Acyclovir-Loaded Solid Lipid Nanoparticles: Optimization, Characterization and Evaluation of Its Pharmacokinetic Profile

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Central Composite Design
2.3. Statistical Analysis
2.4. Verification of the Models
2.5. Preparation of Solid Lipid Nanoparticles
2.6. Size, Zeta Potential and Polydispersity Index (PdI) Analysis
2.7. Drug Entrapment Efficiency (EE)
2.8. Transmission Electron Microscopy (TEM)
2.9. Differential Scanning Calorimetry (DSC)
2.10. In Vitro Release Study
2.11. In Vivo Pharmacokinetic Evaluation
2.11.1. Animal Study
2.11.2. Blood Sample Collection and Plasma Preparation
2.11.3. Ultra Performance Liquid Chromatography (UPLC)
2.11.4. Plasma Protein Precipitation Procedure for Determination of Acyclovir Concentration
2.12. Pharmacokinetic Parameters
2.13. Statistical Analysis
3. Results and Discussion
3.1. Fitting the Response Surface Methodology
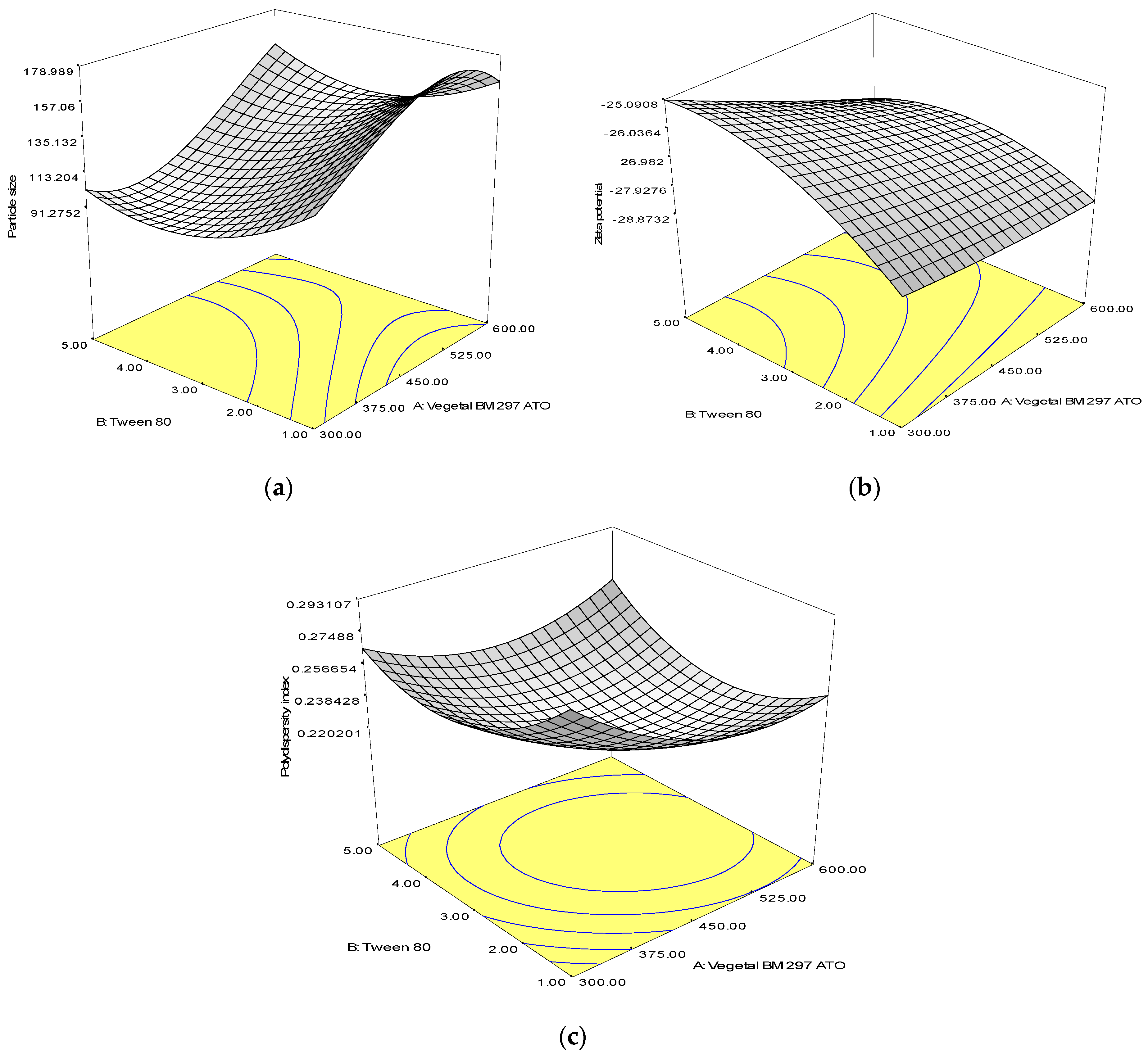
3.2. Response Surface Analysis
3.3. Verification of the Reduced Model
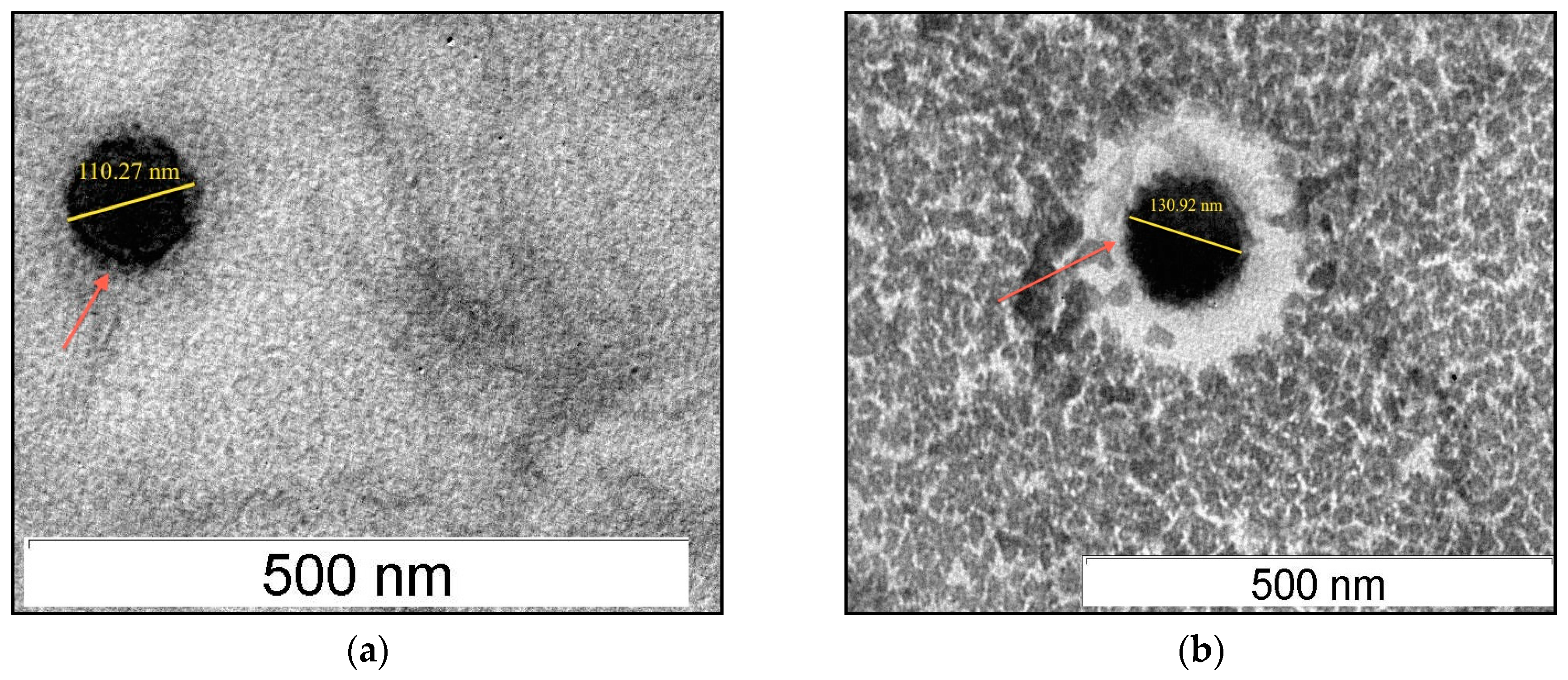
3.4. Physical Characteristics, Morphology and Entrapment Efficiency of Acyclovir-Loaded SLNs
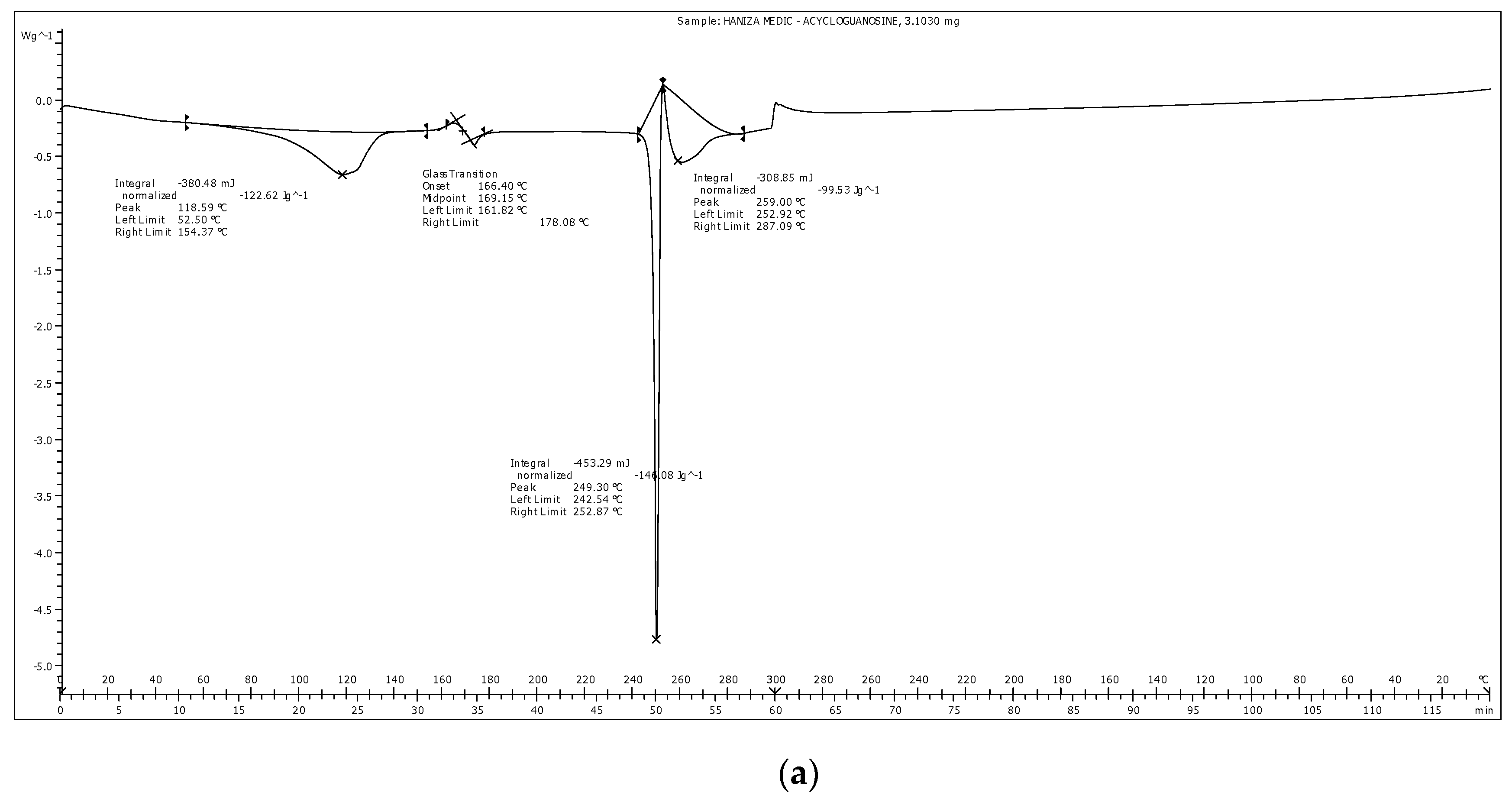
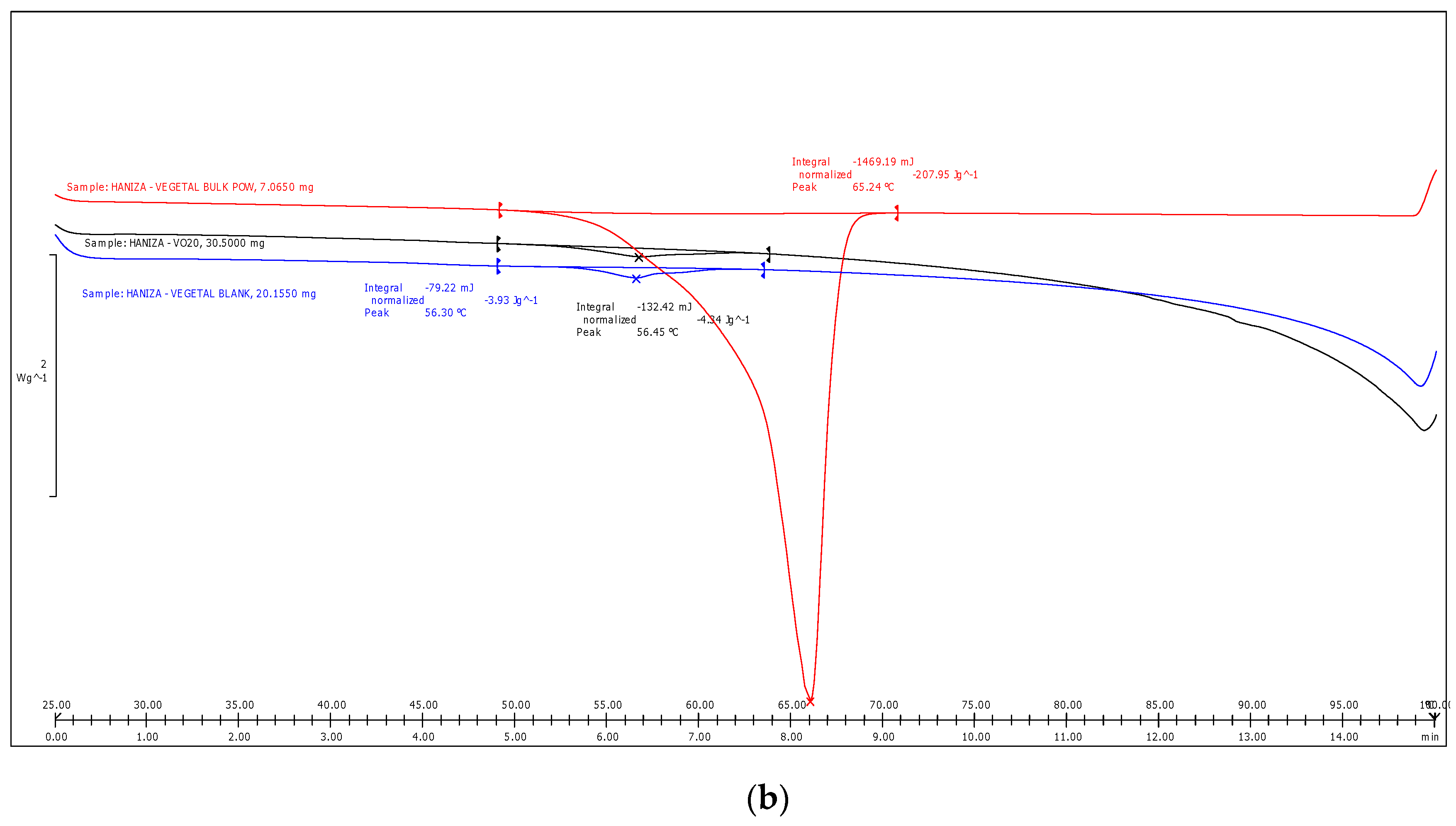
3.5. Differential Scanning Calorimetry Analysis
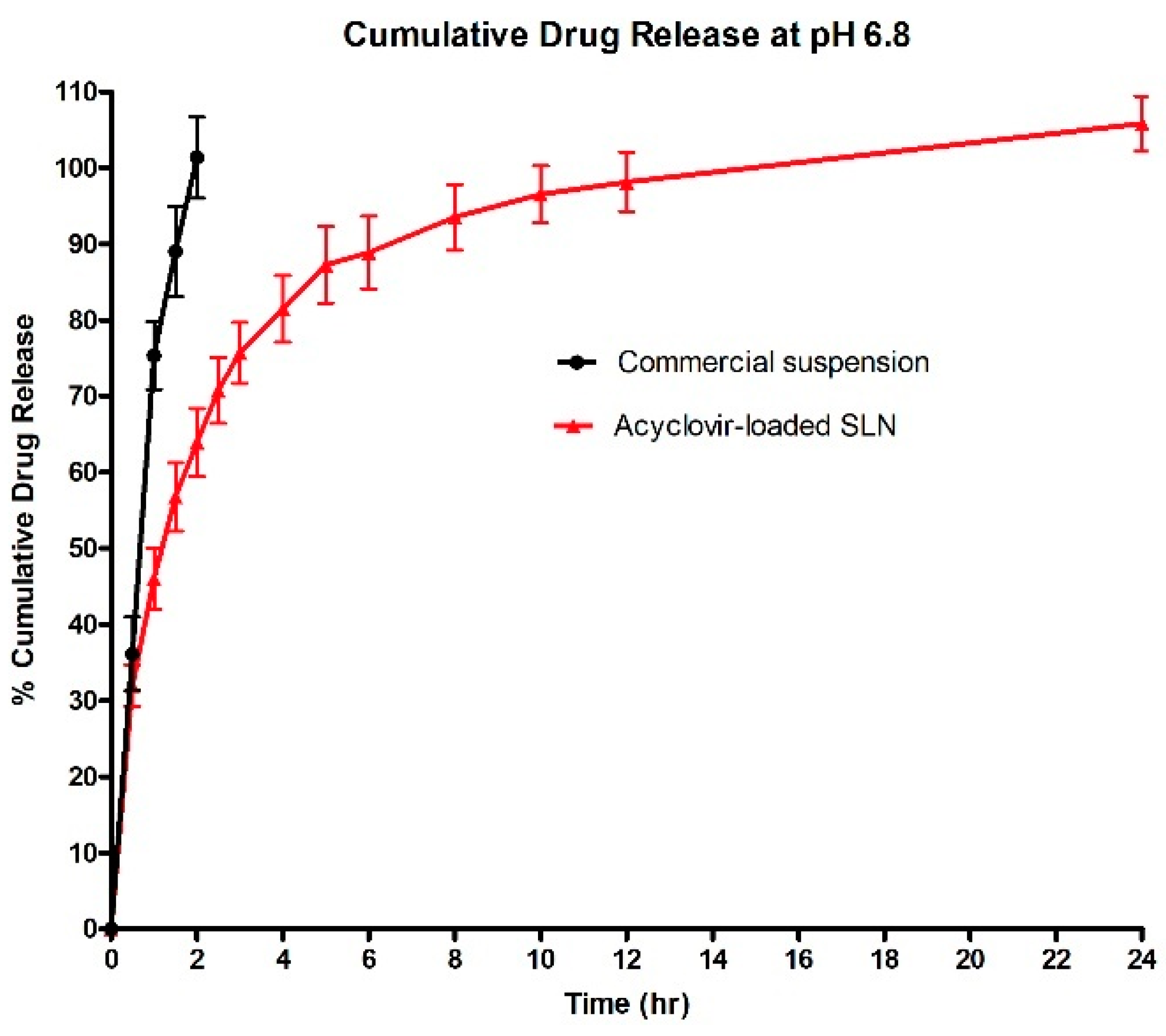
3.6. In Vitro Release Study
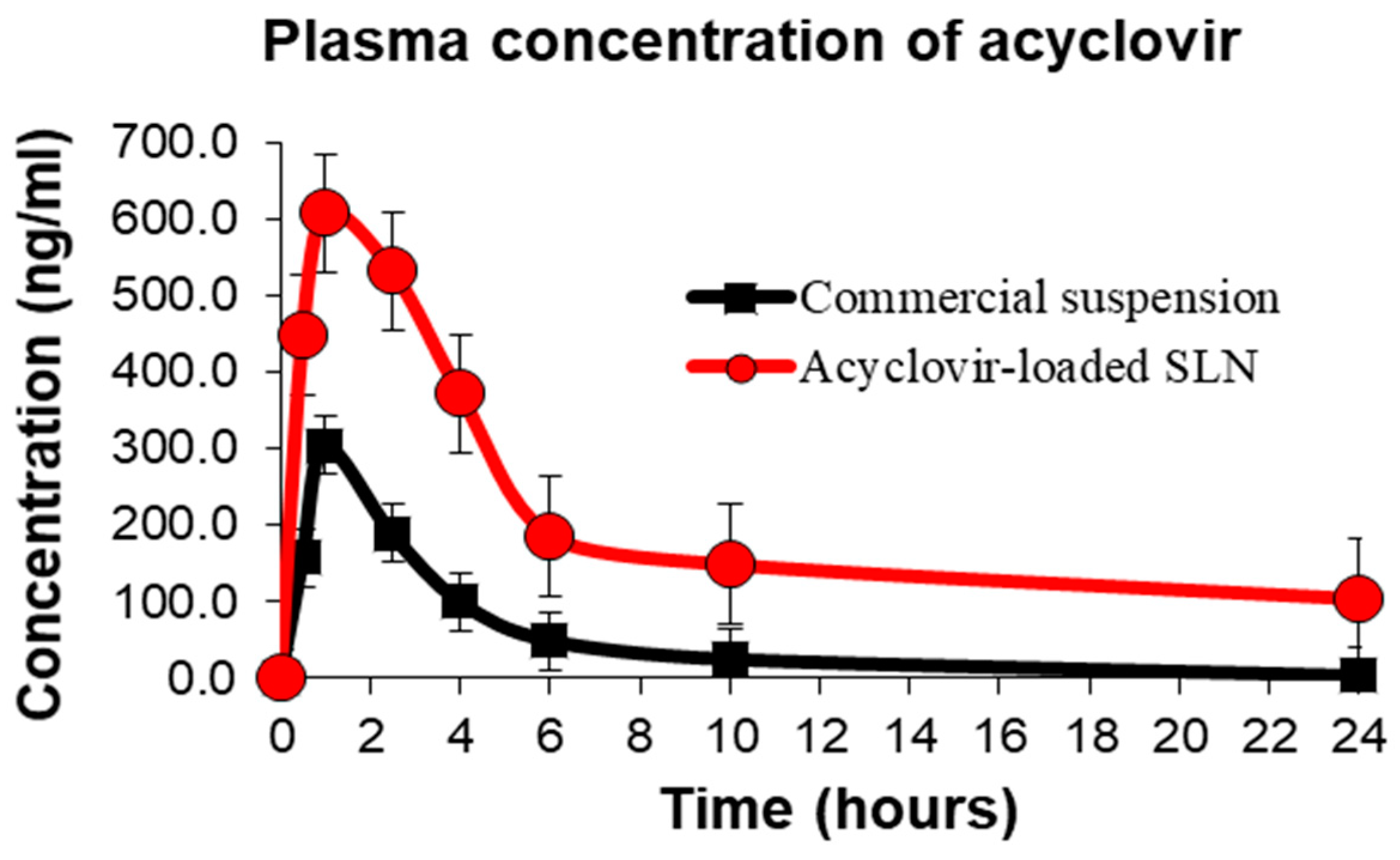
3.7. In Vivo Oral Bioavailability and Pharmacokinetic Evaluation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Du, T.; Zhou, G.; Roizman, B. Modulation of reactivation of latent herpes simplex virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc. Natl. Acad. Sci. USA 2013, 110, 2621–2628. [Google Scholar] [CrossRef]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Heal. Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef]
- Looker, K.J.; Magaret, A.S.; Turner, K.M.E.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS ONE 2015, 10, e114989. [Google Scholar]
