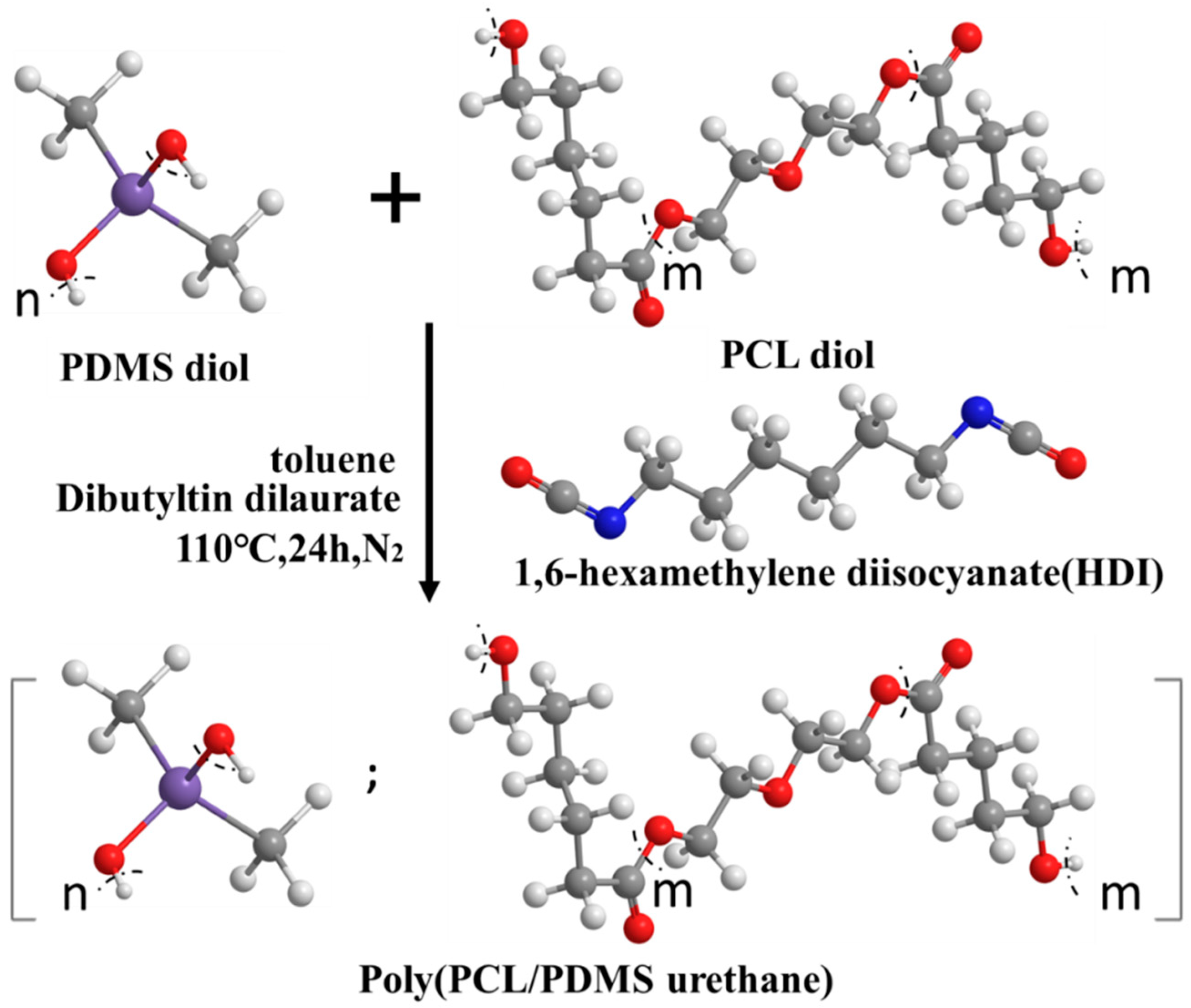
3.1. Molecular Characteristic of PCL–PDMS Copolymer Nanofibres by NMR
The molecular characteristics of the fabricated PCL–PDMS copolymer nanofibres with different ratios were investigated using 1H NMR spectroscopy. Since all of the samples have similar peaks, only the spectrum for PCL
6–PDMS
4 is presented.
