
Data-Driven Modeling of the Cellular Pharmacokinetics of Degradable Chitosan-Based Nanoparticles

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Nanoparticle Preparation, and Image Acquisition
2.2. Nanoparticle Characterization
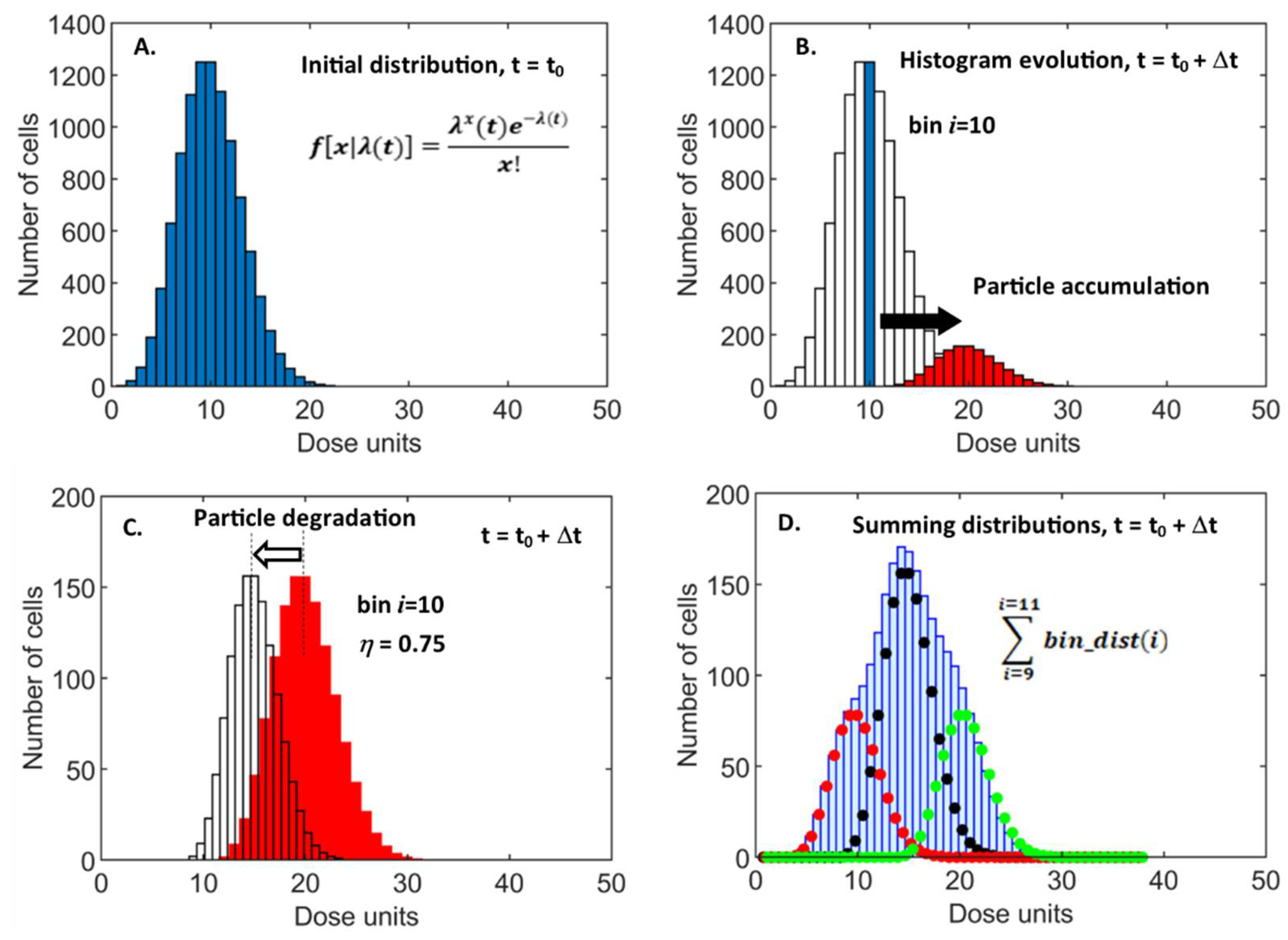
2.3. Nanoparticle Supply Kinetics
2.4. Analytical Models
3. Results