- Álvarez, D.M.; Castillo, E.; Duarte, L.F.; Arriagada, J.; Corrales, N.; Farías, M.A.; Henríquez, A.; Agurto-Muñoz, C.; González, P.A. Current Antivirals and Novel Botanical Molecules Interfering With Herpes Simplex Virus Infection. Front. Microbiol. 2020, 11, 139. [Google Scholar] [CrossRef]
- Kukhanova, M.K.; Korovina, A.N.; Kochetkov, S. Human herpes simplex virus: Life cycle and development of inhibitors. Biochemistry 2014, 79, 1635–1652. [Google Scholar] [CrossRef]
- Smith, J.P.; Weller, S.; Johnson, B.; Nicotera, J.; Luther, J.M.; Haas, D.W. Pharmacokinetics of Acyclovir and Its Metabolites in Cerebrospinal Fluid and Systemic Circulation after Administration of High-Dose Valacyclovir in Subjects with Normal and Impaired Renal Function. Antimicrob. Agents Chemother. 2009, 54, 1146–1151. [Google Scholar] [CrossRef]
- Wald, A.; Benedetti, J.; Davis, G.; Remington, M.; Winter, C.; Corey, L. A randomized, double-blind, comparative trial comparing high- and standard-dose oral acyclovir for first-episode genital herpes infections. Antimicrob. Agents Chemother. 1994, 38, 174–176. [Google Scholar] [CrossRef][Green Version]
- Fleischer, R.; Johnson, M. Acyclovir Nephrotoxicity: A Case Report Highlighting the Importance of Prevention, Detection, and Treatment of Acyclovir-Induced Nephropathy. Case Rep. Med. 2010, 2010, 602783. [Google Scholar] [CrossRef]
- Lu, H.; Han, Y.-J.; Xu, J.-D.; Xing, W.-M.; Chen, J. Proteomic Characterization of Acyclovir-Induced Nephrotoxicity in a Mouse Model. PLoS ONE 2014, 9, e103185. [Google Scholar] [CrossRef]
- Chávez-Iñiguez, J.; Medina-Gonzalez, R.; Aguilar-Parra, L.; Torres-Vázquez, E.J.; Maggiani-Aguilera, P.; Cervantes-Pérez, E.; Garcia-Garcia, G. Oral acyclovir induced hypokalemia and acute tubular necrosis a case report. BMC Nephrol. 2018, 19, 324. [Google Scholar] [CrossRef]
- Perazella, M.A. Crystal-induced acute renal failure. Am. J. Med. 1999, 106, 459–465. [Google Scholar] [CrossRef]
- Leowattana, W. Antiviral Drugs and Acute Kidney Injury (AKI). Infect. Disord. Drug Targets 2019, 19, 375–382. [Google Scholar] [CrossRef]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Lin, C.-H.C.; Lin, Z.-C.; Fang, J.Y. Recent advances in oral delivery of drugs and bioactive natural products using solid lipid nanoparticles as the carriers. J. Food Drug Anal. 2017, 25, 219–234. [Google Scholar] [CrossRef]