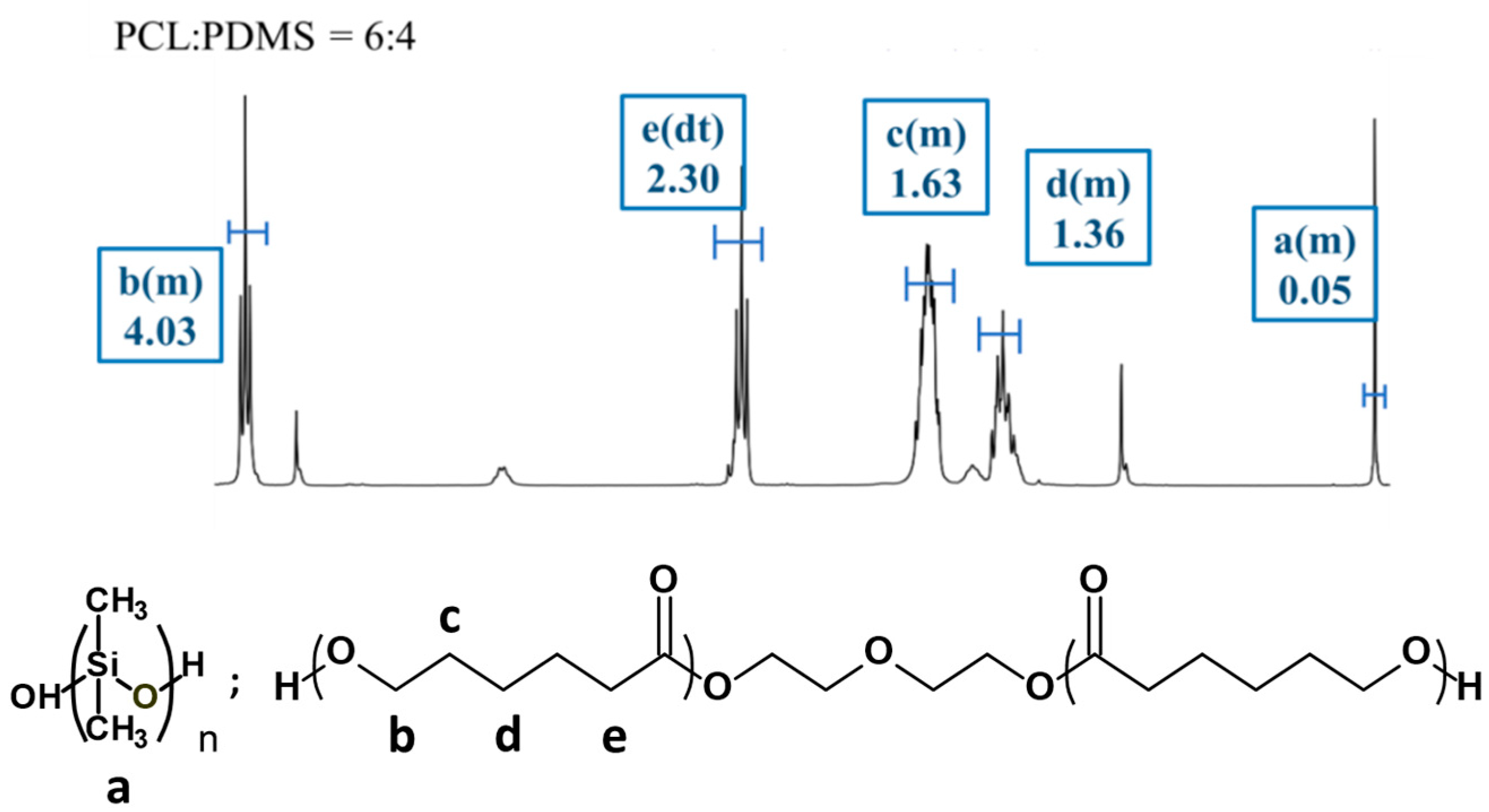
Figure 2 shows the 1H NMR spectra for the PCL
6–PDMS
4 sample and the proton signals associated with both PCL and PDMS are observed. For the PCL segment, the peak detected at 1.63 ppm is attributed to the −(CH2)3−, 2.30 ppm for −CH2CO− and at 4.04 ppm for −OCH2− (methylene protons alpha to the ester group of PCL segments). Meanwhile, methylene protons in the repeated units of PDMS segments were observed at 0.05 ppm peak. The copolymer composition and the molecular weight of all copolymers were determined at 0.05 ppm (PDMS) and 4.04 ppm (PCL).
