Biocompatibility of Biomaterials for Nanoencapsulation: Current Approaches

 ,
,  , , ,
, , ,
Abstract
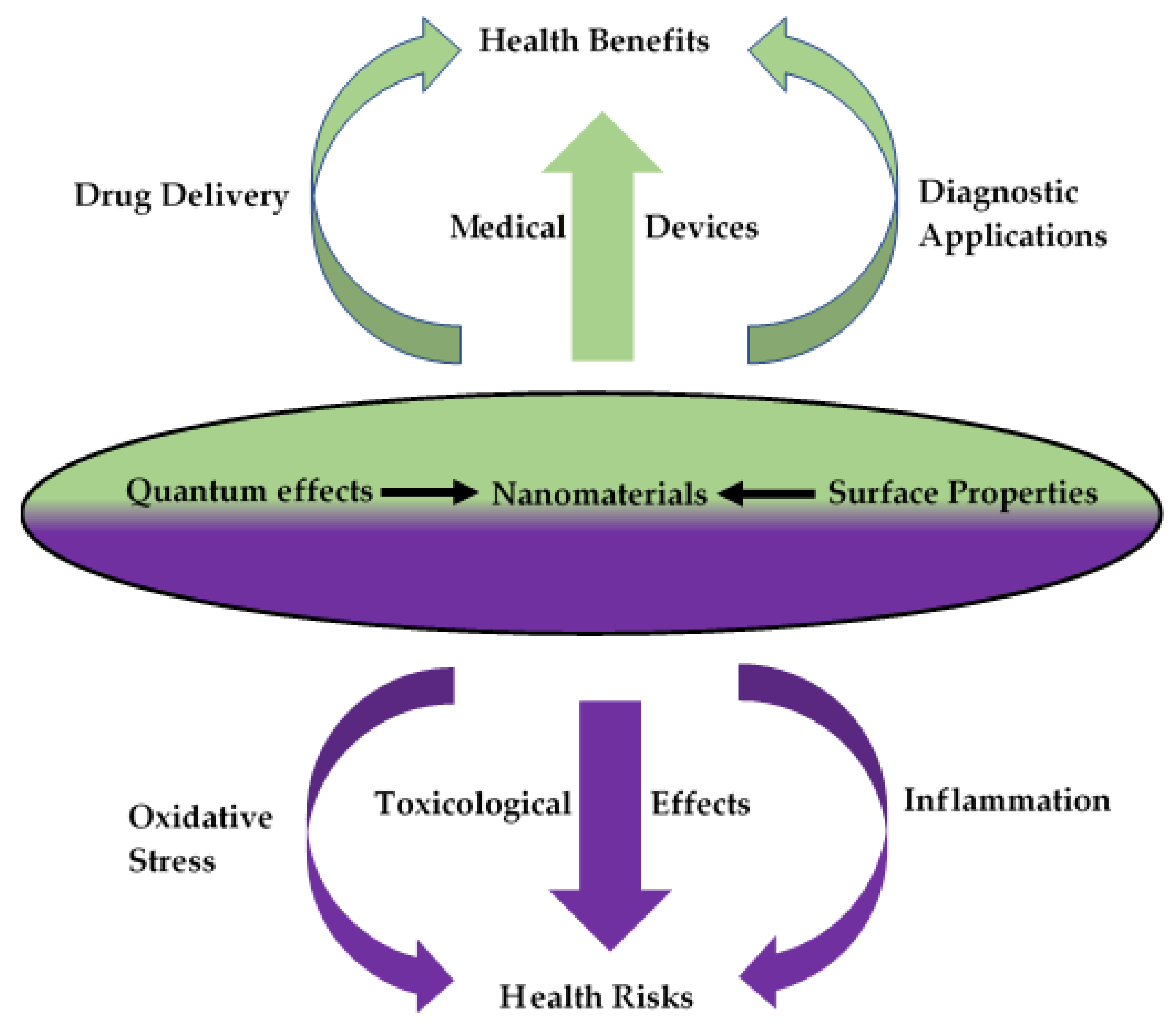
1. Introduction
2. Nanomaterial–Host Interactions
2.1. In Vitro Approaches
2.1.1. Two-Dimensional (2D) Monolayer Cell Culture
Cellular Uptake
Intracellular Trafficking
Bioactivity
2.1.2. Three-Dimensional (3D) Cell Culture
2.2. Ex Vivo Systems and Evaluation
2.3. In Vivo Systems and Evaluation
3. Biocompatibility and Toxicology Testing in Nanotechnology
3.1. Immunotoxicity
3.2. Genotoxicity and Carcinogenicity
3.3. Cytotoxicity
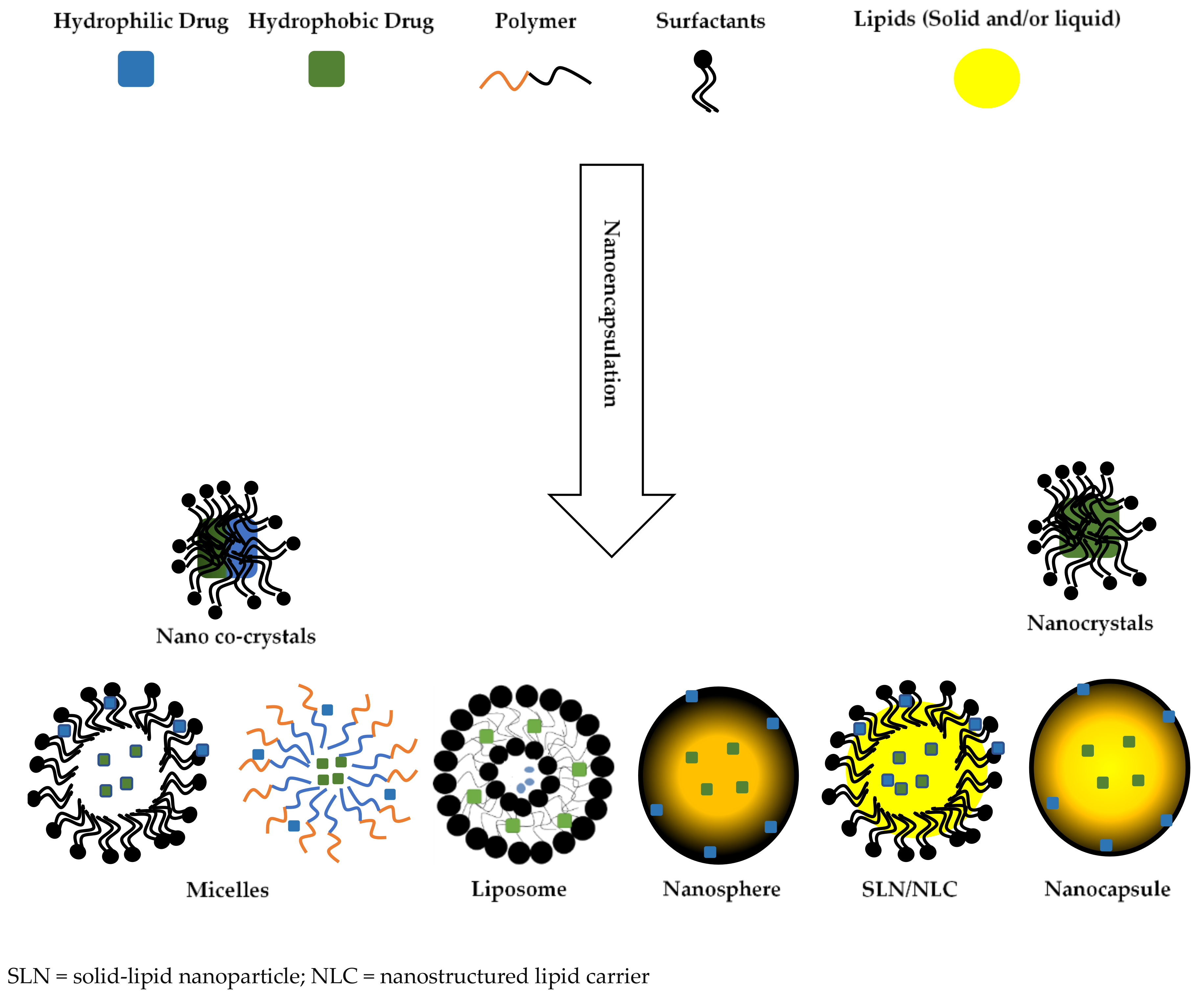
4. Biocompatibility of Biomaterials Used for Nanoencapsulation
4.1. Lipids
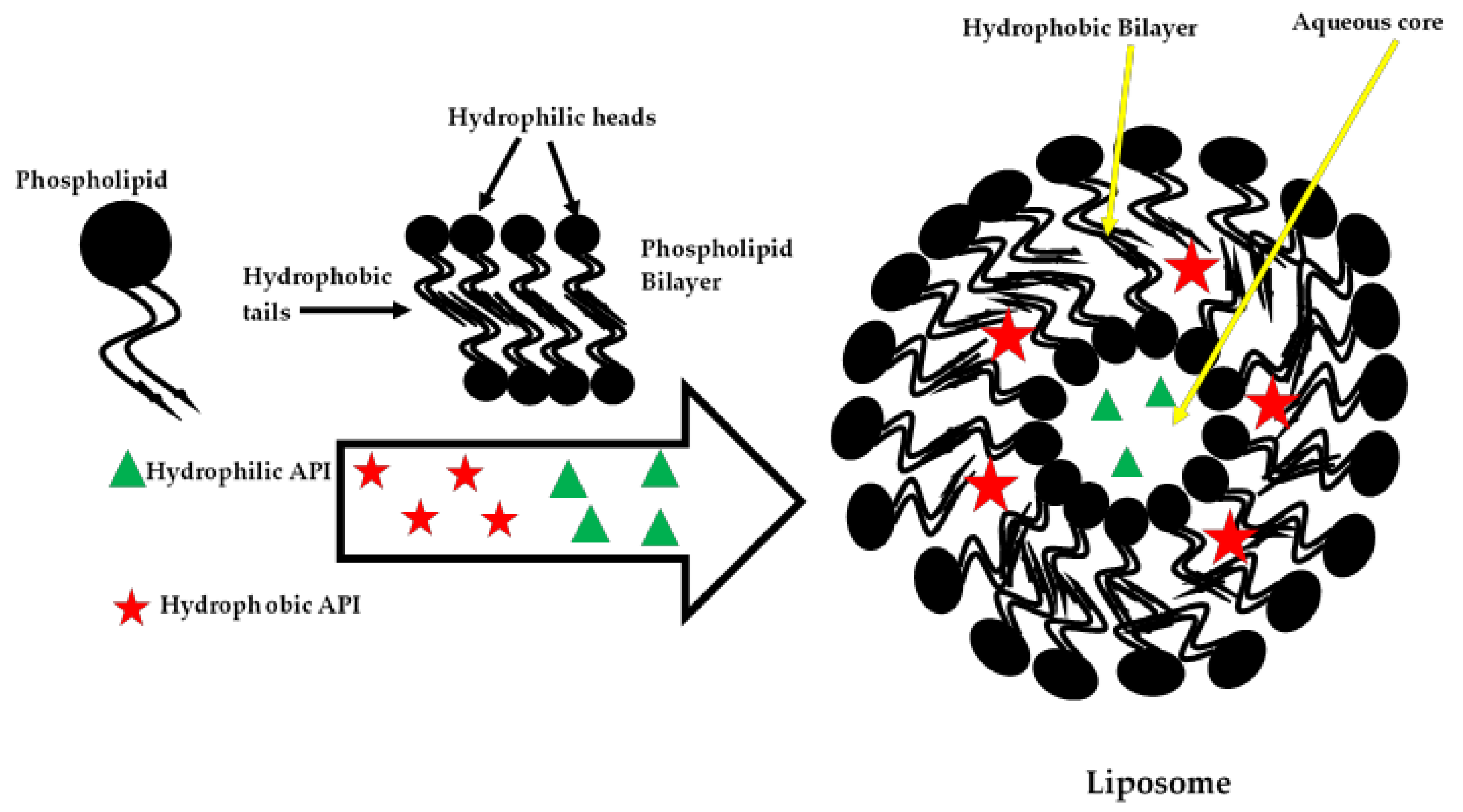
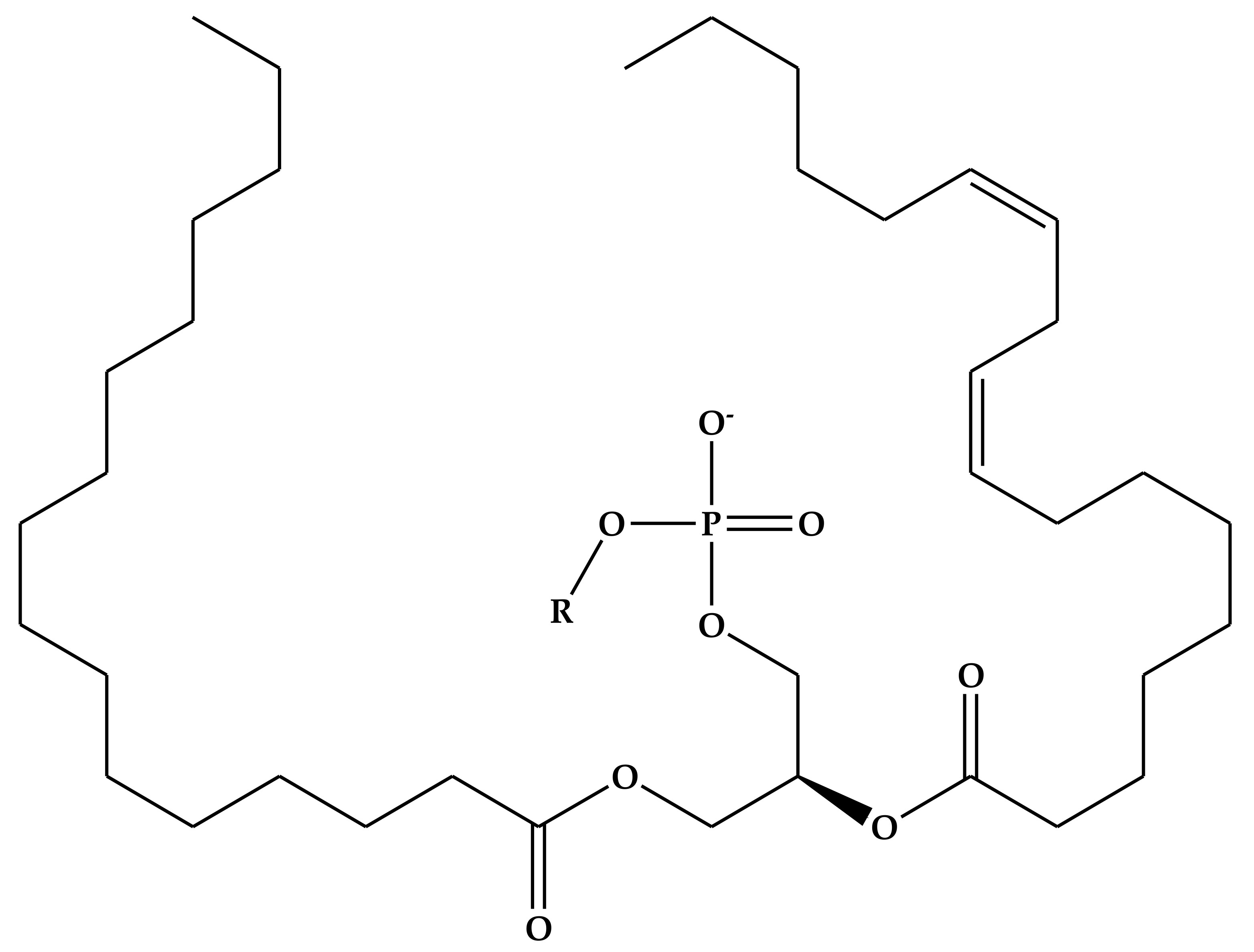
4.2. Phospholipids
4.3. Polymeric Materials
4.4. Non-Ionic Surfactants
4.5. Ionic Surfactants
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The smart drug delivery system and its clinical potential. Theranostics 2016, 6, 1306–1323. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of Nanotechnology on Drug Delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Muller, R.H.; Keck, C.M. Challenges and solutions for the delivery of biotech drugs - A review of drug nanocrystal technology and lipid nanoparticles. J. Biotechnol. 2004, 113, 151–170. [Google Scholar] [CrossRef]
- Kreuter, J. Drug targeting with nanoparticles. Eur. J. Drug Metab. Pharmacokinet. 1994, 19, 253–256. [Google Scholar] [CrossRef]
- Chen, M.L.; John, M.; Lee, S.L.; Tyner, K.M. Development Considerations for Nanocrystal Drug Products. AAPS J. 2017, 19, 642–651. [Google Scholar] [CrossRef]
- Kim, S.; Shi, Y.; Kim, J.Y.; Park, K.; Cheng, J.-X. Overcoming the barriers in micellar drug delivery: Loading efficiency, in vivo stability, and micelle–cell interaction. Expert Opin. Drug Deliv. 2010, 7, 49–62. [Google Scholar] [CrossRef]
- Geeta Vikram Yadav and Sushma, R. Singh Nanosuspension: A Promising Drug Delivery System. Pharmacophore 2012, 3, 217–243. [Google Scholar]
- De Smet, L.; Saerens, L.; De Beer, T.; Carleer, R.; Adriaensens, P.; Van Bocxlaer, J.; Vervaet, C.; Remon, J.P. Formulation of itraconazole nanococrystals and evaluation of their bioavailability in dogs. Eur. J. Pharm. Biopharm. 2014, 87, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sarnes, A.; Kovalainen, M.; Häkkinen, M.R.; Laaksonen, T.; Laru, J.; Kiesvaara, J.; Ilkka, J.; Oksala, O.; Rönkkö, S.; Järvinen, K.; et al. Nanocrystal-based per-oral itraconazole delivery: Superior in vitro dissolution enhancement versus Sporanox® is not realized in in vivo drug absorption. J. Control. Release 2014, 180, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Esfandi, E.; Ramezani, V.; Vatanara, A.; Najafabadi, A.R.; Pouya, S.; Moghaddam, H. Clarithromycin Dissolution Enhancement by Preparation of Aqueous Nanosuspensions Using Sonoprecipitation Technique. Iran. J. Pharm. Res. 2014, 13, 809. [Google Scholar] [PubMed]
- Soisuwan, S.; Teeranachaideekul, V.; Wongrakpanich, A.; Langguth, P.; Junyaprasert, V.B. In vitro performances and cellular uptake of clarithromycin nanocrystals produced by media milling technique. Powder Technol. 2018, 338. [Google Scholar] [CrossRef]
- Soisuwan, S.; Teeranachaideekul, V.; Wongrakpanich, A.; Langguth, P.; Junyaprasert, V.B. Impact of uncharged and charged stabilizers on in vitro drug performances of clarithromycin nanocrystals. Eur. J. Pharm. Biopharm. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Shanthi, N.; Mahato, A.K.; Soni, S.; Rajnikanth, P.S. Preparation of luliconazole nanocrystals loaded hydrogel for improvement of dissolution and antifungal activity. Heliyon 2019, 5, e01688. [Google Scholar] [CrossRef]
- Esmaili, M.; Ghaffari, S.M.; Moosavi-Movahedi, Z.; Atri, M.S.; Sharifizadeh, A.; Farhadi, M.; Yousefi, R.; Chobert, J.M.; Haertlé, T.; Moosavi-Movahedi, A.A. Beta casein-micelle as a nano vehicle for solubility enhancement of curcumin; food industry application. LWT - Food Sci. Technol. 2011, 44, 2166–2172. [Google Scholar] [CrossRef]
- Alkhamis, K.A.; Allaboun, H.; Al-Momani, W.Y. Study of the solubilization of gliclazide by aqueous micellar solutions. J. Pharm. Sci. 2003, 92, 839–846. [Google Scholar] [CrossRef]
- El-Massik, M.A.; Darwish, I.A.; Hassan, E.E.; El-Khordagui, L.K. Development of a dissolution medium for glibenclamide. Int. J. Pharm. 1996, 140, 69–76. [Google Scholar] [CrossRef]
- Talegaonkar, S.; Mishra, P.; Khar, R.; Biju, S. Vesicular systems: An overview. Indian J. Pharm. Sci. 2006, 68, 141. [Google Scholar] [CrossRef]
- Priyanka, K.; Singh, S. A review on skin targeted delivery of bioactives as ultradeformable vesicles: Overcoming the penetration problem. Curr. Drug Targets 2014, 15, 184–198. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, X.; Zhang, T.; Wang, H.; Wu, B. Design and evaluation of injectable niclosamide nanocrystals prepared by wet media milling technique. Drug Dev. Ind. Pharm. 2015, 41, 1416–1424. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Ma, J.; Gao, L.; Wang, X.; Liu, G. Drug nanocrystals: In vivo performances. J. Control. Release 2012, 160, 418–430. [Google Scholar] [CrossRef]
- Tuomela, A.; Hirvonen, J.; Peltonen, L. Stabilizing agents for drug nanocrystals: Effect on bioavailability. Pharmaceutics 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, S.; Feng, S.S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 2012, 33, 4889–4906. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Targeted polymeric micelles for delivery of poorly soluble drugs. Cell. Mol. Life Sci. 2004, 61, 2549–2559. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Leathwood, P.D.; Ryman, B.E. Enzyme entrapment in liposomes. FEBS Lett. 1971, 14, 95–99. [Google Scholar] [CrossRef]