- Rajpoot, K. Solid Lipid Nanoparticles: A Promising Nanomaterial in Drug Delivery. Curr. Pharm. Des. 2019, 25, 3943–3959. [Google Scholar] [CrossRef]
- Jain, A.; Sharma, G.; Thakur, K.; Raza, K.; Shivhare, U.S.; Ghoshal, G.; Katare, O.P. Beta-carotene-Encapsulated Solid Lipid Nanoparticles (BC-SLNs) as Promising Vehicle for Cancer: An Investigative Assessment. AAPS PharmSciTech 2019, 20, 100. [Google Scholar] [CrossRef]
- Cirri, M.; Mennini, N.; Maestrelli, F.; Mura, P.; Ghelardini, C.; Mannelli, L.D.C. Development and in vivo evaluation of an innovative “Hydrochlorothiazide-in Cyclodextrins-in Solid Lipid Nanoparticles” formulation with sustained release and enhanced oral bioavailability for potential hypertension treatment in pediatrics. Int. J. Pharm. 2017, 521, 73–83. [Google Scholar] [CrossRef]
- Severino, P.; Andreani, T.; Macedo, A.S.; Fangueiro, J.F.; Santana, M.H.A.; Silva, A.; Souto, E.B. Current State-of-Art and New Trends on Lipid Nanoparticles (SLN and NLC) for Oral Drug Delivery. J. Drug Deliv. 2011, 2012, 750891. [Google Scholar] [CrossRef]
- Hsu, C.-Y.; Pan, T.-L.; Alalaiwe, A.; Lin, Z.-C.; Fang, J.-Y. Use of Lipid Nanocarriers to Improve Oral Delivery of Vitamins. Nutrients 2019, 11, 68. [Google Scholar] [CrossRef]
- Ganesan, P.; Ramalingam, P.; Karthivashan, G.; Ko, Y.T.; Choi, D.-K. Recent developments in solid lipid nanoparticle and surface-modified solid lipid nanoparticle delivery systems for oral delivery of phyto-bioactive compounds in various chronic diseases. Int. J. Nanomed. 2018, 13, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Nooli, M.; Chella, N.; Kulhari, H.; Shastri, N.R.; Sistla, R. Solid lipid nanoparticles as vesicles for oral delivery of olmesartan medoxomil: Formulation, optimization and in vivo evaluation. Drug Dev. Ind. Pharm. 2017, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Darwis, Y.; Khan, A.A.; Mudassir, J.; Mohtar, N. Advanced drug delivery to the lymphatic system: Lipid-based nanoformulations. Int. J. Nanomed. 2013, 8, 2733–2744. [Google Scholar] [CrossRef]