Table 1 summarised the molecular properties of the PCL–PDMS copolymer nanofibres. Based on the calculated data, the weight ratio of the PCL and PDMS in all copolymer compositions was found to be consistent with the feed ratio, thus, confirming the successful fabrication of the PCL–PDMS copolymer nanofibres.
3.2. PCL–PDMS Copolymer Nanofibres’ Surface Morphology
The PCL–PDMS copolymers were fabricated into nanofibre mesh via the conventional electrospinning process. The copolymer solutions were prepared at 10 wt% (w/w) diluted in HFIP and electro-spun at 10 kV. In general, with an appropriate polymer concentration, polymers with M
n > 10,000 will form continuous fibres during the electrospinning process [
13]. Referring to
Table 1, all the synthesized PCL–PDMS copolymer demonstrated M
n > 10,000 which is within the expected electrospinning fibre-forming range.
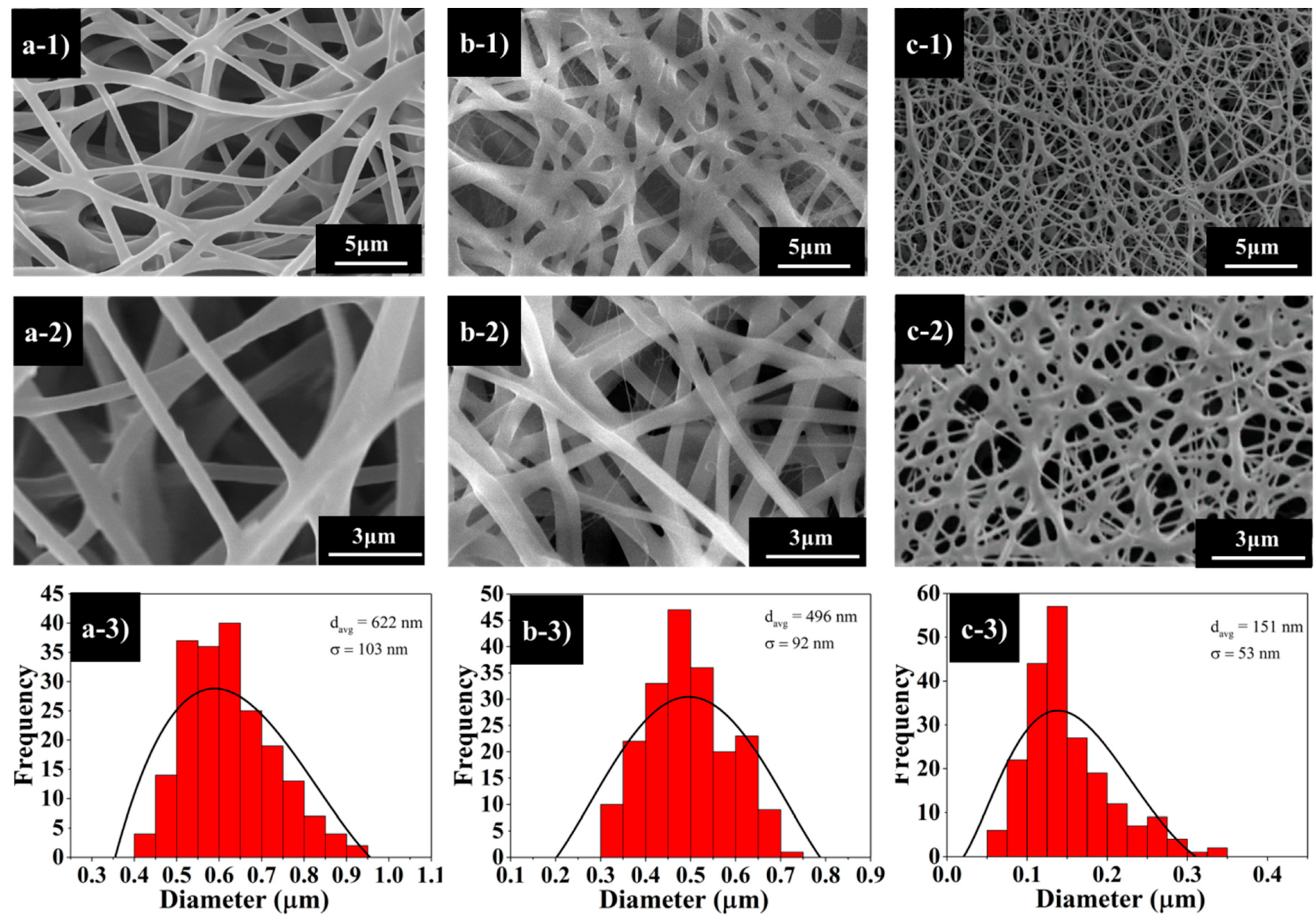
Figure 3 illustrates the SEM images of the fabricated nanofibres from different copolymer composition ratios. The surface morphology of PCL–PDMS copolymer nanofibres show a continuous, smooth and bead-free structure with a random nanofibre orientation.
The PCL
6–PDMS
4 (
Figure 3a) and PCL
7–PDMS
3 (