- Cao, N.; Feng, S.S. Doxorubicin conjugated to d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS): Conjugation chemistry, characterization, in vitro and in vivo evaluation. Biomaterials 2008, 29, 3856–3865. [Google Scholar] [CrossRef]
- Zhang, Z.; Feng, S.S. The drug encapsulation efficiency, in vitro drug release, cellular uptake and cytotoxicity of paclitaxel-loaded poly(lactide)-tocopheryl polyethylene glycol succinate nanoparticles. Biomaterials 2006, 27, 4025–4033. [Google Scholar] [CrossRef]
- Tosi, G.; Costantino, L.; Rivasi, F.; Ruozi, B.; Leo, E.; Vergoni, A.V.; Tacchi, R.; Bertolini, A.; Vandelli, M.A.; Forni, F. Targeting the central nervous system: In vivo experiments with peptide-derivatized nanoparticles loaded with Loperamide and Rhodamine-123. J. Control. Release 2007, 122, 1–9. [Google Scholar] [CrossRef]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 454–477. [Google Scholar] [CrossRef]
- Kreuter, J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv. Rev. 2001, 47, 65–81. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, L.L.; Du, Y.Z.; You, J.; Hu, F.Q.; Zeng, S. Preparation and characteristics of nanostructured lipid carriers for control-releasing progesterone by melt-emulsification. Colloids Surf. B Biointerfaces 2007, 60, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, O.; Erdogan, I.; Köse, M.D.; Kalmaz, G. Nanocarriers for Plant-Derived Natural Compounds. Nanostructures Antimicrob. Ther. Nanostructures Ther. Med. Ser. 2017, 395–412. [Google Scholar] [CrossRef]
- Witika, B.A.; Walker, R.B. Development, manufacture and characterization of niosomes for the delivery for nevirapine. Pharmazie 2019, 74, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomedicine 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Jacobs, C. Buparvaquone mucoadhesive nanosuspension: Preparation, optimisation and long-term stability. Int. J. Pharm. 2002, 237, 151–161. [Google Scholar] [CrossRef]
- Tathe, A.; Ghodke, M.; Nikalje, A.P. A brief review: Biomaterials and their apllication. Int. J. Pharm. Pharm. Sci. 2010, 2, 19–23. [Google Scholar]
- Williams, D. Revisiting the definition of biocompatibility. Med. Device Technol. 2003, 14, 10–13. [Google Scholar]
- Williams, D.F. On the mechanisms of biocompatibility. Biomaterials 2008, 29, 2941–2953. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer - Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Adabi, M.; Naghibzadeh, M.; Adabi, M.; Zarrinfard, M.A.; Esnaashari, S.S.; Seifalian, A.M.; Faridi-Majidi, R.; Tanimowo Aiyelabegan, H.; Ghanbari, H. Biocompatibility and nanostructured materials: Applications in nanomedicine. Artif. Cells Nanomed. Biotechnol. 2017, 45, 833–842. [Google Scholar] [CrossRef]
- Ekkapongpisit, M.; Giovia, A.; Follo, C.; Caputo, G.; Isidoro, C. Biocompatibility, endocytosis, and intracellular trafficking of mesoporous silica and polystyrene nanoparticles in ovarian cancer cells: Effects of size and surface charge groups. Int. J. Nanomed. 2012, 7, 4147–4158. [Google Scholar] [CrossRef]
- Chen, L.; McCrate, J.M.; Lee, J.C.; Li, H. The role of surface charge on the uptake and biocompatibility of hydroxyapatite nanoparticles with osteoblast cells. Nanotechnologyhnology 2011, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Foldvari, M.; Bagonluri, M. Carbon nanotubes as functional excipients for nanomedicines: II. Drug delivery and biocompatibility issues. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 183–200. [Google Scholar] [CrossRef]
- Lewinski, N.; Colvin, V.; Drezek, R. Cytotoxicity of nanopartides. Small 2008, 4, 26–49. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D. Nanomadicine in Cancer Treatment: Drug Targeting and the Safety of the Used Materials for Drug Nanoencapsulation. Biochem. Pharmacol. Open Access 2012, 01. [Google Scholar] [CrossRef]
- Sharma, V.; Shukla, R.K.; Saxena, N.; Parmar, D.; Das, M.; Dhawan, A. DNA damaging potential of zinc oxide nanoparticles in human epidermal cells. Toxicol. Lett. 2009, 185, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Xu, P.; Gullotti, E.; Tong, L.; Highley, C.B.; Errabelli, D.R.; Hasan, T.; Cheng, J.-X.; Kohane, D.S.; Yeo, Y. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Mol. Pharm. 2009, 6, 190–201. [Google Scholar] [CrossRef]
- Kohli, A.G.; Kieler-Ferguson, H.M.; Chan, D.; Szoka, F.C. A robust and quantitative method for tracking liposome contents after intravenous administration. J. Control. Release 2014, 176, 86–93. [Google Scholar] [CrossRef][Green Version]
- Brown, M.; Wittwer, C. Flow Cytometry: Principles and Clinical Applications in Hematology. Clin. Chem. 2000, 46, 1221–1229. [Google Scholar] [CrossRef]
- Harush-Frenkel, O.; Altschuler, Y.; Benita, S. Nanoparticle-cell interactions: Drug delivery implications. Crit. Rev. Ther. Drug Carrier Syst. 2008, 25, 485–544. [Google Scholar] [CrossRef] [PubMed]
- Soldati, T.; Schliwa, M. Powering membrane traffic in endocytosis and recycling. Nat. Rev. Mol. Cell Biol. 2006, 7, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.B.; Mocherla, S.; Heslinga, M.J.; Charoenphol, P.; Eniola-Adefeso, O. Dynamic and cellular interactions of nanoparticles in vascular-targeted drug delivery (review). Mol. Membr. Biol. 2010, 27, 190–205. [Google Scholar] [CrossRef]
