
Multifunctional Nanopolymers for Blood–Brain Barrier Delivery and Inhibition of Glioblastoma Growth through EGFR/EGFRvIII, c-Myc, and PD-1


 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Reagents
2.2. GBM Cell Line
2.3. Fluorescent Staining for BBB Permeation
2.4. Intracranial Tumor Model and Treatment Regimen
2.5. Prevention of Anaphylactic-Like Adverse Effects
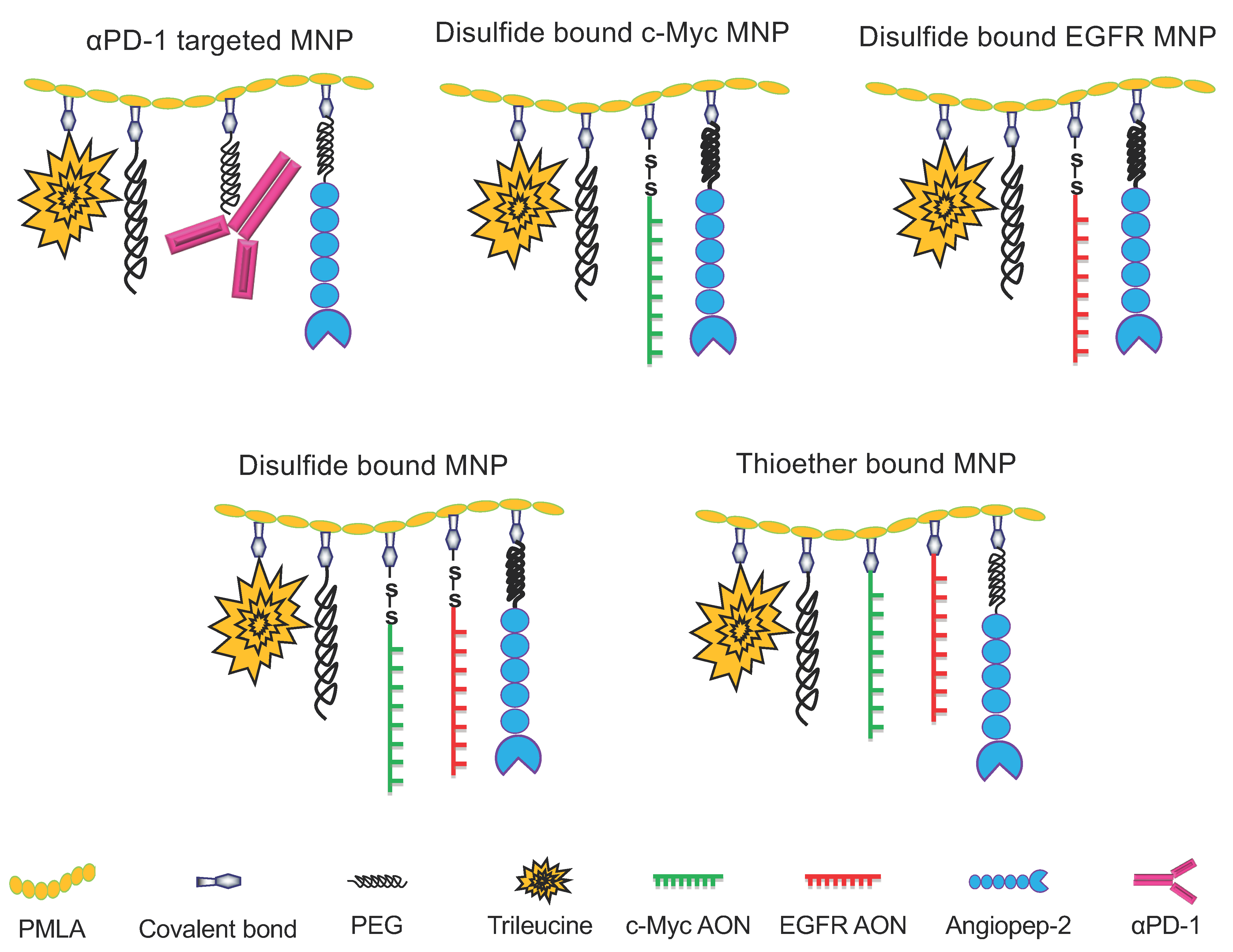
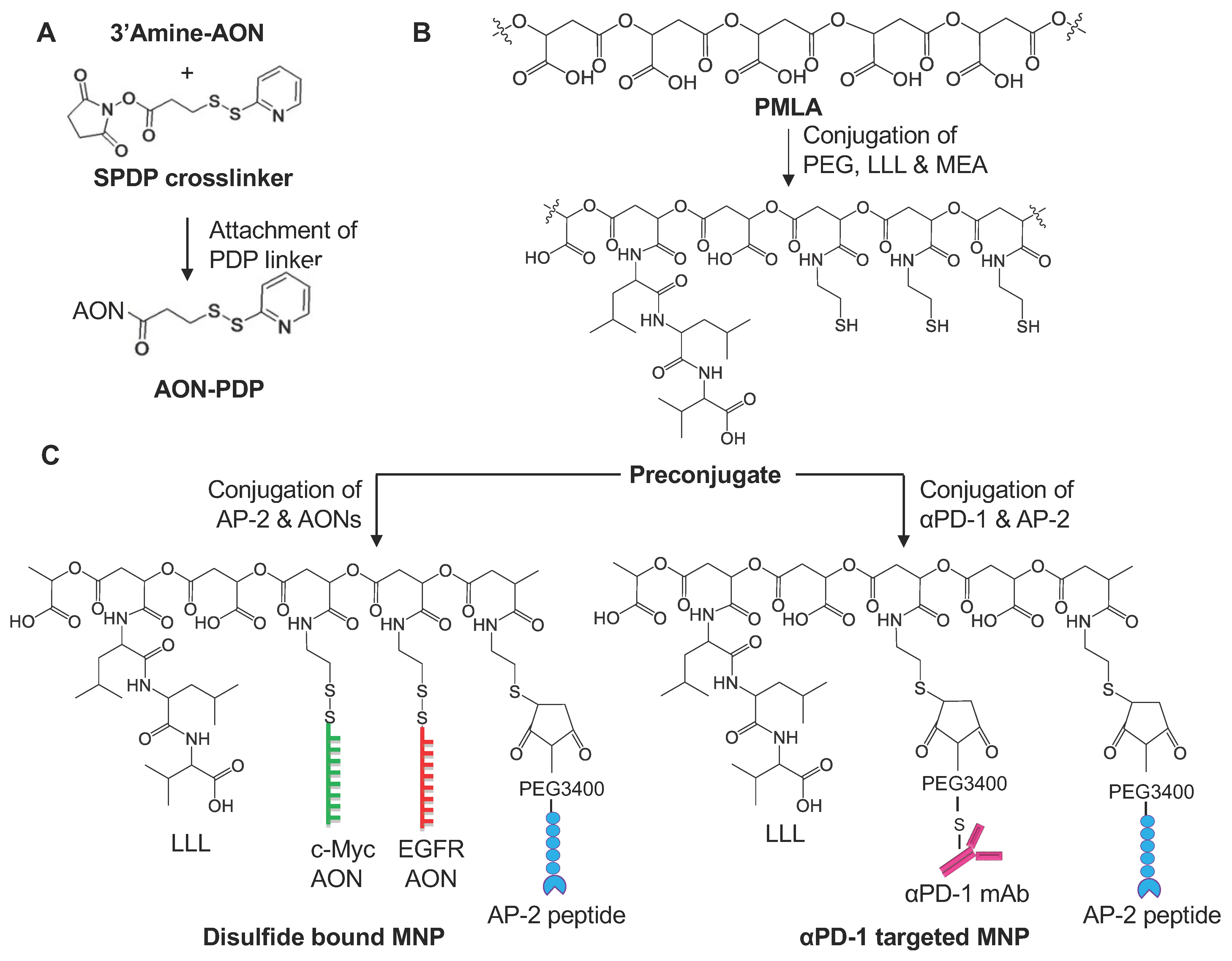
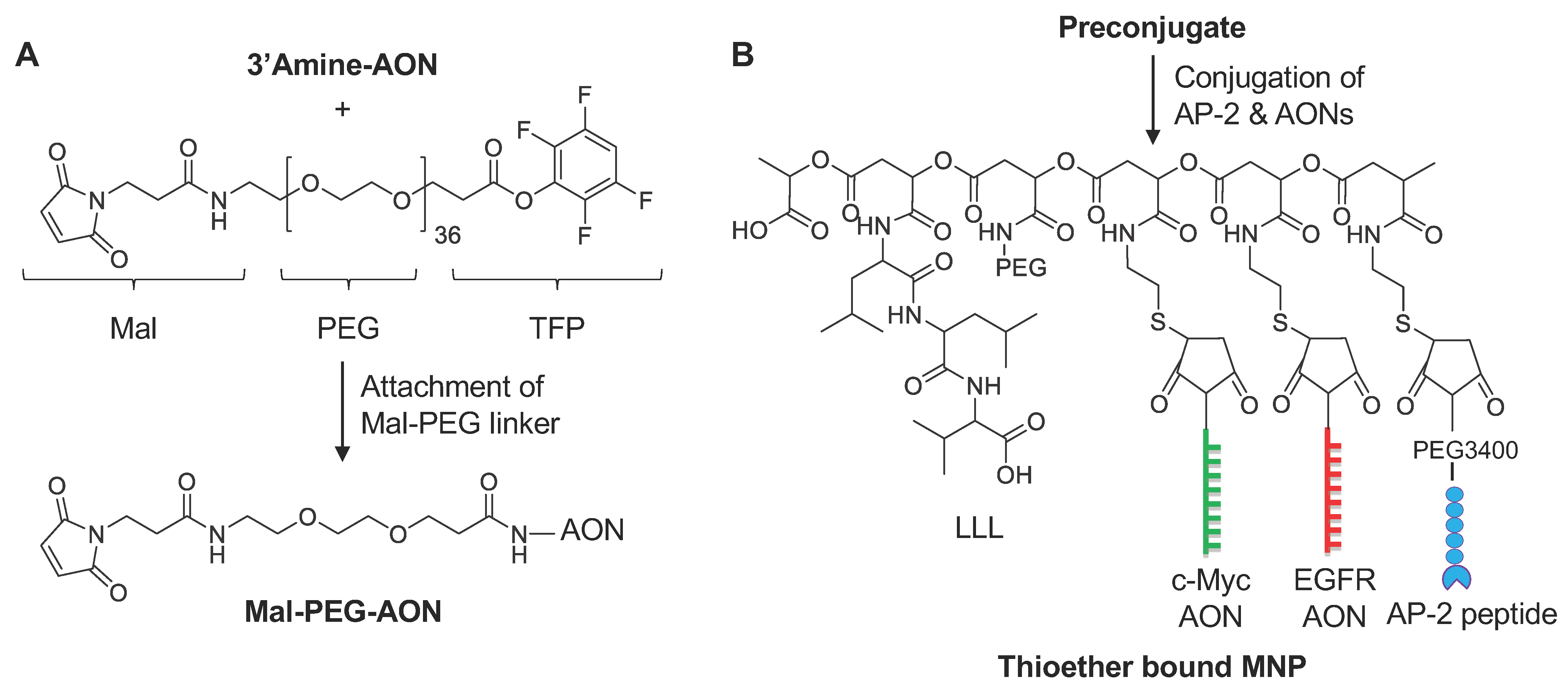
2.6. Synthesis of Novel Nanodrug Variants for Combination Brain Cancer Therapy
2.7. Synthesis of Various Multifunctional Nanoparticles (MNPs)
2.7.1. Synthesis of 3-(2-Pyridyldithio) Propionyl AON (AON-PDP)
2.7.2. Synthesis of S-Succinimidyl-PEG3400-Maleimide mAb
2.7.3. Synthesis of S-Succinimidyl-PEG3400-Maleimide Peptide
2.8. General Procedure for Synthesis of Mal-PEG-AONs
2.9. Physicochemical Characterization of MNPs. Synthesis Monitoring
2.10. Statistical Analysis
3. Results
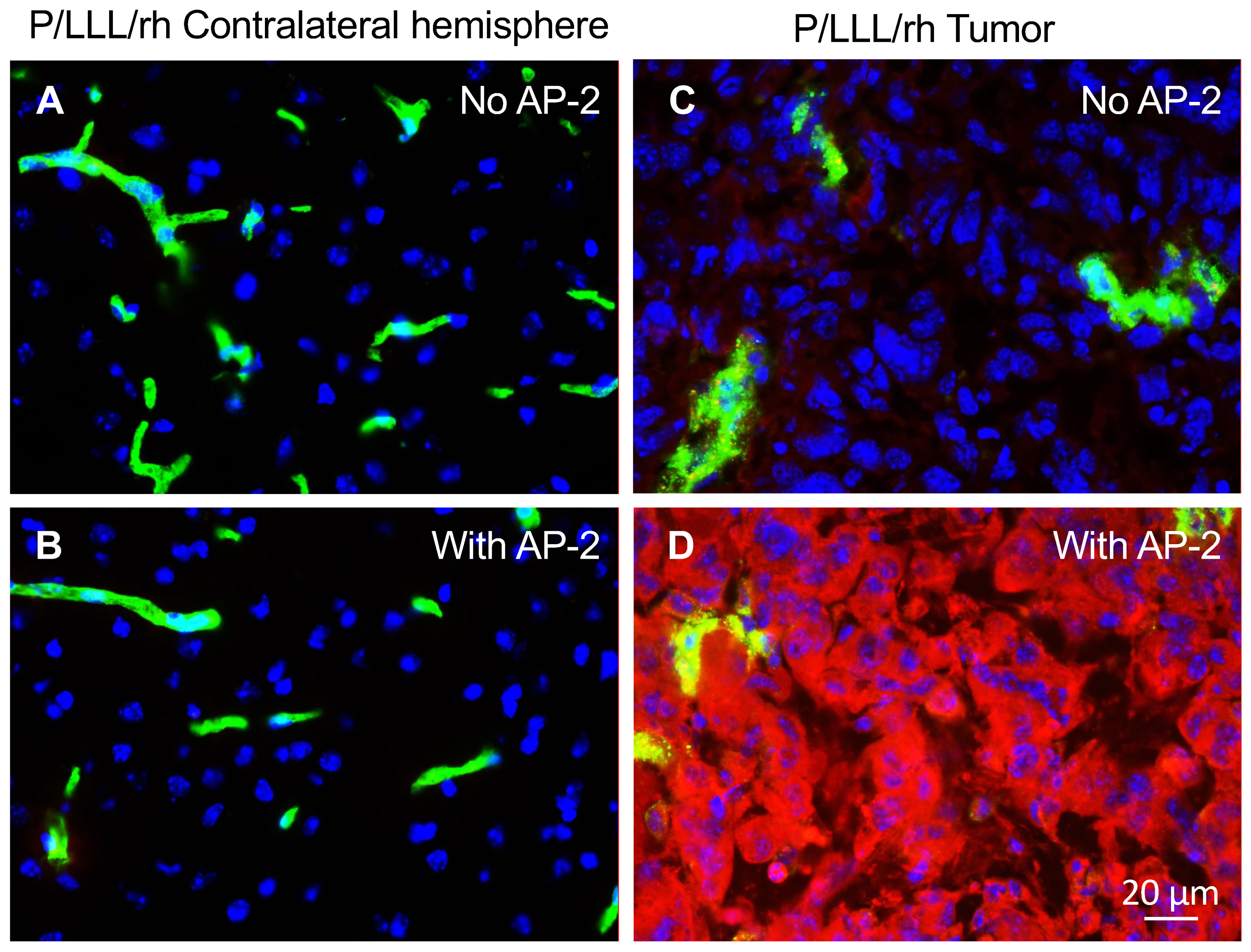
3.1. In Vivo Study of BBB-Crossing Ability of Used MNPs
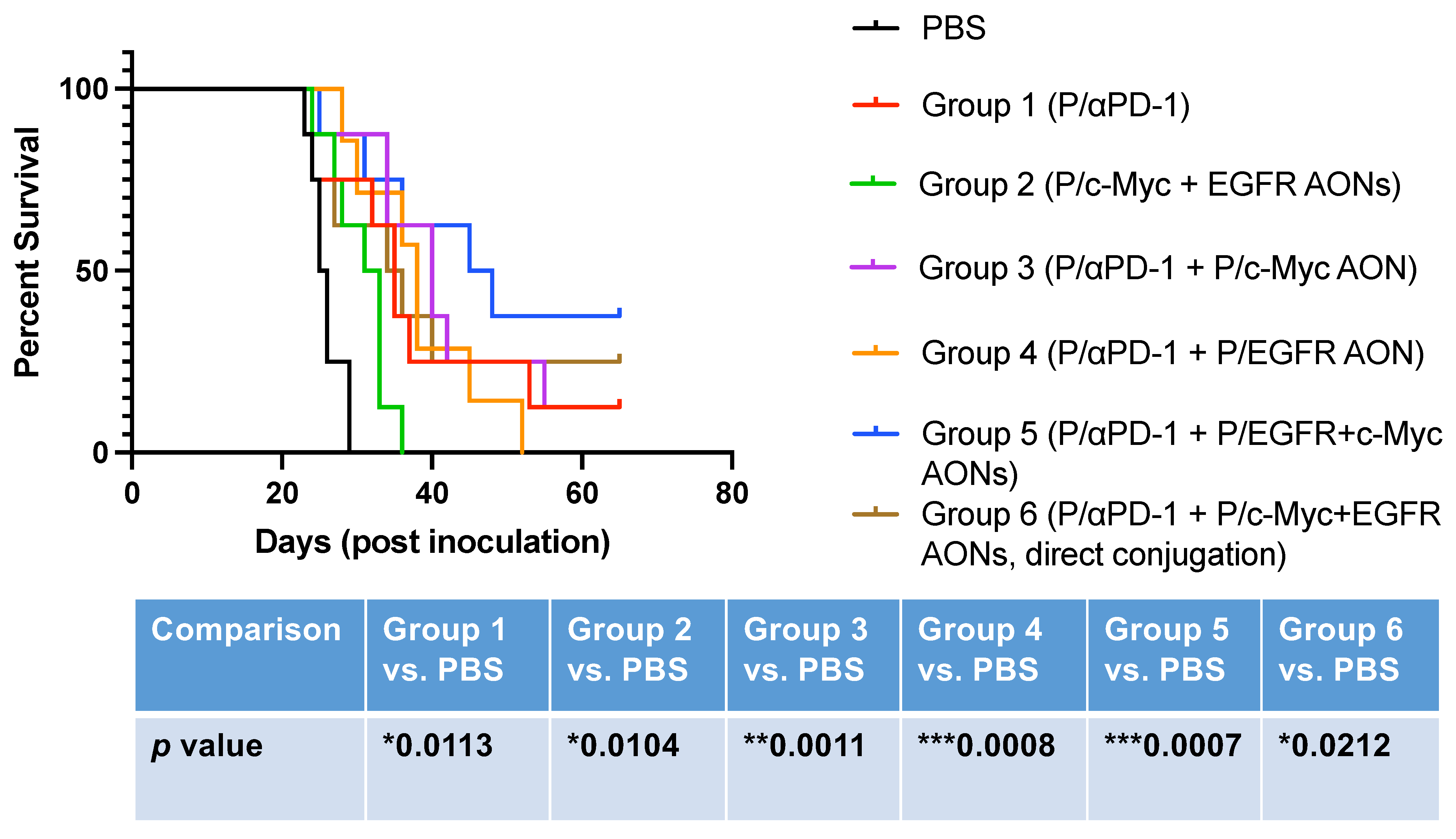
3.2. Brain Tumor Treatment In Vivo
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Tanaka, T.; Morimoto, Y.; Kuranari, Y.; Yamamoto, Y.; Takei, J.; Murayama, Y.; Yoshida, K.; Sasaki, H. Alterations of the tumor microenvironment in glioblastoma following radiation and temozolomide with or without bevacizumab. Ann. Transl. Med. 2020, 8, 297. [Google Scholar] [CrossRef]
- Finch, A.; Solomou, G.; Wykes, V.; Pohl, U.; Bardella, C.; Watts, C. Advances in research of adult gliomas. Int. J. Mol. Sci. 2021, 22, 924. [Google Scholar] [CrossRef]