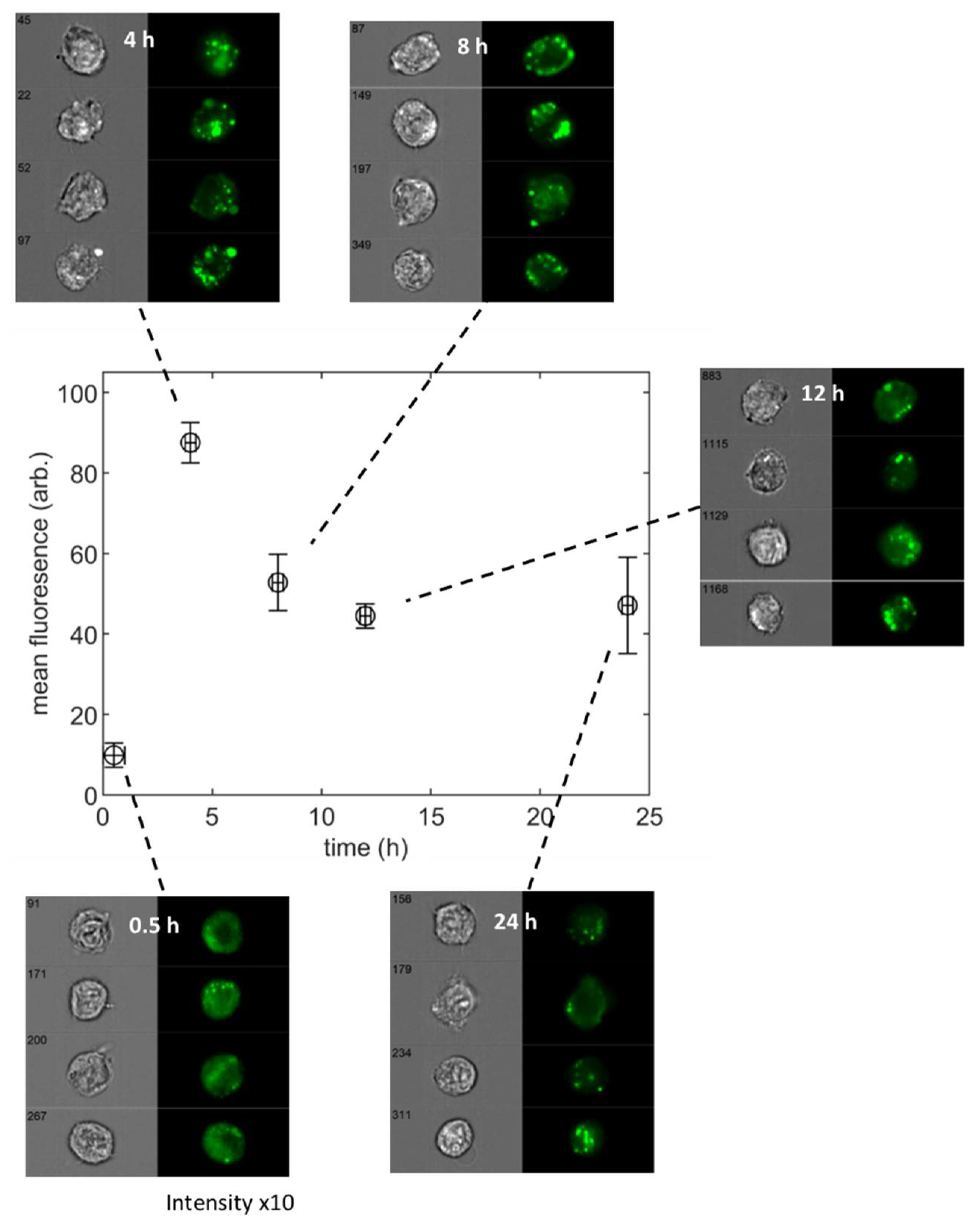
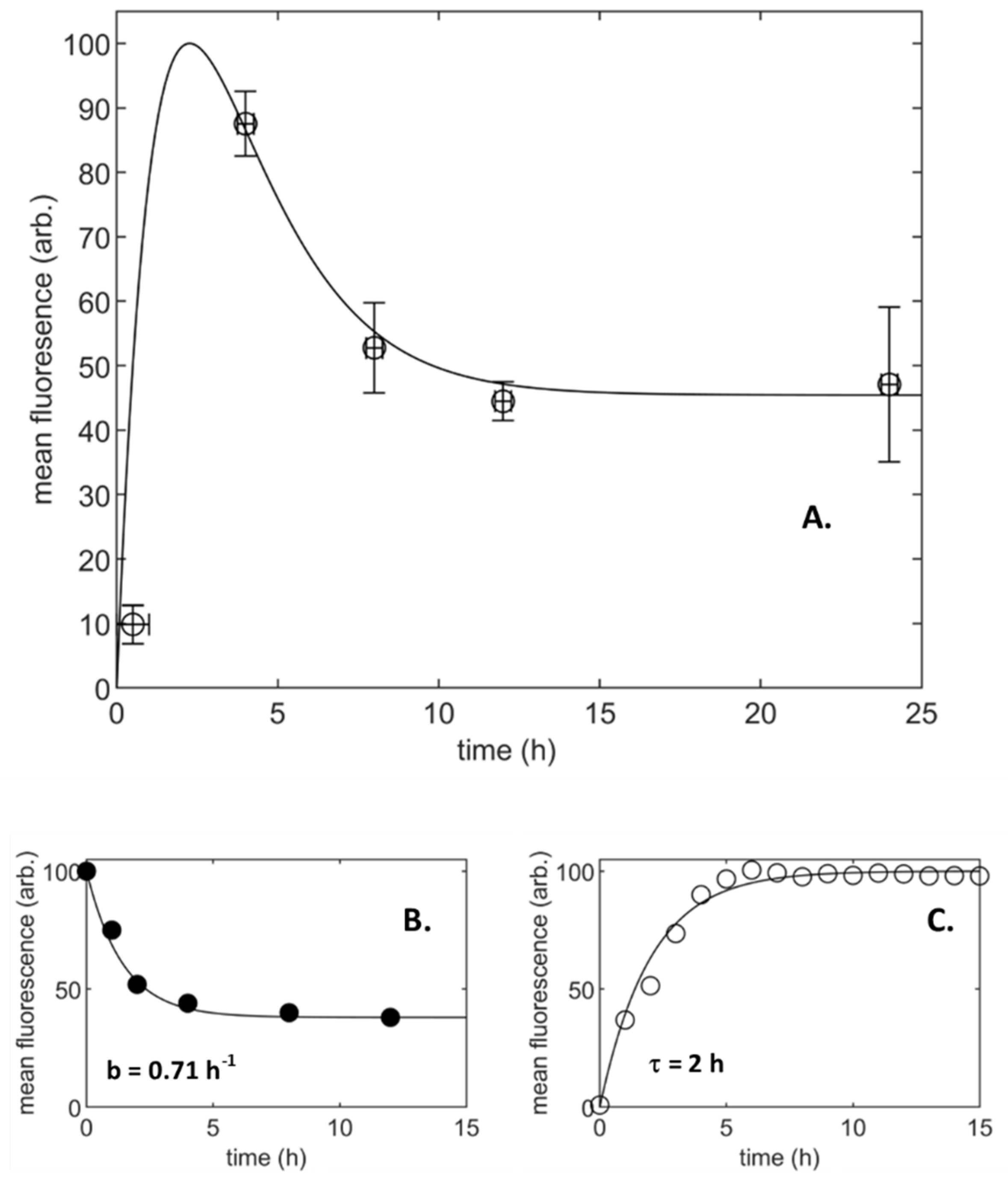
3.1. Population-Averaged Pharmacokinetics
- 1.
- Particle uptake at a constant rate (A1)
- 2.