- Van Deurs, B.; Petersen, O.W.; Olsnes, S.; Sandvig, K. The ways of endocytosis. Int. Rev. Cytol. 1989, 117, 131–177. [Google Scholar] [PubMed]
- Garnett, M.C.; Kallinteri, P. Nanomedicines and nanotoxicology: Some physiological principles. Occup. Med. 2006, 56, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.; Di Fiore, P.P. Endocytosis conducts the cell signaling orchestra. Cell 2006, 124, 897–900. [Google Scholar] [CrossRef]
- Roth, M.G. Clathrin-mediated endocytosis before fluorescent proteins. Nat. Rev. Mol. Cell Biol. 2006, 7, 63–68. [Google Scholar] [CrossRef]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Mahl, D.; Diendorf, J.; Meyer-Zaika, W.; Epple, M. Possibilities and limitations of different analytical methods for the size determination of a bimodal dispersion of metallic nanoparticles. Colloids Surfaces A Physicochem. Eng. Asp. 2011, 377, 386–392. [Google Scholar] [CrossRef]
- Sabharanjak, S.; Mayor, S. Folate receptor endocytosis and trafficking. Adv. Drug Deliv. Rev. 2004, 56, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Mol. Cell 2009, 339, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Carver, L.A.; Schnitzer, J.E. Caveolae: Mining little caves for new cancer targets. Nat. Rev. Cancer 2003, 3, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N.G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Melan, M.A.; Sluder, G. Redistribution and differential extraction of soluble proteins in permeabilized cultured cells. Implications for immunofluorescence microscopy. J. Cell Sci. 1992, 101, 731–743. [Google Scholar] [PubMed]
- Slater, K. Cytotoxicity tests for high-throughput drug discovery. Curr. Opin. Biotechnol. 2001, 12, 70–74. [Google Scholar] [CrossRef]
- Decker, T.; Lohmann-Matthes, M.-L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [Google Scholar] [CrossRef]
- Holback, H.; Yeo, Y. Intratumoral Drug Delivery with Nanoparticulate Carriers. Pharm. Res. 2011, 28, 1819–1830. [Google Scholar] [CrossRef]
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to drug delivery in solid tumors. Tissue Barriers 2014, 2. [Google Scholar] [CrossRef]
- Sun, T.; Jackson, S.; Haycock, J.W.; MacNeil, S. Culture of skin cells in 3D rather than 2D improves their ability to survive exposure to cytotoxic agents. J. Biotechnol. 2006, 122, 372–381. [Google Scholar] [CrossRef]
- Mitra, M.; Mohanty, C.; Harilal, A.; Maheswari, U.K.; Sahoo, S.K.; Krishnakumar, S. A novel in vitro three-dimensional retinoblastoma model for evaluating chemotherapeutic drugs. Mol. Vis. 2012, 18, 1361–1378. [Google Scholar] [PubMed]
- Lee, G.Y.; Kenny, P.A.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Brigger, I.; Dubernet, C.; Couvreur, P. Nanoparticles in cancer therapy and diagnosis. Adv. Drug Deliv. Rev. 2012, 64, 24–36. [Google Scholar] [CrossRef]
- Hosoya, H.; Kadowaki, K.; Matsusaki, M.; Cabral, H.; Nishihara, H.; Ijichi, H.; Koike, K.; Kataoka, K.; Miyazono, K.; Akashi, M.; et al. Engineering fibrotic tissue in pancreatic cancer: A novel three-dimensional model to investigate nanoparticle delivery. Biochem. Biophys. Res. Commun. 2012, 419, 32–37. [Google Scholar] [CrossRef]
- Khademhosseini, A.; Eng, G.; Yeh, J.; Fukuda, J.; Blumling, J.; Langer, R.; Burdick, J.A. Micromolding of photocrosslinkable hyaluronic acid for cell encapsulation and entrapment. J. Biomed. Mater. Res. Part A 2006, 79, 522–532. [Google Scholar] [CrossRef]
- Ho, W.J.; Pham, E.A.; Kim, J.W.; Ng, C.W.; Kim, J.H.; Kamei, D.T.; Wu, B.M. Incorporation of multicellular spheroids into 3-D polymeric scaffolds provides an improved tumor model for screening anticancer drugs. Cancer Sci. 2010, 101, 2637–2643. [Google Scholar] [CrossRef]
- Astashkina, A.I.; Mann, B.K.; Prestwich, G.D.; Grainger, D.W. A 3-D organoid kidney culture model engineered for high-throughput nephrotoxicity assays. Biomaterials 2012, 33, 4700–4711. [Google Scholar] [CrossRef]
- Blake, A.; Pearce, T.; Rao, N.; Johnson, S.; Williams, L. Multilayer PDMS microfluidic chamber for controlling brain slice microenvironment. Lab Chip 2007, 7, 842–849. [Google Scholar] [CrossRef]
- Ng, C.P.; Pun, S.H. A perfusable 3D cell-matrix tissue culture chamber for in situ evaluation of nanoparticle vehicle penetration and transport. Biotechnol. Bioeng. 2008, 99, 1490–1501. [Google Scholar] [CrossRef]
- Lazzari, G.; Couvreur, P.; Mura, S. Multicellular tumor spheroids: A relevant 3D model for the: In vitro preclinical investigation of polymer nanomedicines. Polym. Chem. 2017, 8, 4947–4969. [Google Scholar] [CrossRef]
- Mattek Life Sciences EpiDerm in vitro 3D Tissue | MatTek Life Sciences. Available online: https://www.mattek.com/products/epiderm/#applications (accessed on 9 June 2020).
- Mattek Life Sciences EpiSkin Epidermal Model. Available online: http://www.episkin.com/Episkin (accessed on 9 June 2020).
- Kostadinova, R.; Boess, F.; Applegate, D.; Suter, L.; Weiser, T.; Singer, T.; Naughton, B.; Roth, A. A long-term three dimensional liver co-culture system for improved prediction of clinically relevant drug-induced hepatotoxicity. Toxicol. Appl. Pharmacol. 2013, 268, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Alfaro-Moreno, E.; Nawrot, T.S.; Vanaudenaerde, B.M.; Hoylaerts, M.F.; Vanoirbeek, J.A.; Nemery, B.; Hoet, P.H.M. Co-cultures of multiple cell types mimic pulmonary cell communication in response to urban PM10. Eur. Respir. J. 2008, 32, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Mattek Life Sciences Tissue Models | MatTek Life Sciences. Available online: https://www.mattek.com/products/ (accessed on 9 June 2020).