- Paliwal, R.; Rai, S.; Vaidya, B.; Khatri, K.; Goyal, A.K.; Mishra, N.; Mehta, A.; Vyas, S.P. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomed. Nanotechnol. Boil. Med. 2009, 5, 184–191. [Google Scholar] [CrossRef]
- Shi, L.-L.; Xie, H.; Lu, J.; Cao, Y.; Liu, J.-Y.; Zhang, X.-X.; Zhang, H.; Cui, J.-H.; Cao, Q.-R. Positively Charged Surface-Modified Solid Lipid Nanoparticles Promote the Intestinal Transport of Docetaxel through Multifunctional Mechanisms in Rats. Mol. Pharm. 2016, 13, 2667–2676. [Google Scholar] [CrossRef]
- Bahri-Najafi, R.; Mostafavi, S.A.; Tavakoli, N.; Taymouri, S.; Shahraki, M.-M. Preparation and in vitro-in vivo evaluation of acyclovir floating tablets. Res. Pharm. Sci. 2017, 12, 128–136. [Google Scholar] [CrossRef]
- Jain, S.K.; Jain, R.K.; Chourasia, M.K.; Jain, A.K.; Chalasani, K.B.; Soni, V.; Jain, A. Design and development of multivesicular liposomal depot delivery system for controlled systemic delivery of acyclovir sodium. AAPS PharmSciTech 2005, 6, E35–E41. [Google Scholar] [CrossRef]
- Zainol, S.; Basri, H.; Basri, H.; Shamsuddin, A.F.; Abdul-Gani, S.S.; Karjiban, R.A.; Abdulmalek, E. Formulation Optimization of a Palm-Based Nanoemulsion System Containing Levodopa. Int. J. Mol. Sci. 2012, 13, 13049–13064. [Google Scholar] [CrossRef]
- Padhye, S.G.; Nagarsenker, M.S. Simvastatin Solid Lipid Nanoparticles for Oral Delivery: Formulation Development and In vivo Evaluation. Indian J. Pharm. Sci. 2013, 75, 591–598. [Google Scholar]
- Zirak, M.B.; Pezeshki, A. Effect of Surfactant Concentration on the Particle Size, Stability and Potential Zeta of Beta carotene Nano Lipid Carrier. Int. J. Curr. Microbiol. Appl. Sci. 2015, 4, 924–932. [Google Scholar]