Figure 3b) samples showed cylindrical and uniform nanofibres; meanwhile, the PCL
8–PDMS
2 sample showed a melted fibrous morphology. This might be related to the number-average molecular weight (M
n) of the synthesized copolymer as obtained from the NMR spectroscopy (
Table 1). The molecular weight of the synthesized copolymers decreased with the decreases of the PDMS mixture ratio. The melt fibrous morphology of the PCL
8–PDMS
2 sample indicated an insufficient physical cross-linking with less interconnected junction formed among the fibres. The average diameter of the copolymer nanofibres ranged from 151 nm to 622 nm when the PDMS composition increased from 20% to 40%. A larger nanofibre diameter was achieved with a sample with higher PDMS compositions. The increase in resultant nanofibre diameter was attributed to the molecular weight (M
n) of the sample, where higher M
n will increase the fibre diameter due to the high number of chain entanglement and increased viscosity.
3.3. Thermal Properties of the PCL–PDMS Copolymers
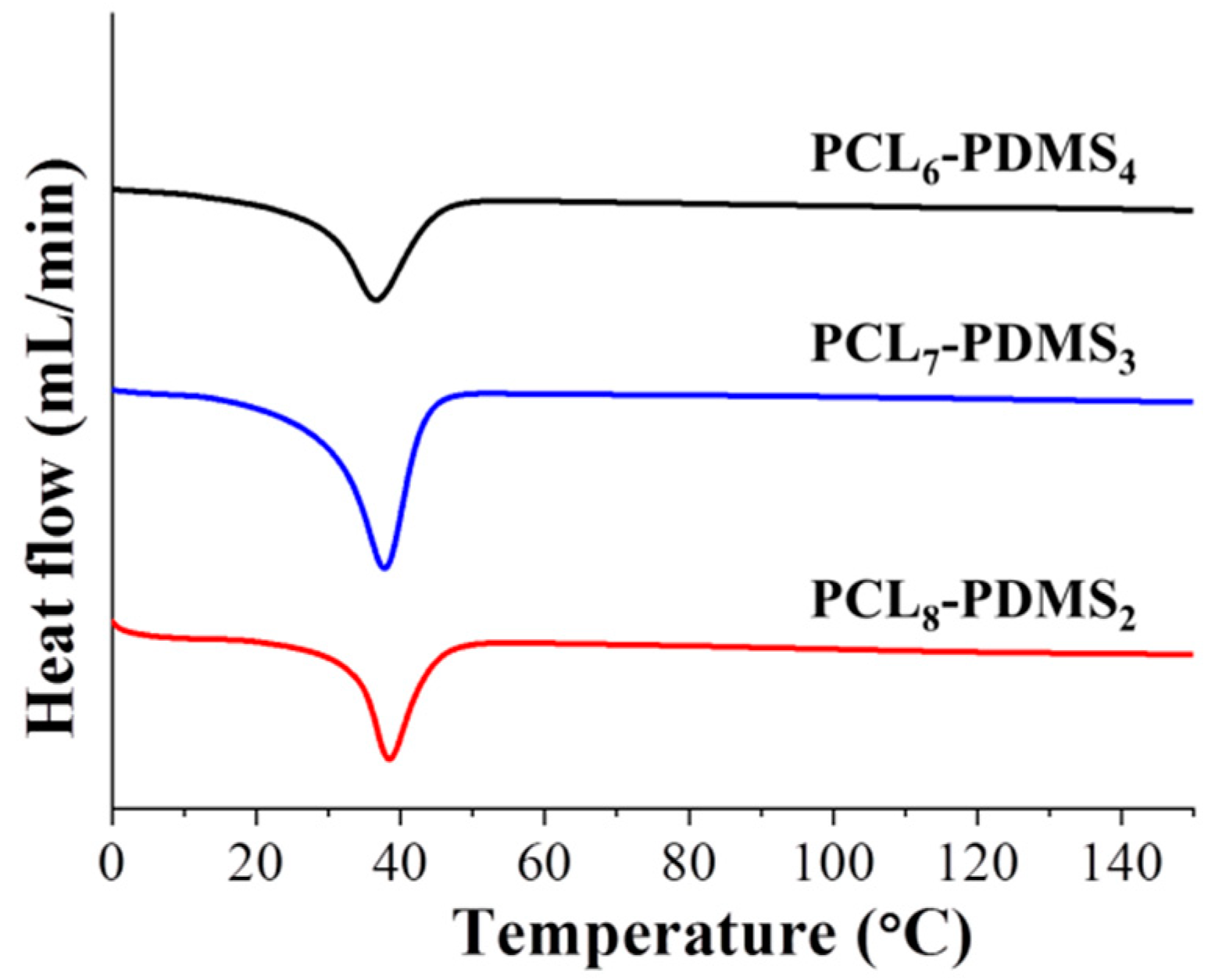
The thermal properties of the copolymer samples were characterised by DSC. The DSC curves after the second heating run were illustrated in
Figure 4. The measurement was made after the second heating run to remove residual solvent and to erase the thermal history of the polymer during the synthesis process. It can be observed that the melting temperatures (T
m) for all PCL–PDMS compositions were similar—approximately 37 °C. The T
m was used as the trigger temperature for shape recovery thus achieving a T
m close to the human body temperature which is very desirable for material intended to be used as biomedical material. The obtained result is consistent with a previous study, where the fabricated shape memory polymer showed a similar T
m around 38 °C [
13].
Although the PCL–PDMS mixture ratio did not display any effect on the melting temperature, however, the crystallinity (X
c) and enthalpy change (∆H
m) showed the opposite effect. Both parameters demonstrated a positive correlation to the amount of PCL segment ratio. For example, the X
c increased from 21.93% to 26.83% when the PCL ratio increased from 60% (PCL
6–PDMS
4) to 80% (PCL
8–PDMS
2) of the copolymer ratio. Besides, the enthalpy change increased from 30.59, 34.82, and 37.42 (J/g) for PCL
6–PDMS
4, PCL
7–PDMS
3, and PCL
8–PDMS
2, respectively. The thermal properties of the fabricated PCL–PDMS copolymer nanofibres are presented in
Table 2.