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Núñez, N.G.; De Feo, D.; Kickingereder, P.V.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimberg, A. The glioblastoma vasculature as a target for cancer therapy. Biochem. Soc. Trans. 2014, 42, 1647–1652. [Google Scholar] [CrossRef]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma stem cells: Signaling, microenvironment, and therapy. Stem Cells Int. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Li, M.; Hall, P.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.S.; Huang, P.; et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778. [Google Scholar] [CrossRef]
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling drugs into the brain: An overview of ligands targeting transcytosis for drug delivery across the blood–brain barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef] [Green Version]
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharm. Exp. Ther. 2008, 324, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Demeule, M.; Currie, J.C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Huile, G.; Shuaiqi, P.; Zhi, Y.; Shijie, C.; Chen, C.; Xinguo, J.; Shun, S.; Zhiqing, P.; Yu, H. A cascade targeting strategy for brain neuroglial cells employing nanoparticles modified with angiopep-2 peptide and EGFP-EGF1 protein. Biomaterials 2011, 32, 8669–8675. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, X.; Gong, M.; Zhang, J. Delivery of a peptide-drug conjugate targeting the blood brain barrier improved the efficacy of paclitaxel against glioma. Oncotarget 2016, 7, 79401–79407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.-X.; Miao, W.-M.; Pang, D.-W.; Wu, J.-S.; Tong, Q.-S.; Li, J.-X.; Luo, J.-Q.; Li, W.-Y.; Du, J.-Z.; Wang, J. Angiopep-2 conjugated nanoparticles loaded with doxorubicin for the treatment of primary central nervous system lymphoma. Biomater. Sci. 2019, 8, 1290–1297. [Google Scholar] [CrossRef]