- Asasutjarit, R.; Lorenzen, S.-I.; Sirivichayakul, S.; Ruxrungtham, K.; Ruktanonchai, U.; Ritthidej, G.C. Effect of Solid Lipid Nanoparticles Formulation Compositions on Their Size, Zeta Potential and Potential for In Vitro pHIS-HIV-Hugag Transfection. Pharm. Res. 2007, 24, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Rachmawati, H.; Rahma, A.; Al Shaal, L.; Müller, R.H.; Keck, C.M. Destabilization Mechanism of Ionic Surfactant on Curcumin Nanocrystal against Electrolytes. Sci. Pharm. 2016, 84, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Mohtar, N.; Khan, N.A.K.; Darwis, Y. Solid Lipid Nanoparticles of Atovaquone Based on 24 Full-Factorial Design. Iran. J. Pharm. Res. IJPR 2015, 14, 989–1000. [Google Scholar] [PubMed]
- Silva, A.; González-Mira, E.; García, M.L.; Egea, M.; Fonseca, J.; Silva, R.; Santos, D.; Souto, E.; Ferreira, D. Preparation, characterization and biocompatibility studies on risperidone-loaded solid lipid nanoparticles (SLN): High pressure homogenization versus ultrasound. Colloids Surf. B Biointerfaces 2011, 86, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, B.; Martins, S.; Ferreira, D.; Souto, E.B. Oral insulin delivery by means of solid lipid nanoparticles. Int. J. Nanomed. 2007, 2, 743–749. [Google Scholar]
- Hu, L.; Tang, X.; Cui, F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J. Pharm. Pharmacol. 2004, 56, 1527–1535. [Google Scholar] [CrossRef]
- Kakkar, V.; Singh, S.; Singla, D.; Kaur, I.P. Exploring solid lipid nanoparticles to enhance the oral bioavailability of curcumin. Mol. Nutr. Food Res. 2010, 55, 495–503. [Google Scholar] [CrossRef]
- Rogošić, M.; Mencer, H.; Gomzi, Z. Polydispersity index and molecular weight distributions of polymers. Eur. Polym. J. 1996, 32, 1337–1344. [Google Scholar] [CrossRef]
- Reis, S.; Neves, A.; Lúcio, M.; Martins, S.; Lima, J.L.F.C. Novel resveratrol nanodelivery systems based on lipid nanoparticles to enhance its oral bioavailability. Int. J. Nanomed. 2013, 8, 177–187. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Müller, R.H.; Shegokar, R.; Keck, C.M. 20 years of lipid nanoparticles (SLN and NLC): Present state of development and industrial applications. Curr. Drug Discov. Technol. 2011, 8, 207–227. [Google Scholar] [CrossRef] [PubMed]
- Emami, J.; Mohiti, H.; Hamishehkar, H.; Varshosaz, J. Formulation and optimization of solid lipid nanoparticle formulation for pulmonary delivery of budesonide using Taguchi and Box-Behnken design. Res. Pharm. Sci. 2015, 10, 17–33. [Google Scholar] [PubMed]