3.4. Mechanical Properties of the PCL–PDMS Copolymer Nanofibres
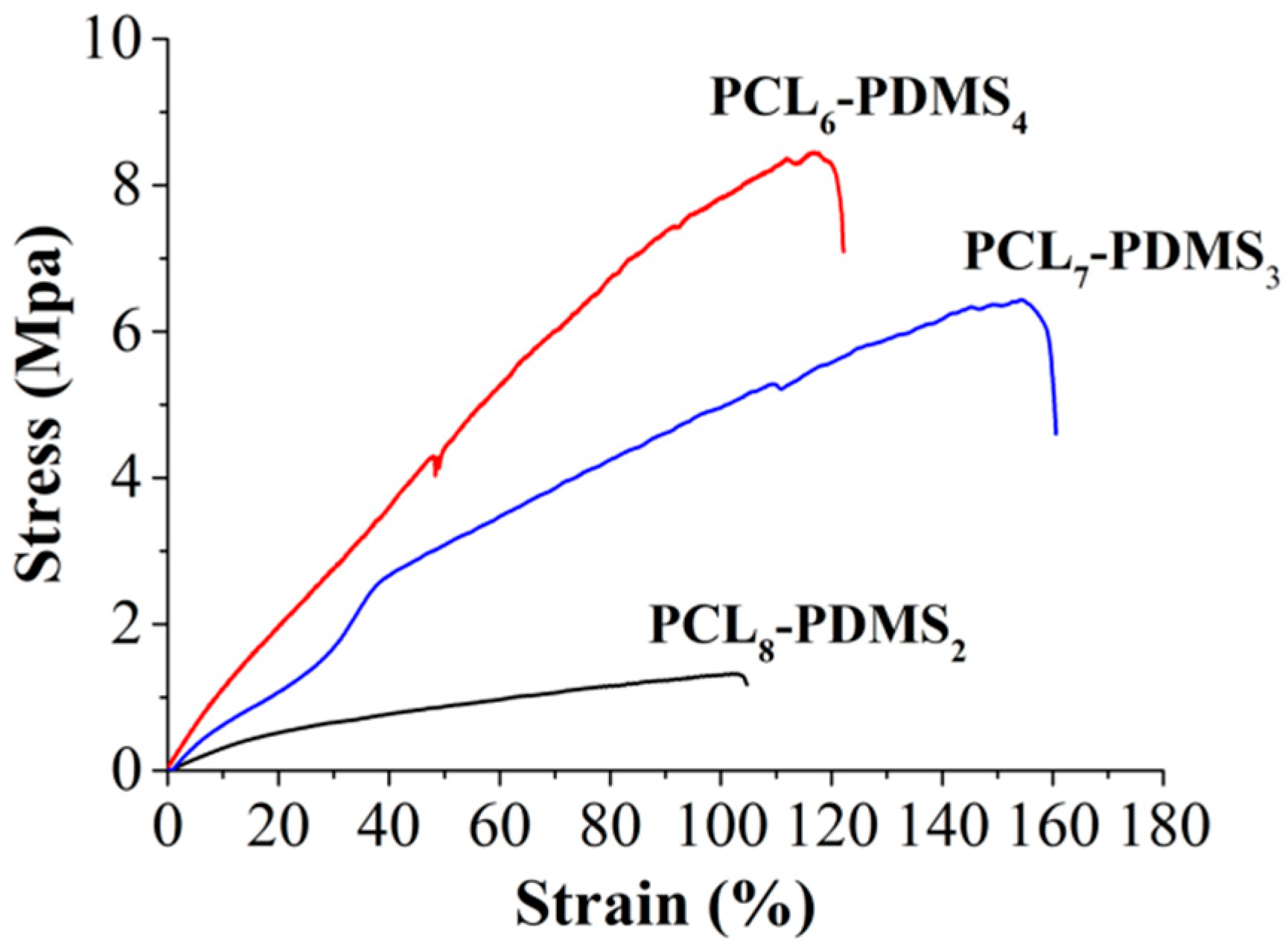
Figure 5 illustrates the stress–strain curve of the copolymer nanofibres during the uniaxial tensile test. The tensile properties of the copolymer nanofibres such as tensile strength, percent of elongation and Young’s modulus are tabulated in
Table 3. The tensile properties of the copolymer nanofibres were highly dependent on the copolymer mixture ratio. The tensile strength of the PCL
8-PDMS
2 is 1.41 MPa, and when the ratio of PDMS content increases to PCL
6-PDMS
4, the tensile strength increases to 6.64 MPa. In addition, the percent of elongation ranging between 122.66% to 154.44% and the modulus ranged from 3.42 MPa to 12.65 MPa with the addition of the PDMS soft segment. The obtained result differed from some published studies that stated that the higher PCL content increases the crystallinity of the copolymer fibres thus increase the tensile properties of the nanofibres [
3]. This result might be attributed to the fabricated nanofibre morphology where the uniformity of the diameter of the PCL
6–PDMS
4 nanofibres could distribute the stress during the tensile test, therefore, providing a more stable structure. Meanwhile, the melted fibrous structure indicating insufficient physical cross-linking with less interconnected junction formed among the fibres [
14] within the PCL
8–PDMS
2 sample caused the reduction in tensile strength. Moreover, it was reported that the addition of the PDMS soft segment content improves the tensile properties of SMP due to the higher soft segment entanglement density [
15]. The fact that the PDMS had low T
g (−125 °C) makes PDMS a particularly effective soft segment which should enhance the deformability [
8].