- Clift, M.J.D.; Gehr, P.; Rothen-Rutishauser, B. Nanotoxicology: A perspective and discussion of whether or not in vitro testing is a valid alternative. Arch. Toxicol. 2011, 85, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Rundén, E.; Seglen, P.O.; Haug, F.M.; Ottersen, O.P.; Wieloch, T.; Shamloo, M.; Laake, J.H. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: Evidence for a MAP kinase-dependent mechanism. J. Neurosci. 1998, 18, 7296–7305. [Google Scholar] [CrossRef]
- OECD. Bovine Corneal Opacity and Permeability Test Method for Identifying i) Chemicals Inducing Serious Eye Damage and ii) Chemicals Not Requiring Classification for Eye Irritation or Serious Eye Damage TG 437; OECD: Paris, France, 2015. [Google Scholar]
- Liberati, T.A.; Randle, M.R.; Toth, L.A. In vitro lung slices: A powerful approach for assessment of lung pathophysiology. Expert Rev. Mol. Diagn. 2010, 10, 501–508. [Google Scholar] [CrossRef]
- Morin, J.-P.; Baste, J.-M.; Gay, A.; Crochemore, C.; Corbière, C.; Monteil, C. Precision cut lung slices as an efficient tool for in vitro lung physio-pharmacotoxicology studies. Xenobiotica 2013, 43, 63–72. [Google Scholar] [CrossRef]
- Schlepütz, M.; Rieg, A.D.; Seehase, S.; Spillner, J.; Perez-Bouza, A.; Braunschweig, T.; Schroeder, T.; Bernau, M.; Lambermont, V.; Schlumbohm, C.; et al. Neurally mediated airway constriction in human and other species: A comparative study using precision-cut lung slices (PCLS). PLoS ONE 2012, 7, e47344. [Google Scholar] [CrossRef]
- Sauer, U.G.; Vogel, S.; Aumann, A.; Hess, A.; Kolle, S.N.; Ma-Hock, L.; Wohlleben, W.; Dammann, M.; Strauss, V.; Treumann, S.; et al. Applicability of rat precision-cut lung slices in evaluating nanomaterial cytotoxicity, apoptosis, oxidative stress, and inflammation. Toxicol. Appl. Pharmacol. 2014, 276, 1–20. [Google Scholar] [CrossRef]
- Lauenstein, L.; Switalla, S.; Prenzler, F.; Seehase, S.; Pfennig, O.; Förster, C.; Fieguth, H.; Braun, A.; Sewald, K. Assessment of immunotoxicity induced by chemicals in human precision-cut lung slices (PCLS). Toxicol. Vitr. 2014, 28, 588–599. [Google Scholar] [CrossRef]
- Kim, Y.H.; Tong, H.; Daniels, M.; Boykin, E.; Krantz, Q.T.; McGee, J.; Hays, M.; Kovalcik, K.; Dye, J.A.; Gilmour, M.I. Cardiopulmonary toxicity of peat wildfire particulate matter and the predictive utility of precision cut lung slices. Part. Fibre Toxicol. 2014, 11, 29. [Google Scholar] [CrossRef]
- Cho, E.J.; Holback, H.; Liu, K.C.; Abouelmagd, S.A.; Park, J.; Yeo, Y. Nanoparticle characterization: State of the art, challenges, and emerging technologies. Mol. Pharm. 2013, 10, 2093–2110. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.P.M.; Chen, A.L.; Foster, A.; Drezek, R. In vivo biodistribution of nanoparticles. Nanomedicine 2011, 6, 815–835. [Google Scholar] [CrossRef] [PubMed]
- Simona, Clichici; Filip, A. In vivo Assessment of Nanomaterials Toxicity. In Nanomaterials - Toxicity and Risk Assessment; IntechOpen: London, UK, 2015; pp. 93–122. [Google Scholar]
- Organization for Economic Cooperation and Development. Test guideline 405. Acute Eye Irritation and Corrosion; OECD Guidelines for the Testing of Chemicals: Paris, France, 2002. [Google Scholar]
- Organization for Economic Cooperation and Development. Test Guideline 434. Acute Dermal Toxicity-Fixed Dose Procedure; OECD Guidelines for the Testing of Chemicals: Paris, France, 2004. [Google Scholar]
- Maneewattanapinyo, P.; Banlunara, W.; Thammacharoen, C.; Ekgasit, S.; Kaewamatawong, T. An evaluation of acute toxicity of colloidal silver nanoparticles. J. Vet. Med. Sci. 2011, 73, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; David, L.; Moldovan, B.; Vulcu, A.; Dreve, S.; Perde-Schrepler, M.; Tatomir, C.; Filip, A.G.; Bolfa, P.; Achim, M.; et al. New nanomaterials for the improvement of psoriatic lesions. J. Mater. Chem. B 2013, 1, 3152–3158. [Google Scholar] [CrossRef]
- Dickinson, A.M.; Godden, J.M.; Lanovyk, K.; Ahmed, S.S. Assessing the safety of nanomedicines: A mini review. Appl. Vitr. Toxicol. 2019, 5, 114–122. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Understanding the correlation between in vitro and in vivo immunotoxicity tests for nanomedicines. J. Control. Release 2013, 172, 456–466. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Whritenour, J.; Ahmed, M.M.; Bibby, L.; Darby, L.; Wang, X.N.; Watson, J.; Dickinson, A.M. Evaluation of a human in vitro skin test for predicting drug hypersensitivity reactions. Toxicol. Appl. Pharmacol. 2019, 369, 39–48. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Wang, X.N.; Fielding, M.; Kerry, A.; Dickinson, I.; Munuswamy, R.; Kimber, I.; Dickinson, A.M. An in vitro human skin test for assessing sensitization potential. J. Appl. Toxicol. 2016, 36, 669–684. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef]
- Di Gioacchino, M.; Petrarca, C.; Lazzarin, F.; Di Giampaolo, L.; Sabbioni, E.; Boscolo, P.; Mariani-Costantini, R.; Bernardini, G. Immunotoxicity of nanoparticles. Int. J. Immunopathol. Pharmacol. 2011, 24, 65S–71S. [Google Scholar]
- Boraschi, D.; Costantino, L.; Italiani, P. Interaction of nanoparticles with immunocompetent cells: Nanosafety considerations. Nanomedicine 2012, 7, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Vanoirbeek, J.A.J.; Hoet, P.H.M. Interactions of nanomaterials with the immune system. Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2012, 4, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Izhaky, D.; Pecht, I. What else can the immune system recognize? Proc. Natl. Acad. Sci. USA 1998, 95, 11509–11510. [Google Scholar] [CrossRef] [PubMed]
- Treuel, L.; Ulrich Nienhaus, G. Nanoparticle interaction with plasma proteins as it relates to biodistribution. In Handbook of Immunological Properties of Engineered Nanomaterials; World Scientific: Singapore, 2016; Volume 2, pp. 1–22. ISBN 9789814699174. [Google Scholar]
- Fadeel, B. Clear and present danger? Engineered nanoparticles and the immune system. Swiss Med. Wkly. 2012, 142, w13609. [Google Scholar] [CrossRef]
- Nystrom, A.M.; Fadeel, B. Safety assessment of nanomaterials: Implications for nanomedicine. J. Control. Release 2012, 161, 403–408. [Google Scholar] [CrossRef]
- Caron, W.P.; Lay, J.C.; Fong, A.M.; La-Beck, N.M.; Kumar, P.; Newman, S.E.; Zhou, H.; Monaco, J.H.; Clarke-Pearson, D.L.; Brewster, W.R.; et al. Translational studies of phenotypic probes for the mononuclear phagocyte system and liposomal pharmacologys. J. Pharmacol. Exp. Ther. 2013, 347, 599–606. [Google Scholar] [CrossRef]
- Drasler, B.; Sayre, P.; Steinhäuser, K.G.; Petri-Fink, A.; Rothen-Rutishauser, B. In vitro approaches to assess the hazard of nanomaterials. NanoImpact 2017, 8, 99–116. [Google Scholar] [CrossRef]
- Dinarello, C.A. Proinflammatory cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef]
- Favre, N.; Bordmann, G.; Rudin, W. Comparison of cytokine measurements using ELISA, ELISPOT and semi-quantitative RT-PCR. J. Immunol. Methods 1997, 204, 57–66. [Google Scholar] [CrossRef]
- Szebeni, J.; Alving, C.R.; Rosivall, L.; Bünger, R.; Baranyi, L.; Bedöcs, P.; Tóth, M.; Barenholz, Y. Animal models of complement-mediated hypersensitivity reactions to liposomes and other lipid-based nanoparticles. J. Liposome Res. 2007, 17, 107–117. [Google Scholar] [CrossRef]
- Merkel, O.M.; Urbanics, R.; Bedocs, P.; Rozsnyay, Z.; Rosivall, L.; Toth, M.; Kissel, T.; Szebeni, J. In vitro and in vivo complement activation and related anaphylactic effects associated with polyethylenimine and polyethylenimine-graft-poly(ethylene glycol) block copolymers. Biomaterials 2011, 32, 4936–4942. [Google Scholar] [CrossRef]
- Neun, B.W.; Dobrovolskaia, M.A. Qualitative Analysis of Total Complement Activation by Nanoparticles. In Characterization of Nanoparticles Intended for Drug Delivery, Methods in Molecular Biology; McNeil, S.E., Ed.; Springer Science and Business Media LLC: Berlin, Germany, 2011; Volume 697, pp. 237–245. ISBN 9781603271981. [Google Scholar]
- Jones, C.F. Frontiers in Nanobiomedical Research: Handbook of Immunological Properties of Engineered Nanomaterials; Yarmush, M., Shi, D., Eds.; World Scientific Publishing Co.Plc. Ltd.: Singapore, 2012; ISBN 9814390259. [Google Scholar]
- Batista-Duharte, A.; Lindblad, E.B.; Oviedo-Orta, E. Progress in understanding adjuvant immunotoxicity mechanisms. Toxicol. Lett. 2011, 203, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Duramad, P.; Tager, I.B.; Holland, N.T. Cytokines and other immunological biomarkers in children’s environmental health studies. Toxicol. Lett. 2007, 172, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Muller, P.Y.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2010, 2, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Fuchsberger, M.; De L. Karlson, T.; Hardy, C.L.; Selomulya, C.; Plebanski, M. Nanoparticles, Immunomodulation and Vaccine Delivery. In Handbook of Immunological Properties of Engineered Nanomaterials; Frontiers in Nanobiomedical Research; World Scientific: Singapore, 2012; Volume 1, pp. 449–475. ISBN 978-981-4390-25-5. [Google Scholar]
- Moyano, D.F.; Goldsmith, M.; Solfiell, D.J.; Landesman-Milo, D.; Miranda, O.R.; Peer, D.; Rotello, V.M. Confocal imaging-guided laser ablation of basal cell carcinomas: An ex vivo study. J. Am. Chem. Soc. 2012, 134, 3965–3967. [Google Scholar] [CrossRef] [PubMed]
- Dusinska, M.; Rundén-Pran, E.; Schnekenburger, J.; Kanno, J. Toxicity tests: In vitro and in vivo. In Adverse Effects of Engineered Nanomaterials: Exposure, Toxicology, and Impact on Human Health, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 51–82. ISBN 9780128091999. [Google Scholar]
- França, A.; Aggarwal, P.; Barsov, E.V.; Kozlov, S.V.; Dobrovolskaia, M.A.; González-Fernández, Á. Macrophage scavenger receptor A mediates the uptake of gold colloids by macrophages in vitro. Nanomedicine 2011, 6, 1175–1188. [Google Scholar] [CrossRef]
- Underhill, D.M.; Goodridge, H.S. Information processing during phagocytosis. Nat. Rev. Immunol. 2012, 12, 492–502. [Google Scholar] [CrossRef]
- Kanno, S.; Furuyama, A.; Hirano, S. A murine scavenger receptor MARCO recognizes polystyrene nanoparticles. Toxicol. Sci. 2007, 97, 398–406. [Google Scholar] [CrossRef]
- Göppert, T.M.; Müller, R.H. Plasma protein adsorption of Tween 80- and poloxamer 188-stabilized solid lipid nanoparticles. J. Drug Target. 2003, 11, 225–231. [Google Scholar] [CrossRef]
- Diederichs, J.E. Plasma protein adsorption patterns on liposomes: Establishment of analytical procedure. Electrophoresis 1996, 17, 607–611. [Google Scholar] [CrossRef]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. “Stealth” corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B. Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Szebeni, J. Hemocompatibility testing for nanomedicines and biologicals: Predictive assays for complement mediated infusion reactions. Eur. J. Nanomed. 2012, 4, 33–53. [Google Scholar] [CrossRef]
- Lu, S.; Duffin, R.; Poland, C.; Daly, P.; Murphy, F.; Drost, E.; Macnee, W.; Stone, V.; Donaldson, K. Efficacy of simple short-term in vitro assays for predicting the potential of metal oxide nanoparticles to cause pulmonary inflammation. Environ. Health Perspect. 2009, 117, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, J.; Zhong, Y.; Zhang, J.; Wang, Z.; Wang, L.; An, Y.; Lin, M.; Gao, Z.; Zhang, D. Biocompatibility of Fe(3)O(4)@Au composite magnetic nanoparticles in vitro and in vivo. Int. J. Nanomed. 2011, 6, 2805–2819. [Google Scholar] [CrossRef]
- ASTM International ASTM Standard E2524-08.Standard Test Method for Analysis of Hemolytic Properties of Nanoparticles. Available online: http://www.astm.org/cgi-bin/resolver.cgi?E2524-08 (accessed on 14 April 2020).