- Regina, A.; Demeule, M.; Che, C.; Lavallee, I.; Poirier, J.; Gabathuler, R.; Beliveau, R.; Castaigne, J.P. Antitumour activity of ANG1005, a conjugate between Paclitaxel and the new brain delivery vector Angiopep. Br. J. Pharmacol. 2008, 155, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, P.; Tang, S.-C.; Brenner, A.J.; Kesari, S.; Piccioni, D.E.; Anders, C.K.; Carrillo, J.A.; Chalasani, P.; Kabos, P.; Puhalla, S.L.; et al. ANG1005, a brain-penetrating peptide–drug conjugate, shows activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases. Clin. Cancer Res. 2020, 26, 2789–2799. [Google Scholar] [CrossRef] [Green Version]
- Israel, L.L.; Braubach, O.; Galstyan, A.; Chiechi, A.; Shatalova, E.S.; Grodzinski, Z.; Ding, H.; Black, K.L.; Ljubimova, J.Y.; Holler, E. A Combination of tri-leucine and Angiopep-2 drives a poly-anionic polymalic acid nanodrug platform across the blood brain barrier. ACS Nano 2019, 13, 1253–1271. [Google Scholar] [PubMed]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood–brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Kelly, N.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; et al. MYC regulates the antitumor immune response through CD47 and PD-L. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Zhao, J.; Xing, Y.; Zhang, X.; Liu, J.; Ouyang, Q.; Chen, J.; Su, F.; Liu, Q.; Song, E. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 2018, 175, 442–457. [Google Scholar] [CrossRef] [Green Version]
- Miyasato, Y.; Takashima, Y.; Takeya, H.; Yano, H.; Hayano, A.; Nakagawa, T.; Makino, K.; Yamanaka, R.; Komohara, Y. The expression of PD-1 ligands and IDO1 by macrophage/microglia in primary central nervous system lymphoma. J. clin. Exp. hematop. 2018, 58, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Baylot, V.; Felsher, D.W. MYC: Master regulator of immune privilege. Trends Immunol. 2017, 38, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Adams, N.M.; Xu, Y.; Cao, J.; Allan, D.S.J.; Carlyle, J.; Chen, X.; Sun, J.C.; Glimcher, L.H. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 2019, 20, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-T.; Patil, R.; Ding, H.; Gangalum, P.R.; Cavenee, W.K.; Furnari, F.; Ljubimov, V.A.; Chesnokova, A.; Galstyan, A.; Kramerov, A.A.; et al. Simultaneous blockade of CK2 and EGFR pathways by tumor-targeted nanobioconjugates significantly improves therapeutic efficacy against glioblastoma multiforme. J. Control Release 2016, 28, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR Family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Ding, H.; Inoue, S.; Ljubimov, A.V.; Patil, R.; Portilla-Arias, J.; Hu, J.; Konda, B.; Wawrowsky, K.A.; Fujita, M.; Karabalin, N.; et al. Inhibition of brain tumor growth by intravenous poly (β-L-malic acid) nanobioconjugate with pH-dependent drug release. Proc. Natl. Acad. Sci. USA 2010, 107, 18143–18148. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Patil, R.; Galstyan, A.; Klymyshyn, D.; Ding, H.; Chesnokova, A.; Cavenee, W.K.; Furnari, F.B.; Ljubimov, V.A.; Shatalova, E.S.; et al. Blockade of a laminin-411-Notch axis with CRISPR/Cas9 or a nanobioconjugate inhibits glioblastoma growth through tumor-microenvironment crosstalk. Cancer Res. 2019, 79, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Portilla-Arias, J.; Patil, R.; Black, K.L.; Ljubimova, J.Y.; Holler, E. The optimization of polymalic acid peptide copolymers for endosomolytic drug delivery. Biomaterials 2011, 32, 5269–5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubimova, J.Y.; Ding, H.; Portilla-Arias, J.; Patil, R.; Gangalum, P.R.; Chesnokova, A.; Inoue, S.; Rekechenetskiy, A.; Nassoura, T.; Black, K.L.; et al. Polymalic acid-based nano biopolymers for targeting of multiple tumor markers: An opportunity for personalized medicine? J. Vis. Exp. 2014, 88, 50668. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.S.; Holler, E. β-poly(L-malate) production by non-growing microplasmodia of Physarum polycephalum: Effects of metabolic intermediates and inhibitors. FEMS Microbiol. Lett. 2000, 193, 69–74. [Google Scholar] [CrossRef]