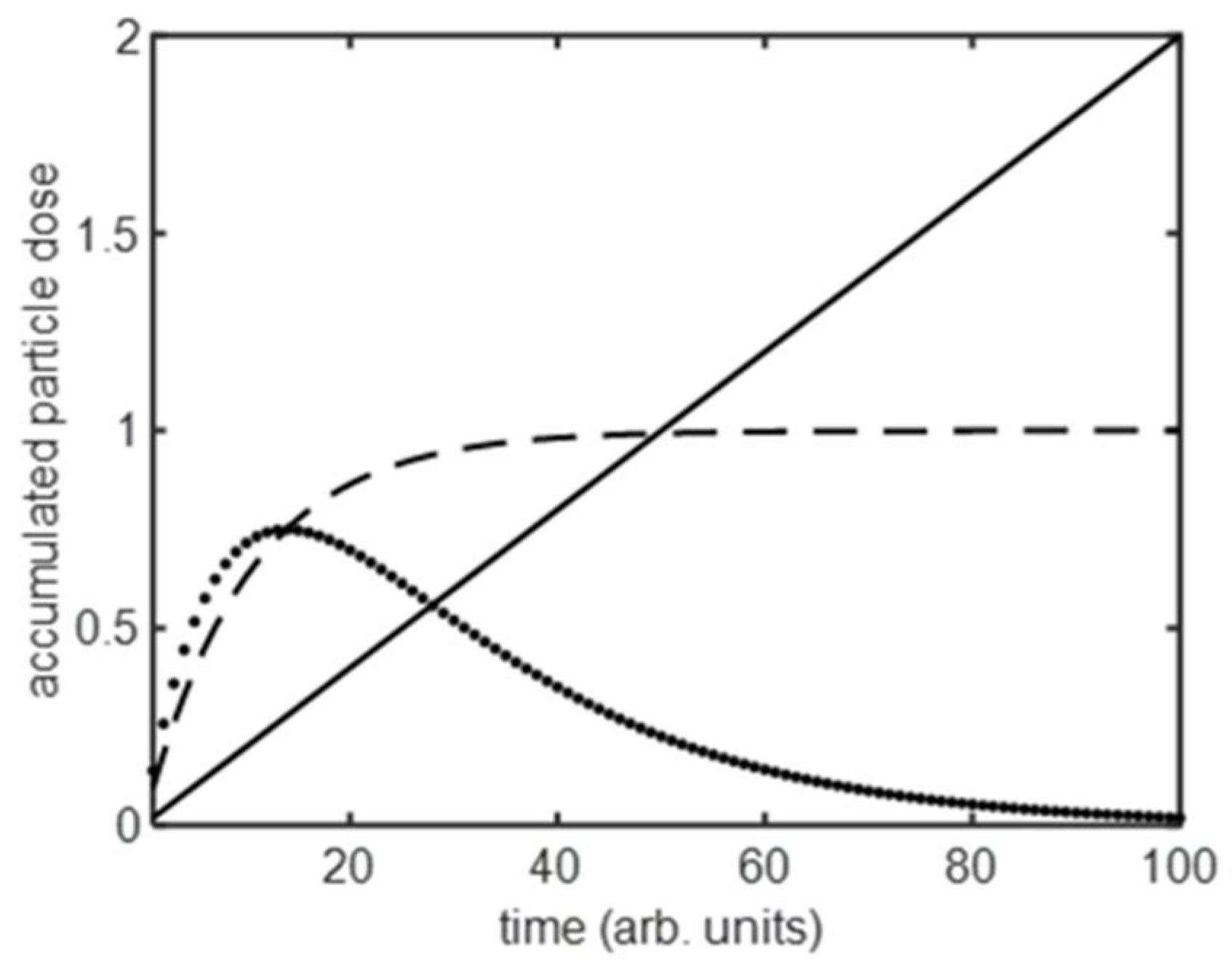
- Particle uptake with a limiting process to supply or accumulation (A2 and A3)
- 3.