- Agashe, H.B.; Dutta, T.; Garg, M.; Jain, N.K. Investigations on the toxicological profile of functionalized fifth-generation poly (propylene imine) dendrimer. J. Pharm. Pharmacol. 2006, 58, 1491–1498. [Google Scholar] [CrossRef]
- Bhadra, D.; Yadav, A.K.; Bhadra, S.; Jain, N.K. Glycodendrimeric nanoparticulate carriers of primaquine phosphate for liver targeting. Int. J. Pharm. 2005, 295, 221–233. [Google Scholar] [CrossRef]
- Asthana, A.; Chauhan, A.S.; Diwan, P.V.; Jain, N.K. Poly(amidoamine) (PAMAM) dendritic nanostructures for controlled site-specific delivery of acidic anti-inflammatory active ingredient. AAPS PharmSciTech 2005, 6, E536–E542. [Google Scholar] [CrossRef]
- Bhadra, D.; Bhadra, S.; Jain, S.; Jain, N.K. A PEGylated dendritic nanoparticulate carrier of fluorouracil. Int. J. Pharm. 2003, 257, 111–124. [Google Scholar] [CrossRef]
- Malik, N.; Wiwattanapatapee, R.; Klopsch, R.; Lorenz, K.; Frey, H.; Weener, J.W.; Meijer, E.W.; Paulus, W.; Duncan, R. Dendrimers: Relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J. Control. Release 2000, 65, 133–148. [Google Scholar] [CrossRef]
- Agrawal, P.; Gupta, U.; Jain, N.K. Glycoconjugated peptide dendrimers-based nanoparticulate system for the delivery of chloroquine phosphate. Biomaterials 2007, 28, 3349–3359. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. In vitro assays for monitoring nanoparticle interaction with components of the immune system. In Fronters in Nanobiological Research:Handbook of Immunological Properties of Engineered Nanomaterials; World Scientific: Singapore, 2016; Volume 1, pp. 581–638. ISBN 9789814699174. [Google Scholar]
- Magdolenova, Z.; Drlickova, M.; Henjum, K.; Rundén-Pran, E.; Tulinska, J.; Bilanicova, D.; Pojana, G.; Kazimirova, A.; Barancokova, M.; Kuricova, M.; et al. Coating-dependent induction of cytotoxicity and genotoxicity of iron oxide nanoparticles. Nanotoxicology 2015, 9, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Najafi-Hajivar, S.; Zakeri-Milani, P.; Mohammadi, H.; Niazi, M.; Soleymani-Goloujeh, M.; Baradaran, B.; Valizadeh, H. Overview on experimental models of interactions between nanoparticles and the immune system. Biomed. Pharmacother. 2016, 83, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Tian, F.; Ozkan, C.S.; Wang, M.; Gao, H. Effect of single wall carbon nanotubes on human HEK293 cells. Toxicol. Lett. 2005, 155, 73–85. [Google Scholar] [CrossRef]
- Kostarelos, K.; Lacerda, L.; Pastorin, G.; Wu, W.; Wieckowski, S.; Luangsivilay, J.; Godefroy, S.; Pantarotto, D.; Briand, J.-P.; Muller, S.; et al. Cellular uptake of functionalized carbon nanotubes is independent of functional group and cell type. Nat. Nanotechnol. 2007, 2, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Pantarotto, D.; Briand, J.P.; Prato, M.; Bianco, A. Translocation of bioactive peptides across cell membranes by carbon nanotubes. Chem. Commun. 2004, 10, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Shi Kam, N.W.; Jessop, T.C.; Wender, P.A.; Dai, H. Nanotube molecular transporters: Internalization of carbon nanotube-protein conjugates into Mammalian cells. J. Am. Chem. Soc. 2004, 126, 6850–6851. [Google Scholar] [CrossRef]
- King, M.A. Detection of dead cells and measurement of cell killing by flow cytometry. J. Immunol. Methods 2000, 243, 155–166. [Google Scholar] [CrossRef]
- Fairbairn, D.W.; Olive, P.L.; O’Neill, K.L. The comet assay: A comprehensive review. Mutat. Res. Genet. Toxicol. 1995, 339, 37–59. [Google Scholar] [CrossRef]
- Krüger, C.T.; Hofmann, M.; Hartwig, A. The in vitro PIG-A gene mutation assay: Mutagenicity testing via flow cytometry based on the glycosylphosphatidylinositol (GPI) status of TK6 cells. Arch. Toxicol. 2015, 89, 2429–2443. [Google Scholar] [CrossRef]
- OECD. Mammalian Erythrocyte Micronucleus, Test No. 474; OECD: Paris, France, 1997. [Google Scholar]
- Laingam, S.; Froscio, S.M.; Humpage, A.R. Flow-cytometric analysis of in vitro micronucleus formation: Comparative studies with WIL2-NS human lymphoblastoid and L5178Y mouse lymphoma cell lines. Mutat. Res. Toxicol. Environ. Mutagen. 2008, 656, 19–26. [Google Scholar] [CrossRef]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Doak, S.H.; Liu, Y.; Chen, C. Genotoxicity and Cancer. In Adverse Effects of Engineered Nanomaterials; Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 243–261. ISBN 9780123869401. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 340, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, P. In Vitro-In Vivo Carcinogenicity. Adv. Biochem. Eng. Biotechnol. 2016, 127–141. [Google Scholar] [CrossRef]
- Ponti, J.; Kinsner-Ovaskainen, A.; Norlen, H.; Altmeyer, S.; Cristina, A.; Bogni, A. Interlaboratory Comparison study of the Colony Forming Efficiency Assay for Assessing Cytotoxicity of Nanomaterials; Publications Office: Luxembourg, 2014; ISBN 9789279446771. [Google Scholar]
- OECD. Guidance Document on the In Vitro Syrian Hamster Embryo (SHE) Cell Transformation Assay; OECD: Paris, France, 2015. [Google Scholar]
- Guadagnini, R.; Halamoda Kenzaoui, B.; Walker, L.; Pojana, G.; Magdolenova, Z.; Bilanicova, D.; Saunders, M.; Juillerat-Jeanneret, L.; Marcomini, A.; Huk, A.; et al. Toxicity screenings of nanomaterials: Challenges due to interference with assay processes and components of classic in vitro tests. Nanotoxicology 2015, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.E.; Gonzalez Carter, D.A.; Motskin, M.; Pienaar, I.S.; Chen, S.; Hu, S.; Ruenraroengsak, P.; Ryan, M.P.; Shaffer, M.S.P.; Dexter, D.T.; et al. High resolution and dynamic imaging of biopersistence and bioreactivity of extra and intracellular MWNTs exposed to microglial cells. Biomaterials 2015, 70, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.; Marfell, B.; Scott, A.; Waterhouse, N. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb–prot087288. [Google Scholar] [CrossRef]
- Stefanowicz-Hajduk, J.; Ochocka, J.R. Real-time cell analysis system in cytotoxicity applications: Usefulness and comparison with tetrazolium salt assays. Toxicol. Reports 2020, 7, 335–344. [Google Scholar] [CrossRef]
- Kroll, A.; Pillukat, M.H.; Hahn, D.; Schnekenburger, J. Current in vitro methods in nanoparticle risk assessment: Limitations and challenges. Eur. J. Pharm. Biopharm. 2009, 72, 370–377. [Google Scholar] [CrossRef]
- Monteiro-Riviere, N.A.; Inman, A.O.; Zhang, L.W. Limitations and relative utility of screening assays to assess engineered nanoparticle toxicity in a human cell line. Toxicol. Appl. Pharmacol. 2009, 234, 222–235. [Google Scholar] [CrossRef]
- Lanone, S.; Boczkowski, J. Biomedical Applications and Potential Health Risks of Nanomaterials: Molecular Mechanisms. Curr. Mol. Med. 2006, 6, 651–663. [Google Scholar] [CrossRef]
- de Vos, P.; Faas, M.M.; Spasojevic, M.; Sikkema, J. Encapsulation for preservation of functionality and targeted delivery of bioactive food components. Int. Dairy J. 2010, 20, 292–302. [Google Scholar] [CrossRef]
- Chau, C.F.; Wu, S.H.; Yen, G.C. The development of regulations for food nanotechnology. Trends Food Sci. Technol. 2007, 18, 269–280. [Google Scholar] [CrossRef]
- Ricaurte, L.; Perea-Flores, M.D.J.; Martinez, A.; Quintanilla-Carvajal, M.X. Production of high-oleic palm oil nanoemulsions by high-shear homogenization (microfluidization). Innov. Food Sci. Emerg. Technol. 2016, 35, 75–85. [Google Scholar] [CrossRef]
- Gonnet, M.; Lethuaut, L.; Boury, F. New trends in encapsulation of liposoluble vitamins. J. Control. Release 2010, 146, 276–290. [Google Scholar] [CrossRef]
- Wibroe, P.P.; Mat Azmi, I.D.; Nilsson, C.; Yaghmur, A.; Moghimi, S.M. Citrem modulates internal nanostructure of glyceryl monooleate dispersions and bypasses complement activation: Towards development of safe tunable intravenous lipid nanocarriers. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1909–1914. [Google Scholar] [CrossRef]
- Yaghmur, A. Nanoencapsulation of food ingredients by cubosomes and hexosomes. In Lipid-Based Nanostructures for Food Encapsulation Purposes; Elsevier: Amsterdam, The Netherlands, 2019; pp. 483–522. [Google Scholar]
- Azmi, I.D.M.; Wibroe, P.P.; Wu, L.P.; Kazem, A.I.; Amenitsch, H.; Moghimi, S.M.; Yaghmur, A. A structurally diverse library of safe-by-design citrem-phospholipid lamellar and non-lamellar liquid crystalline nano-assemblies. J. Control. Release 2016, 239, 1–9. [Google Scholar] [CrossRef]
- Bender, E.A.; Adorne, M.D.; Colomé, L.M.; Abdalla, D.S.P.; Guterres, S.S.; Pohlmann, A.R. Hemocompatibility of poly(ε-caprolactone) lipid-core nanocapsules stabilized with polysorbate 80-lecithin and uncoated or coated with chitosan. Int. J. Pharm. 2012, 426, 271–279. [Google Scholar] [CrossRef]
- Talluri, S.V.; Kuppusamy, G.; Karri, V.V.S.R.; Tummala, S.; Madhunapantula, S.R.V. Lipid-based nanocarriers for breast cancer treatment – comprehensive review. Drug Deliv. 2016, 23, 1291–1305. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surfaces B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Martínez-Ballesta, Mc.; Gil-Izquierdo, Á.; García-Viguera, C.; Domínguez-Perles, R. Nanoparticles and controlled delivery for bioactive compounds: Outlining challenges for new “smart-foods” for health. Foods 2018, 7. [Google Scholar] [CrossRef]
- Marslin, G.; Siram, K.; Liu, X.; Khandelwal, V.K.M.; Shen, X.; Wang, X.; Franklin, G. Solid lipid nanoparticles of albendazole for enhancing cellular uptake and cytotoxicity against u-87 mg glioma cell lines. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.G.; Silva, E.J.; Martins, A.L.L.; Mota, M.F.; Braga, R.C.; Lima, E.M.; Valadares, M.C.; Taveira, S.F.; Marreto, R.N. Development of topotecan loaded lipid nanoparticles for chemical stabilization and prolonged release. Eur. J. Pharm. Biopharm. 2011, 79, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Saini, J.; Bansal, V.; Chandra, A.; Madan, J.; Jain, U.K.; Chandra, R.; Jain, S.M. Bleomycin sulphate loaded nanostructured lipid particles augment oral bioavailability, cytotoxicity and apoptosis in cervical cancer cells. Colloids Surfaces B Biointerfaces 2014, 118, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Mitxelena-Iribarren, O.; Hisey, C.L.; Errazquin-Irigoyen, M.; González-Fernández, Y.; Imbuluzqueta, E.; Mujika, M.; Blanco-Prieto, M.J.; Arana, S. Effectiveness of nanoencapsulated methotrexate against osteosarcoma cells: In vitro cytotoxicity under dynamic conditions. Biomed. Microdevices 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Seabra, C.L.; Nunes, C.; Gomez-Lazaro, M.; Correia, M.; Machado, J.C.; Gonçalves, I.C.; Reis, C.A.; Reis, S.; Martins, M.C.L. Docosahexaenoic acid loaded lipid nanoparticles with bactericidal activity against Helicobacter pylori. Int. J. Pharm. 2017, 519, 128–137. [Google Scholar] [CrossRef]
- Badawi, N.M.; Hteaima, M.H.; El-Say, K.M.; Aattia, D.A.; Ael-Nabarawi, M.A.; Elmazar, M.M. Pomegranate extract-loaded solid lipid nanoparticles: Design, optimization, and in vitro cytotoxicity study. Int. J. Nanomed. 2018, 13, 1313–1326. [Google Scholar] [CrossRef]