3.5. The Shape Memory Behaviour
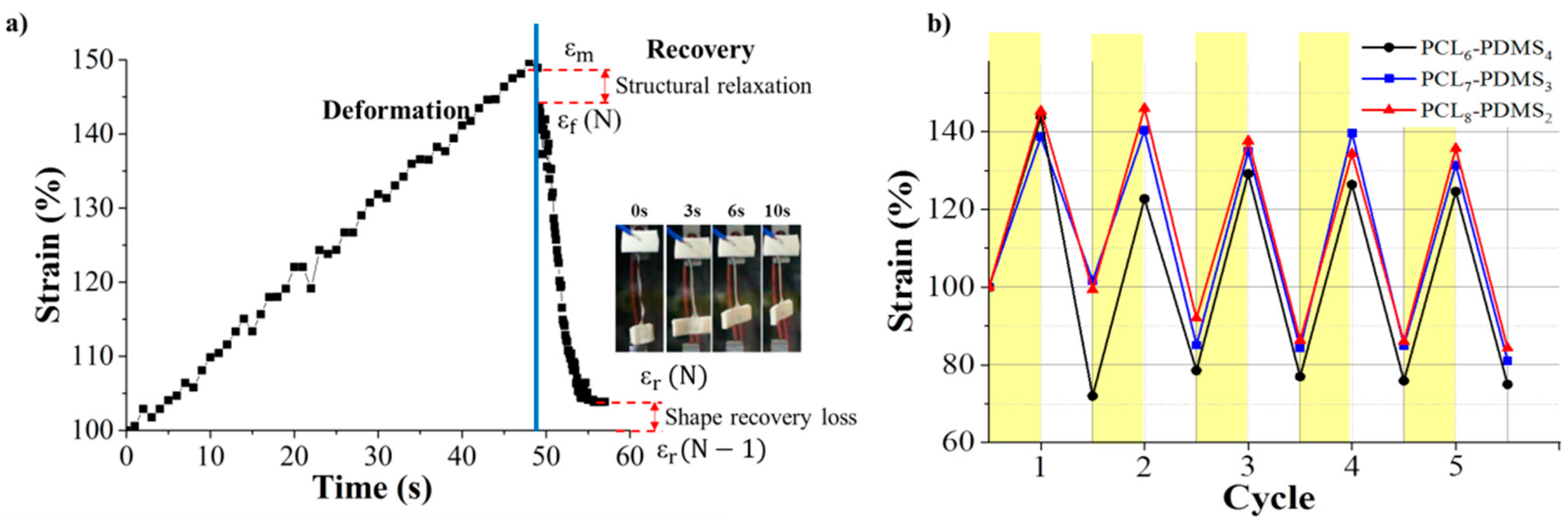
The shape memory behaviour of the PCL–PDMS copolymer nanofibres was evaluated using thermomechanical cycles. The shape memory behaviour of the PCL–PDMS copolymer nanofibres for one complete cycle is shown in
Figure 6a. The cycles involved programming the samples by heating at an elevated temperature, stretching to the desired shape at 40 °C (above T
m) until the shape was fixed and cooling to a temperature lower than T
m (25 °C) so that the chain segment of the samples was in a temporary position before the stress was completely removed. The samples were then re-heated to 40 °C, relaxation occurred, and the temporarily deformed samples recovered to their original permanent shape. The time taken for complete shape recovery of the sample was also recorded. According to
Figure 6a, it was observed that the recovery process started immediately after re-heating and achieved full recovery at approximately 10 s. Such a fast shape recovery of nanofibres in comparison to the film was achieved, due to the high surface area to volume ratio of nanofibres ensuring faster thermal conduction. [
16]. The strain profiles of the five thermomechanical cycles are presented in
Figure 6b. The shape memory performance in terms of shape fixity and recovery ratio for the PCL–PDMS copolymer nanofibres during thermomechanical cycles is tabulated in
Table 4.