- Particle uptake with a limiting process to supply and accumulation (A4)
3.2. Single Cell Pharmacokinetics
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilczewska, A.Z.; Niemirowicz, K.; Markiewicz, K.H.; Car, H. Nanoparticles as drug delivery systems. Pharmacol. Rep. 2012, 64, 1020–1037. [Google Scholar] [CrossRef]
- Yang, J.; Hendricks, W.; Liu, G.; McCaffery, J.M.; Kinzler, K.W.; Huso, D.L.; Vogelstein, B.; Zhou, S. A nanoparticle formulation that selectively transfects metastatic tumors in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 14717–14722. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.L.J.; Benito, J.M.; Mellet, C.O.; Fernández, J.M.G. Molecular nanoparticle-based gene delivery systems. J. Drug Deliv. Sci. Technol. 2017, 42, 18–37. [Google Scholar] [CrossRef]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wang, Z.; Feng, M. Nanocarriers with tunable surface properties to unblock bottlenecks in systemic drug and gene delivery. J. Control. Release 2015, 214, 121–133. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Xu, X.; Bertrand, N.; Pridgen, E.; Swami, A.; Farokhzad, O.C. Interactions of nanomaterials and biological systems: Implications to personalized nanomedicine. Adv. Drug Deliv. Rev. 2012, 64, 1363–1384. [Google Scholar] [CrossRef] [PubMed]
- Hinde, E.; Thammasiraphop, K.; Duong, H.; Yeow, J.; Karagoz, B.; Boyer, C.; Gooding, J.J.; Gaus, K. Pair correlation microscopy reveals the role of nanoparticle shape in intracellular transport and site of drug release. Nat. Nanotechnol. 2016, 12, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, A.; Fenaroli, F.; Monopoli, M.P.; Åberg, C.; Dawson, K.A.; Salvati, A. Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells. ACS Nano 2012, 6, 5845–5857. [Google Scholar] [CrossRef]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef]
- Avgoustakis, K.; Beletsi, A.; Panagi, Z.; Klepetsanis, P.; Karydas, A.G.; Ithakissios, D.S. PLGA–mPEG nanoparticles of cisplatin: In vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control. Release 2002, 79, 123–135. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.; Andresen, T.L. Factors Controlling Nanoparticle Pharmacokinetics: An Integrated Analysis and Perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef]
- Pan, R.; Liu, G.; Li, Y.; Wei, Y.; Li, S.; Tao, L. Size-dependent endocytosis and a dynamic-release model of nanoparticles. Nanoscale 2018, 10, 8269–8274. [Google Scholar] [CrossRef]
- Soares, P.I.; Sousa, A.I.; Silva, J.C.; Ferreira, I.M.; Novo, C.M.; Borges, J.P. Chitosan-based nanoparticles as drug delivery systems for doxorubicin: Optimization and modelling. Carbohydr. Polym. 2016, 147, 304–312. [Google Scholar] [CrossRef]
- Aydın, R.S.T.; Pulat, M. 5-Fluorouracil Encapsulated Chitosan Nanoparticles for pH-Stimulated Drug Delivery: Evaluation of Controlled Release Kinetics. J. Nanomater. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Golovin, Y.; Golovin, D.; Klyachko, N.; Majouga, A.; Kabanov, A. Modeling drug release from functionalized magnetic nanoparticles actuated by non-heating low frequency magnetic field. J. Nanoparticle Res. 2017, 19, 64. [Google Scholar] [CrossRef]
- Son, G.-H.; Lee, B.-J.; Cho, C.-W. Mechanisms of drug release from advanced drug formulations such as polymeric-based drug-delivery systems and lipid nanoparticles. J. Pharm. Investig. 2017, 47, 287–296. [Google Scholar] [CrossRef]