- Evans, L.M.; Cowey, S.L.; Siegal, G.P.; Hardy, R.W. Stearate preferentially induces apoptosis in human breast cancer cells. Nutr. Cancer 2009, 61, 746–753. [Google Scholar] [CrossRef]
- Hagen, R.M.; Rhodes, A.; Ladomery, M.R. Conjugated linoleate reduces prostate cancer viability whereas the effects of oleate and stearate are cell line-dependent. Anticancer Res. 2013, 33, 4395–4400. [Google Scholar]
- Jyoti, K.; Kaur, K.; Pandey, R.S.; Jain, U.K.; Chandra, R.; Madan, J. Inhalable nanostructured lipid particles of 9-bromo-noscapine, a tubulin-binding cytotoxic agent: In vitro and in vivo studies. J. Colloid Interface Sci. 2015, 445, 219–230. [Google Scholar] [CrossRef]
- Sun, M.; Nie, S.; Pan, X.; Zhang, R.; Fan, Z.; Wang, S. Quercetin-nanostructured lipid carriers: Characteristics and anti-breast cancer activities in vitro. Colloids Surfaces B Biointerfaces 2014, 113, 15–24. [Google Scholar] [CrossRef]
- Marcato, P.D.; Caverzan, J.; Rossi-Bergmann, B.; Pinto, E.F.; Machado, D.; Silva, R.A.; Justo, G.Z.; Ferreira, C.V.; Durán, N. Nanostructured polymer and lipid carriers for sunscreen. Biological effects and skin permeation. Proc. J. Nanosci. Nanotechnol. 2011, 11, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Figueiró, F.; Bernardi, A.; Frozza, R.L.; Terroso, T.; Zanotto-Filho, A.; Jandrey, E.H.F.; Moreira, J.C.F.; Salbego, C.G.; Edelweiss, M.I.; Pohlmann, A.R.; et al. Resveratrol-loaded lipid-core nanocapsules treatment reduces in vitro and in vivo glioma growth. J. Biomed. Nanotechnol. 2013, 9, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, G.; Moche, H.; Nieto, A.; Benoit, J.P.; Nesslany, F.; Lagarce, F. Cytotoxicity and genotoxicity of lipid nanocapsules. Toxicol. Vitr. 2017, 41, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Lerata, M.S.; D’Souza, S.; Sibuyi, N.R.S.; Dube, A.; Meyer, M.; Samaai, T.; Antunes, E.M.; Beukes, D.R. Encapsulation of variabilin in stearic acid solid lipid nanoparticles enhances its anticancer activity in vitro. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.D.; Durán, N. Cytotoxicity and Genotoxicity of Solid Lipid Nanoparticles. In Accessing the Erythrocyte Toxicity of Nanomaterials: From Current Methods to Biomolecular Surface Chemistry Interactions; Springer: New York, NY, USA, 2014; pp. 229–244. [Google Scholar]
- Yang, Y.; Li, W.; Kroner, E.; Arzt, E.; Bhushan, B.; Benameur, L.; Wei, L.; Botta, A.; Lu, Y.; Lou, J.; et al. Genotoxicity of Nanoparticles. In Encyclopedia of Nanotechnology; Springer: Amsterdam, The Netherlands, 2012; pp. 952–962. [Google Scholar]
- Da Costa, T.H.M.; Ito, M.K. Phospholipids | Physiology. In Encyclopedia of Food Sciences and Nutrition; Academic Press: Cambridge, MA, USA, 2003; pp. 4523–4531. ISBN 9780122270550. [Google Scholar]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- van Hoogevest, P.; Wendel, A. The use of natural and synthetic phospholipids as pharmaceutical excipients. Eur. J. Lipid Sci. Technol. 2014, 116, 1088–1107. [Google Scholar] [CrossRef]
- Van Hoogevest, P.; Fahr, A. Nanocosmetics. In Nanocosmetics; Cornier, J., Keck, C.M., Van de Voorde, M., Eds.; Springer: New York, NY, USA, 2019; pp. 95–140. ISBN 9783030165734. [Google Scholar]
- Lila, A.S.A.; Ishida, T. Liposomal delivery systems: Design optimization and current applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomedicine 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 1–8. [Google Scholar] [CrossRef]
- Nisini, R.; Poerio, N.; Mariotti, S.; De Santis, F.; Fraziano, M. The multirole of liposomes in therapy and prevention of infectious diseases. Front. Immunol. 2018, 9, 1–23. [Google Scholar] [CrossRef]
- Panahi, Y.; Farshbaf, M.; Mohammadhosseini, M.; Mirahadi, M.; Khalilov, R.; Saghfi, S.; Akbarzadeh, A. Recent advances on liposomal nanoparticles: Synthesis, characterization and biomedical applications. Artif. Cells, Nanomed. Biotechnol. 2017, 45, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Tang, M. Safety of Novel Liposomal Drugs for Cancer Treatment: Advances and Prospects. Chem. Biol. Interact. 2018, 295, 13–19. [Google Scholar] [CrossRef]
- Vangasseri, D.P.; Cui, Z.; Chen, W.; Hokey, D.A.; Falo, L.D.; Huang, L. Immunostimulation of dendritic cells by cationic liposomes. Mol. Membr. Biol. 2006, 23, 385–395. [Google Scholar] [CrossRef]
- Hwang, T.L.; Hsu, C.Y.; Aljuffali, I.A.; Chen, C.H.; Chang, Y.T.; Fang, J.Y. Cationic liposomes evoke proinflammatory mediator release and neutrophil extracellular traps (NETs) toward human neutrophils. Colloids Surfaces B Biointerfaces 2015, 128, 119–126. [Google Scholar] [CrossRef]
- Christensen, D.; Korsholm, K.S.; Andersen, P.; Agger, E.M. Cationic liposomes as vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 513–521. [Google Scholar] [CrossRef]
- Roursgaard, M.; Knudsen, K.B.; Northeved, H.; Persson, M.; Christensen, T.; Kumar, P.E.K.; Permin, A.; Andresen, T.L.; Gjetting, T.; Lykkesfeldt, J.; et al. In vitro toxicity of cationic micelles and liposomes in cultured human hepatocyte (HepG2) and lung epithelial (A549) cell lines. Toxicol. Vitr. 2016, 36, 164–171. [Google Scholar] [CrossRef]
- Dokka, S.; Toledo, D.; Shi, X.; Castranova, V.; Rojanasakul, Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm. Res. 2000, 17, 521–525. [Google Scholar] [CrossRef]
- Postle, A.D.; Heeley, E.L.; Wilton, D.C. A comparison of the molecular species compositions of mammalian lung surfactant phospholipids. Comp. Biochem. Physiol. - A Mol. Integr. Physiol. 2001, 129, 65–73. [Google Scholar] [CrossRef]
- Ingenito, E.P.; Mark, L.; Morris, J.; Espinosa, F.F.; Kamm, R.D.; Johnson, M. Biophysical characterization and modeling of lung surfactant components. J. Appl. Physiol. 1999, 86, 1702–1714. [Google Scholar] [CrossRef] [PubMed]
- Spagnou, S.; Miller, A.D.; Keller, M. Lipidic carriers of siRNA: Differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry 2004, 43, 13348–13356. [Google Scholar] [CrossRef]
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Nanomedicine based approaches for the delivery of siRNA in cancer. J. Intern. Med. 2010, 267, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Monpara, J.; Kanthou, C.; Tozer, G.M.; Vavia, P.R. Rational Design of Cholesterol Derivative for Improved Stability of Paclitaxel Cationic Liposomes. Pharm. Res. 2018, 35, 1–17. [Google Scholar] [CrossRef]
- Carstens, M.G.; Camps, M.G.M.; Henriksen-Lacey, M.; Franken, K.; Ottenhoff, T.H.M.; Perrie, Y.; Bouwstra, J.A.; Ossendorp, F.; Jiskoot, W. Effect of vesicle size on tissue localization and immunogenicity of liposomal DNA vaccines. Vaccine 2011, 29, 4761–4770. [Google Scholar] [CrossRef]
- Desjardins, R.; Krzystyniak, K.; Thérien, H.M.; Banska, W.; Tancrede, P.; Fournier, M. Immunoactivating potential of multilamellar liposome vesicles (MLV) in murine popliteal lymph node (PLN) test. Int. J. Immunopharmacol. 1995, 17, 367–374. [Google Scholar] [CrossRef]
- Szebeni, J.; Bedocs, P.; Rozsnyay, Z.; Weiszhár, Z.; Urbanics, R.; Rosivall, L.; Cohen, R.; Garbuzenko, O.; Báthori, G.; Tóth, M.; et al. Liposome-induced complement activation and related cardiopulmonary distress in pigs: Factors promoting reactogenicity of Doxil and AmBisome. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 176–184. [Google Scholar] [CrossRef]
- Kuznetsova, N.R.; Sevrin, C.; Lespineux, D.; Bovin, N.V.; Vodovozova, E.L.; Mészáros, T.; Szebeni, J.; Grandfils, C. Hemocompatibility of liposomes loaded with lipophilic prodrugs of methotrexate and melphalan in the lipid bilayer. J. Control. Release 2012, 160, 394–400. [Google Scholar] [CrossRef]
- Isalomboto Nkanga, C.; Murhimalika Bapolisi, A.; Ikemefuna Okafor, N.; Werner Maçedo Krause, R. General Perception of Liposomes: Formation, Manufacturing and Applications. In Liposomes - Advances and Perspectives; Catala, A., Ed.; IntechOpen: London, UK, 2019; pp. 1–24. [Google Scholar]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm, F.R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [Google Scholar] [CrossRef]
- Zheng, S.; Beissinger, R.; Sherwood, R.L.; McCormick, D.L.; Lasic, D.D.; Martin, F.J. Rationale for the Development of Red Blood Cell Substitutes. J. Liposome Res. 1993, 3, 575–588. [Google Scholar] [CrossRef]
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Moein Moghimi, S.; Hamad, I.; Andresen, T.L.; Jørgensen, K.; Szebeni, J.; Moein Moghimi, S.; Hamad, I.; Andresen, T.L.; Jørgensen, K.; Szebeni, J. Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. FASEB J. 2006, 20, 2591–2593. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef]
- Kaminskas, L.M.; Mcleod, V.M.; Porter, C.J.H.; Boyd, B.J. Differences in Colloidal Structure of PEGylated Nanomaterials Dictate the Likelihood of Accelerated Blood Clearance. J. Pharm. Sci. 2011, 100, 5069–5077. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, E.; Sun, X.; Huang, P.; Long, H.; Wang, H.; Yu, X.; Zheng, C.; Huang, Y. Pluronic L61 as a long-circulating modifier for enhanced liposomal delivery of cancer drugs. Polym. Chem. 2013, 4, 2958–2962. [Google Scholar] [CrossRef]
- Caddeo, C.; Pucci, L.; Gabriele, M.; Carbone, C.; Fernàndez-Busquets, X.; Valenti, D.; Pons, R.; Vassallo, A.; Fadda, A.M.; Manconi, M. Stability, biocompatibility and antioxidant activity of PEG-modified liposomes containing resveratrol. Int. J. Pharm. 2018, 538, 40–47. [Google Scholar] [CrossRef]
- Bokrova, J.; Marova, I.; Matouskova, P.; Pavelkova, R. Fabrication of novel PHB-liposome nanoparticles and study of their toxicity in vitro. J. Nanoparticle Res. 2019, 21, 1–12. [Google Scholar] [CrossRef]
- Chan, J.; Valencia, P.M.; Zhang, L.; Langer, R.; Farokhzad, O.C. Polymeric Nanoparticles for Drug Delivery. In Cancer Nanotechnology, Methods in Molecular Biology; Springer Science and Business Media LLC: Berlin, Germany, 2010; pp. 163–175. ISBN 978-1-60761-608-5. [Google Scholar]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Wu, B.; Wu, Y.; Lu, W.F.; Fuh, J.Y.H. Polycaprolactone/Pluronic F127 Tissue Engineering Scaffolds via Electrohydrodynamic Jetting for Gastro Intestinal Repair. Procedia CIRP 2017, 65, 184–188. [Google Scholar] [CrossRef]
- Lee, C.H.; Li, Y.J.; Huang, C.C.; Lai, J.Y. Poly(ϵ-caprolactone) nanocapsule carriers with sustained drug release: Single dose for long-term glaucoma treatment. Nanoscale 2017, 9, 11754–11764. [Google Scholar] [CrossRef] [PubMed]
