] [CrossRef]
- Slack, G.A. Nonmetallic crystals with high thermal conductivity. J. Phys. Chem. Solids 1973, 34, 321–335. [Google Scholar] [CrossRef]
- Shalímova, K. Fisica de los Semiconductors; Energoatomizdat: Madrid, Spain, 1982. [Google Scholar]
- Morelli, D.T.; Heremans, J.P. Thermal Conductivity of Germanium, Silicon, and Carbon Nitrides. Appl. Phys. Lett. 2002, 81, 5126–5128. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PbBiTeS | PbBiTeSe | PbBiTe | ||
|---|---|---|---|---|
| Bulk | a | 0.4230 | 0.4300 | 0.4430 |
| c | 3.954 | 4.042 | 4.156 | |
| Slab thickness | 1.037 | 1.072 | 1.127 | |
| 0.3029 | 0.3051 | 0.3070 | ||
| 0.2987 | 0.3085 | 0.3248 | ||
| 0.2958 | 0.3046 | 0.3210 | ||
| Nanosheet | a | 0.4210 | 0.4280 | 0.4410 |
| c | 1.041 | 1.074 | 1.128 | |
| 0.3018 | 0.3030 | 0.3048 | ||
| 0.2987 | 0.3082 | 0.3244 | ||
| 0.2959 | 0.3047 | 0.3203 |
| B | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PbBiTeS | 30.33 | 50.2 | 18.2 | 23.8 | 14.1 | 41.1 | 27.9 | 21.2 | −0.023/0.073 | −3.22 |
| PbBiTeSe | 28.75 | 49.5 | 14.5 | 22.7 | 13.5 | 40.2 | 27.0 | 21.2 | −0.024/0.073 | −3.09 |
| PbBiTe | 26.05 | 44.5 | 13.0 | 20.7 | 13.1 | 36.4 | 35.6 | 19.6 | −0.028/0.085 | −2.93 |
| Strains | PbBiTeS | PbBiTeSe | PbBiTe | ||||||
|---|---|---|---|---|---|---|---|---|---|
| a | c | a | c | a | c | ||||
| −3% | 4.09 | 10.76 | 8.28 | 4.15 | 11.10 | 10.41 | 4.27 | 11.65 | 9.93 |
| −2% | 4.13 | 10.64 | 3.19 | 4.20 | 10.98 | 4.99 | 4.32 | 11.53 | 4.90 |
| −1% | 4.17 | 10.52 | 0.5 | 4.24 | 10.86 | 1.84 | 4.36 | 11.40 | 1.92 |
| 0% | 4.21 | 10.41 | 0 | 4.28 | 10.74 | 0 | 4.41 | 11.28 | 0 |
| 1% | 4.26 | 10.29 | 0.7 | 4.33 | 10.62 | 1.72 | 4.45 | 11.16 | 1.70 |
| 2% | 4.30 | 10.17 | 3.68 | 4.37 | 10.50 | 4.41 | 4.49 | 11.04 | 4.31 |
| 3% | 4.34 | 10.05 | 8.74 | 4.41 | 10.38 | 8.90 | 4.54 | 10.91 | 8.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Tian, J.; Boulet, P.; Record, M.-C. First-Principle Investigations on the Electronic and Transport Properties of PbBi2Te2X2 (X = S/Se/Te) Monolayers. Nanomaterials 2021, 11, 2979. https://doi.org/10.3390/nano11112979
Ma W, Tian J, Boulet P, Record M-C. First-Principle Investigations on the Electronic and Transport Properties of PbBi2Te2X2 (X = S/Se/Te) Monolayers. Nanomaterials. 2021; 11(11):2979. https://doi.org/10.3390/nano11112979
Chicago/Turabian StyleMa, Weiliang, Jing Tian, Pascal Boulet, and Marie-Christine Record. 2021. "First-Principle Investigations on the Electronic and Transport Properties of PbBi2Te2X2 (X = S/Se/Te) Monolayers" Nanomaterials 11, no. 11: 2979. https://doi.org/10.3390/nano11112979
APA StyleMa, W., Tian, J., Boulet, P., & Record, M.-C. (2021). First-Principle Investigations on the Electronic and Transport Properties of PbBi2Te2X2 (X = S/Se/Te) Monolayers. Nanomaterials, 11(11), 2979. https://doi.org/10.3390/nano11112979