- Li, M.; Al-Jamal, K.T.; Kostarelos, K.; Reineke, J. Physiologically Based Pharmacokinetic Modeling of Nanoparticles. ACS Nano 2010, 4, 6303–6317. [Google Scholar] [CrossRef] [PubMed]
- Carlander, U.; Li, D.; Jolliet, O.; Emond, C.; Johanson, G. Toward a general physiologically-based pharmacokinetic model for intravenously injected nanoparticles. Int. J. Nanomed. 2016, ume 11, 625–640. [Google Scholar] [CrossRef]
- Lin, Z.; Monteiro-Riviere, N.A.; Riviere, J.E. A physiologically based pharmacokinetic model for polyethylene glycol-coated gold nanoparticles of different sizes in adult mice. Nanotoxicology 2015, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yaehne, K.; Tekrony, A.; Clancy, A.; Gregoriou, Y.; Walker, J.; Dean, K.; Nguyen, T.; Doiron, A.; Rinker, K.; Jiang, X.Y.; et al. Nanoparticle Accumulation in Angiogenic Tissues: Towards Predictable Pharmacokinetics. Small 2013, 9, 3118–3127. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Åberg, C.; Santos, T.; Varela, J.A.; Pinto, P.; Lynch, I.; Dawson, K.A. Experimental and theoretical comparison of intracellular import of polymeric nanoparticles and small molecules: Toward models of uptake kinetics. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Soininen, S.K.; Vellonen, K.-S.; Heikkinen, A.T.; Auriola, S.; Ranta, V.-P.; Urtti, A.; Ruponen, M. Intracellular PK/PD Relationships of Free and Liposomal Doxorubicin: Quantitative Analyses and PK/PD Modeling. Mol. Pharm. 2016, 13, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Donahue, N.D.; Acar, H.; Wilhelm, S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv. Drug Deliv. Rev. 2019, 143, 68–96. [Google Scholar] [CrossRef]
- Lopes, C.D.; Gomes, C.P.; Neto, E.; Sampaio, P.; Aguiar, P.; Pêgo, A.P. Microfluidic-based platform to mimic the in vivo peripheral administration of neurotropic nanoparticles. Nanomedicine 2016, 11, 3205–3221. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.; Gonçalves, N.P.; Gomes, C.; Saraiva, M.J.; Pêgo, A.P. BDNF gene delivery mediated by neuron-targeted nanoparticles is neuroprotective in peripheral nerve injury. Biomaterials 2017, 121, 83–96. [Google Scholar] [CrossRef]
- Gomes, C.; Varela-Moreira, A.; Leiro, V.; Lopes, C.; Moreno, P.; Lazaro, M.G.; Pêgo, A.P. A high-throughput bioimaging study to assess the impact of chitosan-based nanoparticle degradation on DNA delivery performance. Acta Biomater. 2016, 46, 129–140. [Google Scholar] [CrossRef]
- Ware, M.J.; Godin, B.; Singh, N.; Majithia, R.; Shamsudeen, S.; Serda, R.E.; Meissner, K.E.; Rees, P.; Summers, H.D. Analysis of the Influence of Cell Heterogeneity on Nanoparticle Dose Response. ACS Nano 2014, 8, 6693–6700. [Google Scholar] [CrossRef]
- Lunov, O.; Zablotskii, V.; Syrovets, T.; Röcker, C.; Tron, K.; Nienhaus, G.U.; Simmet, T. Modeling receptor-mediated endocytosis of polymer-functionalized iron oxide nanoparticles by human macrophages. Biomaterials 2011, 32, 547–555. [Google Scholar] [CrossRef]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Hans, M.L.; Lowman, A.M. Biodegradable nanoparticles for drug delivery and targeting. Curr. Opin. Solid State Mater. Sci. 2002, 6, 319–327. [Google Scholar] [CrossRef]
- Oh, N.; Park, J.H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014, 9, 51–63. [Google Scholar]
- Tancini, B.; Buratta, S.; Delo, F.; Sagini, K.; Chiaradia, E.; Pellegrino, R.M.; Emiliani, C.; Urbanelli, L. Lysosomal Exocytosis: The Extracellular Role of an Intracellular Organelle. Membranes 2020, 10, 406. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.F.; Johnson, H.E.; Granick, S. A Simple Kinetic Model of Polymer Adsorption and Desorption. Science 1993, 262, 2010–2012. [Google Scholar] [CrossRef] [PubMed]