- Cockle, J.V.; Rajani, K.; Zaidi, S.; Kottke, T.; Thompson, J.; Diaz, R.M.; Shim, K.; Peterson, T.; Parney, I.; Short, S.; et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro-Oncology 2016, 18, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.; Jo, A.; Giedt, J.; Vinegoni, C.; Yang, K.S.; Peruzzi, P.; Chiocca, E.A.; Breakefield, X.O.; Lee, H.; Weissleder, R. Characterization of single microvesicles in plasma from glioblastoma patients. Neuro-Oncology 2019, 21, 606–615. [Google Scholar] [CrossRef]
- Ding, H.; Patil, R.; Portilla-Arias, J.; Black, K.L.; Ljubimova, J.Y.; Holler, E. Quantitative analysis of PMLA nanoconjugate components after backbone cleavage. Int. J. Mol. Sci. 2015, 16, 8607–8620. [Google Scholar] [CrossRef] [Green Version]
- Ljubimova, J.Y.; Portilla-Arias, J.; Patil, R.; Ding, H.; Inoue, S.; Markman, J.; Rekechenetskiy, A.; Konda, B.; Gangalum, P.R.; Chesnokova, A.; et al. Toxicity and efficacy evaluation of multiple targeted polymalic acid conjugates for triple-negative breast cancer treatment. J. Drug Target. 2013, 21, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Kramerov, A.A.; Shah, R.; Ding, H.; Holler, E.; Turjman, S.; Rabinowitz, Y.S.; Ghiam, S.; Maguen, E.; Svendsen, C.N.; Saghizadeh, M.; et al. Novel nanopolymer RNA therapeutics normalize human diabetic corneal wound healing and epithelial stem cells. Nanomed. Nanotechnol. Biol. Med. 2021, 32, 102332. [Google Scholar] [CrossRef]
- Ljubimova, J.Y.; Fujita, M.; Khazenzon, N.M.; Lee, B.-S.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Holler, E. Nanoconjugate based on polymalic acid for tumor targeting. Chem. Biol. Interact. 