- Westesen, K.; Siekmann, B.; Koch, M.H. Investigations on the physical state of lipid nanoparticles by synchrotron radiation X-ray diffraction. Int. J. Pharm. 1993, 93, 189–199. [Google Scholar] [CrossRef]
- Vivek, K.; Reddy, H.; Murthy, R.S.R. Investigations of the effect of the lipid matrix on drug entrapment, in vitro release, and physical stability of olanzapine-loaded solid lipid nanoparticles. AAPS PharmSciTech 2007, 8, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Souto, E.; Anselmi, C.; Centini, M.; Müller, R. Preparation and characterization of n-dodecyl-ferulate-loaded solid lipid nanoparticles (SLN®). Int. J. Pharm. 2005, 295, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, E.; Souto, E.B.; Müller, R.H. Physicochemical investigations on the structure of drug-free and drug-loaded solid lipid nanoparticles (SLN) by means of DSC and 1H NMR. Die Pharm. 2005, 60, 508–513. [Google Scholar]
- Sullender, W.M.; Arvin, A.M.; Diaz, P.S.; Connor, J.D.; Straube, R.; Dankner, W.; Levin, M.J.; Weller, S.; Blum, M.R.; Chapman, S. Pharmacokinetics of acyclovir suspension in infants and children. Antimicrob. Agents Chemother. 1987, 31, 1722–1726. [Google Scholar] [CrossRef][Green Version]
- Bhosale, U.; Devi, K.; Choudhary, S. Development and in vitro-in vivo evaluation of oral drug delivery system of acyclovir loaded PLGA nanoparticles. Int. J. Drug Deliv. 2013, 5, 331–343. [Google Scholar]
- Parr, A.; Hidalgo, I.J.; Bode, C.; Brown, W.; Yazdanian, M.; Gonzalez, M.A.; Sagawa, K.; Miller, K.; Jiang, W.; Stippler, E.S. The Effect of Excipients on the Permeability of BCS Class III Compounds and Implications for Biowaivers. Pharm. Res. 2015, 33, 167–176. [Google Scholar] [CrossRef]
- Harde, H.; Das, M.; Kushwah, V. Solid lipid nanoparticles: An oral bioavailability enhancer vehicle. Expert Opin. Drug Deliv. 2011, 8, 1407–1424. [Google Scholar] [CrossRef]
- Yang, Z.-G.; Meng, H.; Zhang, X.; Li, X.-D.; Lv, W.-L.; Zhang, Q. [Effect of quercetin on the acyclovir intestinal absorption]. Beijing Da Xue Xue Bao Yi Xue Ban 2004, 36, 309–312. [Google Scholar] [PubMed]