- Valencia, P.M.; Pridgen, E.M.; Perea, B.; Gadde, S.; Sweeney, C.; Kantoff, P.W.; Bander, N.H.; Lippard, S.J.; Langer, R.; Karnik, R.; et al. Synergistic cytotoxicity of irinotecan and cisplatin in dual-drug targeted polymeric nanoparticles. Nanomedicine 2013, 8, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Jain, S.K.; Ganesh, N.; Barve, J.; Beg, A.M. Design and development of ligand-appended polysaccharidic nanoparticles for the delivery of oxaliplatin in colorectal cancer. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Chopra, M.; Bernela, M.; Kaur, P.; Manuja, A.; Kumar, B.; Thakur, R. Alginate/gum acacia bipolymeric nanohydrogels-Promising carrier for Zinc oxide nanoparticles. Int. J. Biol. Macromol. 2015, 72, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fan, X.; Li, L. PH-sensitive polymeric micelles formed by doxorubicin conjugated prodrugs for co-delivery of doxorubicin and paclitaxel. Carbohydr. Polym. 2016, 137, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.R.P.; Jain, N.J.N.; Sharma, R.K.R.; Bahadur, P. Effect of additives on the micellization of PEO/PPO/PEO block copolymer F127 in aqueous solution. Colloids Surfaces A Physicochem. Eng. Asp. 2001, 178, 57–69. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, Y.; Chen, Y.; Yu, S.; Hao, J.; Luo, J.; Sha, X.; Fang, X. Enhanced antitumor efficacy by Paclitaxel-loaded Pluronic P123/F127 mixed micelles against non-small cell lung cancer based on passive tumor targeting and modulation of drug resistance. Eur. J. Pharm. Biopharm. 2010, 75, 341–353. [Google Scholar] [CrossRef]
- Zili, Z.; Sfar, S.; Fessi, H. Preparation and characterization of poly-caprolactone nanoparticles containing griseofulvin. Int. J. Pharm. 2005, 294, 261–267. [Google Scholar] [CrossRef]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Guterres, S.S.; Fessi, H.; Barratt, G.; Devissaguet, J.P.; Puisieux, F. Poly (DL-lactide) nanocapsules containing diclofenac: I. Formulation and stability study. Int. J. Pharm. 1995, 113, 57–63. [Google Scholar] [CrossRef]
- Zhang, K.; Tang, X.; Zhang, J.; Lu, W.; Lin, X.; Zhang, Y.; Tian, B.; Yang, H.; He, H. PEG-PLGA copolymers: Their structure and structure-influenced drug delivery applications. J. Control. Release 2014, 183, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Gao, X.; Su, L.; Xia, H.; Gu, G.; Pang, Z.; Jiang, X.; Yao, L.; Chen, J.; Chen, H. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 2011, 32, 8010–8020. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Suresh, P.K.; Desmukh, R. Design of Eudragit RL 100 nanoparticles by nanoprecipitation method for ocular drug delivery. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Seremeta, K.P.; Chiappetta, D.A.; Sosnik, A. Poly(ε-caprolactone), Eudragit® RS 100 and poly(ε-caprolactone)/Eudragit® RS 100 blend submicron particles for the sustained release of the antiretroviral efavirenz. Colloids Surfaces B Biointerfaces 2013, 102, 441–449. [Google Scholar] [CrossRef]
- Dongqin, B.; Chenyi, Y.; Luo, J.; Décoppet, J.; Zhang, F.; Zakeeruddin, M.; Li, X.; Grätzel, M.; Hagfeldt, A. Polymer-templated nucleation and crystal growth of perovskite films for solar cells. Nat. Energy 2016, 1, 1–8. [Google Scholar]
- Lopedota, A.; Trapani, A.; Cutrignelli, A.; Chiarantini, L.; Pantucci, E.; Curci, R.; Manuali, E.; Trapani, G. The use of Eudragit®RS 100/cyclodextrin nanoparticles for the transmucosal administration of glutathione. Eur. J. Pharm. Biopharm. 2009, 72, 509–520. [Google Scholar] [CrossRef]
- Grabowski, N.; Hillaireau, H.; Vergnaud, J.; Tsapis, N.; Pallardy, M.; Kerdine-Römer, S.; Fattal, E. Surface coating mediates the toxicity of polymeric nanoparticles towards human-like macrophages. Int. J. Pharm. 2015, 482, 75–83. [Google Scholar] [CrossRef]
- Ahlin, P.; Kristl, J.; Kristl, A.; Vrečer, F. Investigation of polymeric nanoparticles as carriers of enalaprilat for oral administration. Int. J. Pharm. 2002, 239, 113–120. [Google Scholar] [CrossRef]
- Mu, L.; Feng, S.S. Vitamin E TPGS used as emulsifier in the solvent evaporation/extraction technique for fabrication of polymeric nanospheres for controlled release of paclitaxel (Taxol®). J. Control. Release 2002, 80, 129–144. [Google Scholar] [CrossRef]
- Barichello, J.M.; Morishita, M.; Takayama, K.; Nagai, T. Absorption of insulin from Pluronic F-127 gels following subcutaneous administration in rats. Int. J. Pharm. 1999, 184, 189–198. [Google Scholar] [CrossRef]
- Shah, V.; Taratula, O.; Garbuzenko, O.B.; Patil, M.L.; Savla, R.; Zhang, M.; Minko, T. Genotoxicity of Different Nanocarriers: Possible Modifications for the Delivery of Nucleic Acids. Curr. Drug Discov. Technol. 2013, 10, 8–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Porto, I.C.C.M. Polymer biocompatibility. In Polymerization; IntechOpen: London, UK, 2012; pp. 47–62. ISBN 978-953-51-0745-3. [Google Scholar]
- Nallamuthu, I.; Devi, A.; Khanum, F. Chlorogenic acid loaded chitosan nanoparticles with sustained release property, retained antioxidant activity and enhanced bioavailability. Asian J. Pharm. Sci. 2015, 10, 203–211. [Google Scholar] [CrossRef]
- Patel, B.K.; Parikh, R.H.; Aboti, P.S. Development of Oral Sustained Release Rifampicin Loaded Chitosan Nanoparticles by Design of Experiment. J. Drug Deliv. 2013, 2013, 370938. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, S.; Paliwal, R.; Paliwal, S.R.; Vyas, S.P. Hyaluronic acid modified chitosan nanoparticles for effective management of glaucoma: Development, characterization, and evaluation. J. Drug Target. 2010, 18, 292–302. [Google Scholar] [CrossRef]
- Friedl, H.E.; Dünnhaupt, S.; Waldner, C.; Bernkop-Schnürch, A. Preactivated thiomers for vaginal drug delivery vehicles. Biomaterials 2013, 34, 7811–7818. [Google Scholar] [CrossRef]
- Kolawole, O.M.; Lau, W.M.; Khutoryanskiy, V.V. Chitosan/β-glycerophosphate in situ gelling mucoadhesive systems for intravesical delivery of mitomycin-C. Int. J. Pharm. X 2019, 1. [Google Scholar] [CrossRef]
- Campos, E.; Branquinho, J.; Carreira, A.S.; Carvalho, A.; Coimbra, P.; Ferreira, P.; Gil, M.H. Designing polymeric microparticles for biomedical and industrial applications. Eur. Polym. J. 2013, 49, 2005–2021. [Google Scholar] [CrossRef]
- VandeVord, P.J.; Matthew, H.W.T.; DeSilva, S.P.; Mayton, L.; Wu, B.; Wooley, P.H. Evaluation of the biocompatibility of a chitosan scaffold in mice. J. Biomed. Mater. Res. 2002, 59, 585–590. [Google Scholar] [CrossRef]
- Rao, S.B.; Sharma, C.P. Use of chitosan as a biomaterial: Studies on its safety and hemostatic potential. J. Biomed. Mater. Res. 1997, 34, 21–28. [Google Scholar] [CrossRef]
- Tomihata, K.; Ikada, Y. In vitro and in vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials 1997, 18, 567–575. [Google Scholar] [CrossRef]
- Rodrigues, S.; Dionísio, M.; López, C.R.; Grenha, A. Biocompatibility of Chitosan Carriers with Application in Drug Delivery. J. Funct. Biomater. 2012, 3, 615–641. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.; Mohanty, S.; Mallick, R.; Jacob, B.; Sonawane, A. Toxicity and antibacterial assessment of chitosancoated silver nanoparticles on human pathogens and macrophage cells. Int. J. Nanomed. 2012, 7, 1805–1818. [Google Scholar] [CrossRef]
- Gammon, J.M.; Tostanoski, L.H.; Adapa, A.R.; Chiu, Y.-C.; Jewell, C.M. Controlled delivery of a metabolic modulator promotes regulatory T cells and restrains autoimmunity. J. Control. Release 2015, 210, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-based biocompatible nanoparticles for next-generation tolerogenic vaccines against autoimmune disease. Int. J. Mol. Sci. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef]
- Diwan, M.; Elamanchili, P.; Cao, M.; Samuel, J. Dose Sparing of CpG Oligodeoxynucleotide Vaccine Adjuvants by Nanoparticle Delivery. Curr. Drug Deliv. 2005, 1, 405–412. [Google Scholar] [CrossRef]
- Husseini, G.A.; Pitt, W.G. Micelles and nanoparticles for ultrasonic drug and gene delivery. Adv. Drug Deliv. Rev. 2008, 60, 1137–1152. [Google Scholar] [CrossRef]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 2012, 64, 72–82. [Google Scholar] [CrossRef]
- Allemann, E.; Brasseur, N.; Benrezzak, O.; Rousseau, J.; Kudrevich, S.V.; Boyle, R.W.; Leroux, J.-C.; Gurny, R.; Van Lier, J.E. PEG-coated Poly(lactic acid) Nanoparticles for the Delivery of Hexadecafluoro Zinc Phthalocyanine to EMT-6 Mouse Mammary Tumours. J. Pharm. Pharmacol. 1995, 47, 382–387. [Google Scholar] [CrossRef]
- Duncan, R.; Izzo, L. Dendrimer biocompatibility and toxicity. Adv. Drug Deliv. Rev. 2005, 57, 2215–2237. [Google Scholar] [CrossRef]
- Neerman, M.F.; Zhang, W.; Parrish, A.R.; Simanek, E.E. In vitro and in vivo evaluation of a melamine dendrimer as a vehicle for drug delivery. Int. J. Pharm. 2004, 281, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Aillon, K.L.; Xie, Y.; El-Gendy, N.; Berkland, C.J.; Forrest, M.L. Effects of nanomaterial physicochemical properties on in vivo toxicity. Adv. Drug Deliv. Rev. 2009, 61, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Koziara, J.M.; Lockman, P.R.; Allen, D.D.; Mumper, R.J. The blood-brain barrier and brain drug delivery. J. Nanosci. Nanotechnol. 2006, 6, 2712–2735. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J.; Alyautdin, R.N.; Kharkevich, D.A.; Ivanov, A.A. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). Brain Res. 1995, 674, 171–174. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Rego, A.C.; Oliveira, C.R. Cellular and molecular mechanisms involved in the neurotoxicity of opioid and psychostimulant drugs. Brain Res. Rev. 2008, 58, 192–208. [Google Scholar] [CrossRef]
- Voigt, N.; Henrich-Noack, P.; Kockentiedt, S.; Hintz, W.; Tomas, J.; Sabel, B.A. Toxicity of polymeric nanoparticles in vivo and in vitro. J. Nanoparticle Res. 2014, 16, 1–22. [Google Scholar] [CrossRef]
- Lockman, P.R.; Koziara, J.M.; Mumper, R.J.; Allen, D.D. Nanoparticle Surface Charges Alter Blood–Brain Barrier Integrity and Permeability. J. Drug Target. 2004, 12, 635–641. [Google Scholar] [CrossRef]
- Ali, I.; Saifullah, S.; Ahmed, F.; Ullah, S.; Imkan, I.; Hussain, K.; Imran, M.; Shah, M.R. Synthesis of long-tail nonionic surfactants and their investigation for vesicle formation, drug entrapment, and biocompatibility. J. Liposome Res. 2019. [Google Scholar] [CrossRef]
- Negm, N.A.; Kandile, N.G.; Mohamad, M.A. Synthesis, characterization and surface activity of new eco-friendly schiff bases vanillin derived cationic surfactants. J. Surfactants Deterg. 2011, 14, 325–331. [Google Scholar] [CrossRef]
- Chen, M.; Hu, X.; Fu, M. Novel synthesis of a new surfactant 4-((4-bromophenyl)(dodecyl)amino)-4- oxobutanoic acid containing a benzene ring using a copper catalyst cross-coupling reaction and its properties. J. Surfactants Deterg. 2013, 16, 581–585. [Google Scholar] [CrossRef]
- Kalhapure, R.S.; Akamanchi, K.G. Oleic acid based heterolipid synthesis, characterization and application in self-microemulsifying drug delivery system. Int. J. Pharm. 2012, 425, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Masotti, A.; Vicennati, P.; Alisi, A.; Marianecci, C.; Rinaldi, F.; Carafa, M.; Ortaggi, G. Novel Tween® 20 derivatives enable the formation of efficient pH-sensitive drug delivery vehicles for human hepatoblastoma. Bioorganic Med. Chem. Lett. 2010, 20, 3021–3025. [Google Scholar] [CrossRef] [PubMed]
- Witika, B.A. The Development, Manufacture and Characterisation of Niosomes Intended To Deliver Nevirapine To the Brain. Master‘s Thesis, Rhodes University, Grahamstown, South Africa, 2017. [Google Scholar]
- Gasco, M.R. Lipid nanoparticles: Perspectives and challenges. Adv. Drug Deliv. Rev. 2007, 59, 377–378. [Google Scholar] [CrossRef] [PubMed]
- Vidlářová, L.; Romero, G.B.; Hanuš, J.; Štěpánek, F.; Müller, R.H. Nanocrystals for dermal penetration enhancement - Effect of concentration and underlying mechanisms using curcumin as model. Eur. J. Pharm. Biopharm. 2016, 104, 216–225. [Google Scholar] [CrossRef]