2008, 171, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Helguera, G.; Rodríguez, J.A.; Markman, J.; Luria-Pérez, R.; Gangalum, P.R.; Portilla-Arias, J.; Inoue, S.; Daniels-Wells, T.R.; Black, K.L.; et al. Polymalic acid nanoconjugate for simultaneous inhibition of tumor growth and immunostimulation in HER2/neu-positive breast cancer. J. Control. Release 2013, 171, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.S.; Vert, M.; Holler, E. Water-soluble aliphatic polyesters: Poly(malic acid)s. In Biopolymers-Polyesters I: Biological Systems and Biotechnological Production; Wiley-VCH: Weinheim, Germany, 2001; Volume 3a. [Google Scholar]
- Inoue, S.; Patil, R.; Portilla-Arias, J.; Ding, H.; Konda, B.; Espinoza, A.; Mongayt, D.; Elramsisy, A.; Phillips, H.W.; Black, K.L.; et al. Synthesis of nanobioconjugate for direct targeting tumor-cell and inhibition of EGFR expression in triple negative breast cancer. PLoS ONE 2012, 7, e31070. [Google Scholar] [CrossRef] [Green Version]
- Nag, A.; Mitra, G.; Ghosh, P.C. A colorimetric assay for estimation of polyethylene glycol and polyethylene glycolated protein using ammonium ferrothiocyanate. Anal. Biochem. 1996, 237, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, M.P.; Saiz, E. Combination of SEC/MALS experimental procedures and theoretical analysis for studying the solution properties of macromolecules. J. Biochem. Biophys. Methods 2003, 56, 95–116. [Google Scholar] [CrossRef]
- Fujita, M.; Lee, B.-S.; Khazenzon, N.M.; Penichet, M.L.; Wawrowsky, K.A.; Patil, R.; Ding, H.; Holler, E.; Black, K.L.; Ljubimova, J.Y. Brain tumor tandem targeting using a combination of monoclonal antibodies attached to biopoly(β-L-malic acid). J. Control. Release 2007, 122, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Fan, W.; Lau, J.; Deng, L.; Shen, Z.; Chen, X. Emerging blood–brain-barrier-crossing nanotechnology for brain cancer theranostics. Chem. Soc. Rev. 2019, 48, 2967–3014. [Google Scholar] [CrossRef]