- Jonker, C.T.H.; Deo, C.; Zager, P.J.; Tkachuk, A.N.; Weinstein, A.M.; Rodriguez-Boulan, E.; Lavis, L.D.; Schreiner, R. Accurate measurement of fast endocytic recycling kinetics in real time. J. Cell Sci. 2019, 133. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, A.; Salvati, A.; Santos-Martinez, M.J.; Radomski, M.W.; Dawson, K.A.; Åberg, C. Nanoparticle Adhesion to the Cell Membrane and Its Effect on Nanoparticle Uptake Efficiency. J. Am. Chem. Soc. 2013, 135, 1438–1444. [Google Scholar] [CrossRef]
- Pires, L.R.; Oliveira, H.; Barrias, C.C.; Sampaio, P.; Pereira, A.J.; Maiato, H.; Simões, S.; Pêgo, A.P. Imidazole-grafted chitosan-mediated gene delivery: In vitro study on transfection, intracellular trafficking and degradation. Nanomedicine 2011, 6, 1499–1512. [Google Scholar] [CrossRef]
- Summers, H.D.; Rees, P.; Holton, M.D.; Brown, M.R.; Chappell, S.C.; Smith, P.J.; Errington, R.J. Statistical analysis of nanoparticle dosing in a dynamic cellular system. Nat. Nanotechnol. 2011, 6, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Rees, P.; Wills, J.W.; Brown, R.; Barnes, C.M.; Summers, H.D. The origin of heterogeneous nanoparticle uptake by cells. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Gaylor, D.W. The use of Haber’s Law in standard setting and risk assessment. Toxicology 2000, 149, 17–19. [Google Scholar] [CrossRef]
- Stukalin, E.B.; Aifuwa, I.; Kim, J.S.; Wirtz, D.; Sun, S.X. Age-dependent stochastic models for understanding population fluctuations in continuously cultured cells. J. R. Soc. Interface 2013, 10, 20130325. [Google Scholar] [CrossRef]
- Alonso, A.A.; Molina, I.; Theodoropoulos, C. Modeling Bacterial Population Growth from Stochastic Single-Cell Dynamics. Appl. Environ. Microbiol. 2014, 80, 5241–5253. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Amir, A. The Effects of Stochasticity at the Single-Cell Level and Cell Size Control on the Population Growth. Cell Syst. 2017, 5, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Editorial. Join the dialogue. Nat. Nanotechnol. 2012, 7, 545. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Editorial. The dialogue continues. Nat. Nanotechnol. 2013, 8, 69. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Summers, H.D.; Gomes, C.P.; Varela-Moreira, A.; Spencer, A.P.; Gomez-Lazaro, M.; Pêgo, A.P.; Rees, P. Data-Driven Modeling of the Cellular Pharmacokinetics of Degradable Chitosan-Based Nanoparticles. Nanomaterials 2021, 11, 2606. https://doi.org/10.3390/nano11102606
Summers HD, Gomes CP, Varela-Moreira A, Spencer AP, Gomez-Lazaro M, Pêgo AP, Rees P. Data-Driven Modeling of the Cellular Pharmacokinetics of Degradable Chitosan-Based Nanoparticles. Nanomaterials. 2021; 11(10):2606. https://doi.org/10.3390/nano11102606
Chicago/Turabian StyleSummers, Huw D., Carla P. Gomes, Aida Varela-Moreira, Ana P. Spencer, Maria Gomez-Lazaro, Ana P. Pêgo, and Paul Rees. 2021. "Data-Driven Modeling of the Cellular Pharmacokinetics of Degradable Chitosan-Based Nanoparticles" Nanomaterials 11, no. 10: 2606. https://doi.org/10.3390/nano11102606
APA StyleSummers, H. D., Gomes, C. P., Varela-Moreira, A., Spencer, A. P., Gomez-Lazaro, M., Pêgo, A. P., & Rees, P. (2021). Data-Driven Modeling of the Cellular Pharmacokinetics of Degradable Chitosan-Based Nanoparticles. Nanomaterials, 11(10), 2606. https://doi.org/10.3390/nano11102606