- Palmberger, T.F.; Hombach, J.; Bernkop-Schnürch, A. Thiolated chitosan: Development and in vitro evaluation of an oral delivery system for acyclovir. Int. J. Pharm. 2008, 348, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Takano, M. Intestinal efflux transporters and drug absorption. Expert Opin. Drug Metab. Toxicol. 2008, 4, 923–939. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Tang, J.; Li, M.; Ren, J.; Zheng, N.; Wu, L. Curcumin-loaded solid lipid nanoparticles with Brij78 and TPGS improvedin vivooral bioavailability andin situintestinal absorption of curcumin. Drug Deliv. 2014, 23, 459–470. [Google Scholar] [CrossRef]
- Shono, Y.; Nishihara, H.; Matsuda, Y.; Furukawa, S.; Okada, N.; Fujita, T.; Yamamoto, A. Modulation of Intestinal P-Glycoprotein Function by Cremophor EL and Other Surfactants by an In Vitro Diffusion Chamber Method Using the Isolated Rat Intestinal Membranes. J. Pharm. Sci. 2004, 93, 877–885. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Yao, M.; Morrison, R.A.; Chong, S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch. Pharmacal Res. 2003, 26, 768–772. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Coded Levels | ||||
|---|---|---|---|---|---|
| Axial(−α) | Low | Centre | High | Axial(+α) | |
| Biogapress Vegetal 297 ATO (mg) | 237.87 | 300.00 | 450.00 | 600.00 | 662.13 |
| Tween 80 (w/w) | 0.17 | 1.00 | 3.00 | 5.00 | 5.83 |
| Size, R1 | Equation: 118.82 + 33.92A − 32.74B − 1.82A2 + 20.85B2 + 5.13AB R2 value: 0.9992 p-value: <0.0001 |
| Zeta Potential, R2 | Equation: −26.78 − 0.93A + 0.96B+0.084A2 − 0.87B2 − 0.58AB R2 value: 0.9492 p-value: 0.0002 |
| Polydispersity Index, R3 | Equation: 0.22 − 0.01A − 0.004B + 0.02A2 + 0.025B2 + 0.01AB R2 value: 0.9460 p-value: 0.0003 |
| Variables | Size | Zeta Potential | PdI | ||||
|---|---|---|---|---|---|---|---|
| F Value | p-Value | F Value | p-Value | F Value | p-Value | ||
| Main Effects | A | 575.69 | <0.0001 | 26.16 | 0.0002 | 17.57 | 0.0041 |
| B | 2681.96 | <0.0001 | 42.55 | 0.0003 | 1.51 | 0.2592 | |