- Ljubimova, J.Y.; Sun, T.; Mashouf, L.; Ljubimov, A.V.; Israel, L.L.; Ljubimov, V.A.; Falahatian, V.; Holler, E. Covalent nanodelivery systems for selective imaging and treatment of brain tumors. Adv. Drug. Deliv. Rev. 2017, 113, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. CSF, blood-brain barrier, and brain drug delivery. Expert Opin. Drug. Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Villaseñor, R.; Ozmen, L.; Messaddeq, N.; Grüninger, F.; Loetscher, H.; Keller, A.; Betsholtz, C.; Freskgård, P.-O.; Collin, L. Trafficking of endogenous immunoglobulins by endothelial cells at the blood-brain barrier. Sci. Rep. 2016, 6, 25658. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 183, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, Z.; Kloepper, J.; Ren, J.; Tay, R.E.; Kazer, S.W.; Kiner, E.; Krishnan, S.; Posada, J.M.; Ghosh, M.; Mamessier, E.; et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Fares, J.; Ulasov, I.; Timashev, P.; Lesniak, M.S. Emerging principles of brain immunology and immune checkpoint blockade in brain metastases. Brain 2021, 144, 1046–1066. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumor barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Multifunctional Nano Polymers (MNPs) | Abbreviation | Hydrodynamic Diameter (nm) | ζ Potential (mV) |
|---|---|---|---|
| PMLA/PEG5000(2%)/LLL(40%)/ AP-2(2%)/αPD-1(0.2%) | αPD-1 MNP | 15.2 (±1.7) | −9.5 (±0.4) |
| PMLA/PEG5000(2%)/LLL(40%)/ AP-2(2%)/c-Myc+EGFR AON(2.0%) | AON-SS MNP | 8.9 (±0.9) | −9.2 (±0.8) |
| PMLA/PEG5000(2%)/LLL(40%)/ AP-2(2%)/c-Myc+EGFR AON-thioether(2%) | AON-Thioether MNP | 10.6 (±0.9) | −10.1 (±0.7) |
| PMLA/PEG5000(2%)/LLL(40%)/ AP-2(2%)/c-Myc AON(2.0%) | c-Myc-AON MNP | 9.6 (±0.9) | −8.3 (±0.5) |
| PMLA/PEG5000(2%)/LLL(40%)/ AP-2(2%)/EGFR AON(2.0%) | EGFR-AON MNP | 9.3 (±1.1) | −9.4 (±0.9) |
| Group | MNP Composition |
|---|---|
| 1 | P/mPEG5000(2%)/LLL(40%)/AP-2*(2%)/αPD-1**(0.2%) |
| 2 | P/mPEG5000(2%)/LLL(40%/AP-2(2%)/EGFR/EGFRvIII AON***(2%)/c-Myc AON***(2%) |
| 3 | P/mPEG5000(2%)/LLL(40%)/AP-2(2%)/αPD-1(0.2%) co-administered with P/mPEG5000(2%)/LLL(40%)/AP-2(2%)/c-Myc AON(2%) |