- Makoni, P.A.; Kasongo, K.W.; Walker, R.B. Short term stability testing of efavirenz-loaded solid lipid nanoparticle (SLN) and nanostructured lipid carrier (NLC) dispersions. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef]
- Manosroi, A.; Jantrawut, P.; Khositsuntiwong, N.; Manosroi, W.; Manosroi, J. Novel elastic nanovesicles for cosmeceutical and pharmaceutical applications. Chiang Mai J. Sci. 2009, 36, 168–178. [Google Scholar]
- Agarwal, R.; Katare, O.P.; Vyas, S.P. Preparation and in vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int. J. Pharm. 2001, 228, 43–52. [Google Scholar] [CrossRef]
- Sreya, M.; Krishna Sailaja, A. Preparation and evaluation of diclofenac sodium niosomal formulations. J. Bionanoscience 2017, 11, 489–496. [Google Scholar] [CrossRef]
- Witika, B.A.; Smith, V.J.; Walker, R.B. Quality by design optimization of cold sonochemical synthesis of zidovudine-lamivudine nanosuspensions. Pharmaceutics 2020, 12, 1–20. [Google Scholar] [CrossRef]
- Witika, B.A.; Smith, V.J.; Walker, R.B. A comparative study of the effect of different stabilizers on the critical quality attributes of self-assembling nano co-crystals. Pharmaceutics 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Abdelbary, G.; El-gendy, N. Niosome-Encapsulated Gentamicin for Ophthalmic Controlled Delivery. AAPS PharmSciTech 2008, 9, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, P.; Shanmugam, S.; Lee, W.M.S.; Lee, W.M.S.; Kim, J.O.J.S.; Oh, D.H.; Kim, D.D.; Kim, J.O.J.S.; Yoo, B.K.; Choi, H.G.; et al. Formulation and in vitro Assessment of Minoxidil Niosomes for Enhanced Skin Delivery. Int. J. Pharm. 2009, 377, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Manconi, M.; Valenti, D.; Sinico, C.; Lai, F.; Loy, G.; Fadda, A.M. Niosomes as carriers for tretinoin: II. Influence of vesicular incorporation on tretinoin photostability. Int. J. Pharm. 2003, 260, 261–272. [Google Scholar] [CrossRef]
- Sun, H.; Yang, R.; Wang, J.; Yang, X.; Tu, J.; Xie, L.; Li, C.; Lao, Q.; Sun, C. Component-based biocompatibility and safety evaluation of polysorbate 80. RSC Adv. 2017, 7, 15127–15138. [Google Scholar] [CrossRef]
- Ali, I.; Shah, M.R.; Yousuf, S.; Ahmed, S.; Shah, K.; Javed, I. Hemolytic and cellular toxicology of a sulfanilamide-based nonionic surfactant: A niosomal carrier for hydrophobic drugs. Toxicol. Res. 2018, 7, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Ullah, S.; Shah, M.R.; Shoaib, M.; Imran, M.; Elhissi, A.M.A.; Ahmad, F.; Ali, I.; Shah, S.W.A. Development of a biocompatible creatinine-based niosomal delivery system for enhanced oral bioavailability of clarithromycin. Drug Deliv. 2016, 23, 3480–3491. [Google Scholar] [CrossRef] [PubMed]
- Gharbavi, M.; Manjili, H.K.; Amani, J.; Sharafi, A.; Danafar, H. In vivo and in vitro biocompatibility study of novel microemulsion hybridized with bovine serum albumin as nanocarrier for drug delivery. Heliyon 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Sagiri, S.S.; Behera, B.; Sudheep, T.; Pal, K. Effect of composition on the properties of tween-80-span-80-based organogels. Des. Monomers Polym. 2012, 15, 253–273. [Google Scholar] [CrossRef]
- Katiyar, S.S.; Kushwah, V.; Dora, C.P.; Patil, R.Y.; Jain, S. Design and Toxicity Evaluation of Novel Fatty Acid-Amino Acid-Based Biocompatible Surfactants. AAPS PharmSciTech 2019, 20, 1–10. [Google Scholar] [CrossRef]
- Ali, I.; Shah, M.R.; Imran, M. Shafiullah Synthesis of Sulfur-Based Biocompatible Nonionic Surfactants and Their Nano-Vesicle Drug Delivery. J. Surfactants Deterg. 2017, 20, 1367–1375. [Google Scholar] [CrossRef]
- Teeranachaideekul, V.; Junyaprasert, V.B.; Souto, E.B.; Müller, R.H. Development of ascorbyl palmitate nanocrystals applying the nanosuspension technology. Int. J. Pharm. 2008, 354, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Y.; Ma, Y.; Hou, X.S.; Zhang, W.J.; Wang, P.; Chen, H.; Li, B.; Zhang, C.; Ding, Y. Co-delivery of antineoplastic and protein drugs by chitosan nanocapsules for a collaborative tumor treatment. Carbohydr. Polym. 2017, 157, 1470–1478. [Google Scholar] [CrossRef]
- Knowlton, J.; Pearce, S. Handbook of Cosmetic Science & Technology; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9781856171977. [Google Scholar]
- Petkovic, M.; Ferguson, J.L.; Gunaratne, H.Q.N.; Ferreira, R.; Leitão, M.C.; Seddon, K.R.; Rebelo, L.P.N.; Pereira, C.S. Novel biocompatible cholinium-based ionic liquids—toxicity and biodegradability. Green Chem. 2010, 12, 643–664. [Google Scholar] [CrossRef]
- Hauss, D.J. Oral lipid-based formulations. Adv. Drug Deliv. Rev. 2007, 59, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Li, S.; Yin, R.; Liu, H.; Xu, L. Effect of surfactants on the formation and characterization of a new type of colloidal drug delivery system: Nanostructured lipid carriers. Colloids Surfaces A Physicochem. Eng. Asp. 2008, 315, 210–216. [Google Scholar] [CrossRef]
- Callender, S.P.; Mathews, J.A.; Kobernyk, K.; Wettig, S.D. Microemulsion utility in pharmaceuticals: Implications for multi-drug delivery. Int. J. Pharm. 2017, 526, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Gehlot, P.S.; Kulshrestha, A.; Bharmoria, P.; Damarla, K.; Chokshi, K.; Kumar, A. Surface-Active Ionic Liquid Cholinium Dodecylbenzenesulfonate: Self-Assembling Behavior and Interaction with Cellulase. ACS Omega 2017, 2, 7451–7460. [Google Scholar] [CrossRef]
- de la Harpe, K.M.; Kondiah, P.P.D.; Choonara, Y.E.; Marimuthu, T.; du Toit, L.C.; Pillay, V. The Hemocompatibility of Nanoparticles: A Review of Cell-Nanoparticle Interactions and Hemostasis. Cells 2019, 8. [Google Scholar] [CrossRef]
- Buzea, C.; Pacheco, I.I.; Robbie, K. Nanomaterials and nanoparticles: Sources and toxicity. Biointerphases 2007, 2, MR17–MR71. [Google Scholar] [CrossRef]
- Greish, K.; Thiagarajan, G.; Herd, H.; Price, R.; Bauer, H.; Hubbard, D.; Burckle, A.; Sadekar, S.; Yu, T.; Anwar, A.; et al. Size and surface charge significantly influence the toxicity of silica and dendritic nanoparticles. Nanotoxicology 2012, 6, 713–723. [Google Scholar] [CrossRef]
- Doktorovová, S.; Kovačević, A.B.; Garcia, M.L.; Souto, E.B. Preclinical safety of solid lipid nanoparticles and nanostructured lipid carriers: Current evidence from in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2016, 108, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Doktorovova, S.; Silva, A.M.; Gaivão, I.; Souto, E.B.; Teixeira, J.P.; Martins-Lopes, P. Comet assay reveals no genotoxicity risk of cationic solid lipid nanoparticles. J. Appl. Toxicol. 2014, 34, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Doktorovova, S.; Shegokar, R.; Rakovsky, E.; Gonzalez-Mira, E.; Lopes, C.M.; Silva, A.M.; Martins-Lopes, P.; Muller, R.H.; Souto, E.B. Cationic solid lipid nanoparticles (cSLN): Structure, stability and DNA binding capacity correlation studies. Int. J. Pharm. 2011, 420, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Doktorovova, S.; Shegokar, R.; Martins-Lopes, P.; Silva, A.M.; Lopes, C.M.; Muller, R.H.; Souto, E.B. Modified Rose Bengal assay for surface hydrophobicity evaluation of cationic solid lipid nanoparticles (cSLN). Eur. J. Pharm. Sci. 2012, 45, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Fangueiro, J.F.; Andreani, T.; Egea, M.A.; Garcia, M.L.; Souto, S.B.; Silva, A.M.; Souto, E.B. Design of cationic lipid nanoparticles for ocular delivery: Development, characterization and cytotoxicity. Int. J. Pharm. 2014, 461, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Gaidamauskas, E.; Cleaver, D.P.; Chatterjee, P.B.; Crans, D.C. Effect of Micellar and Reverse Micellar Interface on Solute Location: 2,6-Pyridinedicarboxylate in CTAB Micelles and CTAB and AOT Reverse Micelles. Langmuir 2010, 26, 13153–13161. [Google Scholar] [CrossRef]
- Severino, P.; Santana, M.H.A.; Souto, E.B. Optimizing SLN and NLC by 2(2) full factorial design: Effect of homogenization technique. Mater. Sci. Eng. C. Mater. Biol. Appl. 2012, 32, 1375–1379. [Google Scholar] [CrossRef]
- Severino, P.; Szymanski, M.; Favaro, M.; Azzoni, A.R.; Chaud, M.V.; Santana, M.H.A.; Silva, A.M.; Souto, E.B. Development and characterization of a cationic lipid nanocarrier as non-viral vector for gene therapy. Eur. J. Pharm. Sci. 2015, 66, 78–82. [Google Scholar] [CrossRef]
- Silva, A.M.; Martins-Gomes, C.; Coutinho, T.E.; Fangueiro, J.F.; Sanchez-Lopez, E.; Pashirova, T.N.; Andreani, T.; Souto, E.B. Soft cationic nanoparticles for drug delivery: Production and cytotoxicity of solid lipid nanoparticles (SLNs). Appl. Sci. 2019, 9. [Google Scholar] [CrossRef]
- Santos de Almeida, T.; Júlio, A.; Saraiva, N.; Fernandes, A.S.; Araújo, M.E.M.; Baby, A.R.; Rosado, C.; Mota, J.P. Choline- versus imidazole-based ionic liquids as functional ingredients in topical delivery systems: Cytotoxicity, solubility, and skin permeation studies. Drug Dev. Ind. Pharm. 2017, 43, 1858–1865. [Google Scholar] [CrossRef]
- Jaitely, V.; Karatas, A.; Florence, A.T. Water-immiscible room temperature ionic liquids (RTILs) as drug reservoirs for controlled release. Int. J. Pharm. 2008, 354, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, S.; Liu, M.; Hui, J.; Yang, B.; Tao, L.; Wei, Y. Surfactant-dispersed nanodiamond: Biocompatibility evaluation and drug delivery applications. Toxicol. Res. 2013, 2, 335–342. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituent (R) | Phospholipid | Net Charge at Neutral pH |
|---|---|---|
| Hydrogen | Phosphatidic acid (PA) | −1 |
| Ethanolamine | Phosphatidylethanolamine (PE) | 0 |
| Choline | Phosphatidylcholine (PC) | 0 |
| Serine | Phosphatidylserine (PS) | −1 |
| Glycerol | Phosphatidylglycerol (PG) | −1 |
| Inositol | Phosphatidylinositol (PI) | −1 |
| Phosphatidylglycerol | Cadiolipin (CL) | −2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witika, B.A.; Makoni, P.A.; Matafwali, S.K.; Chabalenge, B.; Mwila, C.; Kalungia, A.C.; Nkanga, C.I.; Bapolisi, A.M.; Walker, R.B. Biocompatibility of Biomaterials for Nanoencapsulation: Current Approaches. Nanomaterials 2020, 10, 1649. https://doi.org/10.3390/nano10091649
Witika BA, Makoni PA, Matafwali SK, Chabalenge B, Mwila C, Kalungia AC, Nkanga CI, Bapolisi AM, Walker RB. Biocompatibility of Biomaterials for Nanoencapsulation: Current Approaches. Nanomaterials. 2020; 10(9):1649. https://doi.org/10.3390/nano10091649
Chicago/Turabian StyleWitika, Bwalya A., Pedzisai A. Makoni, Scott K. Matafwali, Billy Chabalenge, Chiluba Mwila, Aubrey C. Kalungia, Christian I. Nkanga, Alain M. Bapolisi, and Roderick B. Walker. 2020. "Biocompatibility of Biomaterials for Nanoencapsulation: Current Approaches" Nanomaterials 10, no. 9: 1649. https://doi.org/10.3390/nano10091649
APA StyleWitika, B. A., Makoni, P. A., Matafwali, S. K., Chabalenge, B., Mwila, C., Kalungia, A. C., Nkanga, C. I., Bapolisi, A. M., & Walker, R. B. (2020). Biocompatibility of Biomaterials for Nanoencapsulation: Current Approaches. Nanomaterials, 10(9), 1649. https://doi.org/10.3390/nano10091649