| Quadratic Effects | A2 | 14.45 | 0.0126 | 0.30 | 0.5990 | 43.01 | 0.0003 |
| B2 | 1892.21 | <0.0001 | 32.44 | 0.0007 | 66.87 | <0.0001 | |
| Interaction Effect | AB | 65.72 | 0.0005 | 8.22 | 0.0241 | 6.03 | 0.0437 |
| Responses | Predicted | Observed |
|---|---|---|
| Particle Size (nm) | 130.00 | 122.72 ± 2.15 |
| Polydispersity Index | 0.22 | 0.23 ± 0.01 |
| Zeta Potential (mV) | −27.09 | −24.37 ± 1.07 |
| Parameters | Acyclovir Suspension | Acyclovir-Loaded SLNs |
|---|---|---|
| Cmax (ng/mL) | 303.50 ± 26.70 | 607.00 ± 71.64 * |
| Tmax (h) | 1.00 ± 0.00 | 1.25 ± 0.25 |
| AUC0-24 (h·ng·mL−1) | 1243.75 ± 125.90 | 4899.50 ± 321.30 * |
| AUC0–∞(h·ng·mL−1) | 1341.67 ± 133.40 | 5683.43 ± 368.70 * |
| Ke (h−1) | 0.37 ± 0.05 | 0.14 ± 0.02 |
| T1/2 (h) | 2.06 ± 0.29 | 5.26 ± 0.55 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, H.; Bello, R.O.; Adam, S.K.; Alias, E.; Meor Mohd Affandi, M.M.R.; Shamsuddin, A.F.; Basir, R. Acyclovir-Loaded Solid Lipid Nanoparticles: Optimization, Characterization and Evaluation of Its Pharmacokinetic Profile. Nanomaterials 2020, 10, 1785. https://doi.org/10.3390/nano10091785
Hassan H, Bello RO, Adam SK, Alias E, Meor Mohd Affandi MMR, Shamsuddin AF, Basir R. Acyclovir-Loaded Solid Lipid Nanoparticles: Optimization, Characterization and Evaluation of Its Pharmacokinetic Profile. Nanomaterials. 2020; 10(9):1785. https://doi.org/10.3390/nano10091785
Chicago/Turabian StyleHassan, Haniza, Ramatu Omenesa Bello, Siti Khadijah Adam, Ekram Alias, Meor Mohd Redzuan Meor Mohd Affandi, Ahmad Fuad Shamsuddin, and Rusliza Basir. 2020. "Acyclovir-Loaded Solid Lipid Nanoparticles: Optimization, Characterization and Evaluation of Its Pharmacokinetic Profile" Nanomaterials 10, no. 9: 1785. https://doi.org/10.3390/nano10091785
APA StyleHassan, H., Bello, R. O., Adam, S. K., Alias, E., Meor Mohd Affandi, M. M. R., Shamsuddin, A. F., & Basir, R. (2020). Acyclovir-Loaded Solid Lipid Nanoparticles: Optimization, Characterization and Evaluation of Its Pharmacokinetic Profile. Nanomaterials, 10(9), 1785. https://doi.org/10.3390/nano10091785