| 4 | P/mPEG5000(2%)/LLL(40%)/AP-2(2%)/αPD-1(0.2%) co-administered with P/mPEG5000(2%)/LLL(40%)/AP-2(2%)/αPD-1 (0.2%)/EGFR/EGFRvIII AON(2%) |
| 5 | P/mPEG5000(2%)/LLL(40%)/AP-2(2%)/αPD-1(0.2%) co-administered with P/mPEG5000(2%)/LLL(40%/AP-2(2%)/EGFR/EGFRvIII AON(2%)/c-Myc AON(2%) |
| 6 | P/mPEG5000(2%)/LLL(40%)/AP-2(2%)/αPD-1(0.2%) co-administered with P/mPEG5000(2%)/LLL(40%/AP-2(2%)/EGFR/EGFRvIII AON****(2%)/ c-Myc AON****(2%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, R.; Sun, T.; Rashid, M.H.; Israel, L.L.; Ramesh, A.; Davani, S.; Black, K.L.; Ljubimov, A.V.; Holler, E.; Ljubimova, J.Y. Multifunctional Nanopolymers for Blood–Brain Barrier Delivery and Inhibition of Glioblastoma Growth through EGFR/EGFRvIII, c-Myc, and PD-1. Nanomaterials 2021, 11, 2892. https://doi.org/10.3390/nano11112892
Patil R, Sun T, Rashid MH, Israel LL, Ramesh A, Davani S, Black KL, Ljubimov AV, Holler E, Ljubimova JY. Multifunctional Nanopolymers for Blood–Brain Barrier Delivery and Inhibition of Glioblastoma Growth through EGFR/EGFRvIII, c-Myc, and PD-1. Nanomaterials. 2021; 11(11):2892. https://doi.org/10.3390/nano11112892
Chicago/Turabian StylePatil, Rameshwar, Tao Sun, Mohammad Harun Rashid, Liron L. Israel, Arshia Ramesh, Saya Davani, Keith L. Black, Alexander V. Ljubimov, Eggehard Holler, and Julia Y. Ljubimova. 2021. "Multifunctional Nanopolymers for Blood–Brain Barrier Delivery and Inhibition of Glioblastoma Growth through EGFR/EGFRvIII, c-Myc, and PD-1" Nanomaterials 11, no. 11: 2892. https://doi.org/10.3390/nano11112892
APA StylePatil, R., Sun, T., Rashid, M. H., Israel, L. L., Ramesh, A., Davani, S., Black, K. L., Ljubimov, A. V., Holler, E., & Ljubimova, J. Y. (2021). Multifunctional Nanopolymers for Blood–Brain Barrier Delivery and Inhibition of Glioblastoma Growth through EGFR/EGFRvIII, c-Myc, and PD-1. Nanomaterials, 11(11), 2892. https://doi.org/10.3390/nano11112892