The PCL–PDMS copolymer nanofibres exhibited excellent shape fixity ratio which was above 83% for the first tensile cycle and exceeded 90% for subsequent cycles. The shape fixity of the copolymer nanofibres was lower in the first cycle because of the segment-chain orientation and relaxation effects [
17,
18]. As such, the shape memory nanofibres should be pre-conditioned to fully utilise the shape memory potential [
13,
17,
18]. For subsequent cycles, the shape fixity of all samples surpassed 90%. After the fifth cycle, all samples maintained an excellent shape fixity performance up to 98% for PCL
6–PDMS
4. The samples were capable of maintaining the deformed shape and were not weakened regardless of the copolymer mixture ratio as proven by the excellent fixity ratio. The excellent fixity shown by the copolymer PCL–PDMS nanofibres was associated with the physical cross-links formed through inter- or intra-polymeric chain attractions such as hydrogen bonding or dipole–dipole interaction. Moreover, the degree of crystallinity for all samples obtained from the DSC was higher than 20% (
Table 2) which allows for a high strain fixity ratio [
19]. On the other hand, the shape recovery ratio for all samples was higher than 100%.
Our result shows the recovery ratio exceeded 100% which means that the length of the recovered SMP after heating was shorter than the original permanent shape. This might be due to the super-contraction behaviour of the nanofibre. The pre-orientation of the nanofibre during the electrospinning process leads to the super-contraction behaviour. At the temporary state, the nanofibres were pre-stretched causes a certain degree of molecular orientation and electrostatic repulsion. When the nanofibre was heated to recover the permanent shape, thermal expansion induced re-crystallization of the PCL. Rapid crystallization of PCL segments during the cooling process formed a more compact structure of the fibre [
20]. The thermal expansion effect is neglected in the discussion. Besides, the shape recovery > 100% were reported in the previous reports for PCL–PDMS SMP [
18,
20]. The super-contraction behaviour of the SMP helps to maintain the shape after several cycles and prevent the fibre from sagging. In one report, the super-contraction behaviour of SMP helps to heal macroscopic damages as the fibres exert a higher compressive force on the damage interfaces due to the tendency to shrink further [
16]. Since the thermomechanical cycles are repeatable, therefore, the capability to heal macroscopic damages may be repeatable. The PCL
6–PDMS
4 nanofibres with the largest diameter and amorphous domain yield the highest shape recovery in the first cycle (163.41%). Based on the result, it can be concluded that both the shape fixity and recovery ratio was independent of the number of cycles.
Furthermore, the nanofibres’ surface morphology during deformation and fixation after shape recovery was assessed, and the SEM images are presented in
Figure 7. Different nanofibres structures are observed before and after the deformation. Initially, the fibres show a randomly oriented structure (
Figure 3); however, after the temporary stretch (
Figure 7a–c), the fibres tend to align along the strain direction. The samples maintained continuity and no broken fibres were detected, indicating the flexibility of the samples. Once the samples were re-heated, the directed nanofibres recovered to their original random orientation. The average nanofibres’ diameter increased in comparison to the initial state (
Figure 3) after temporary deformation with the sample PCL
6–PDMS
4 showing the highest diameter increase from 348 nm to 1048 nm. Following the shape recovery, the average fibre diameter recovered to around 1143 nm, 1152 nm and 654 nm for PCL
6–PDMS
4, PCL
7–PDMS
3 and PCL
8–PDMS
2, respectively.
3.6. Cytotoxicity and Biocompatibility Study
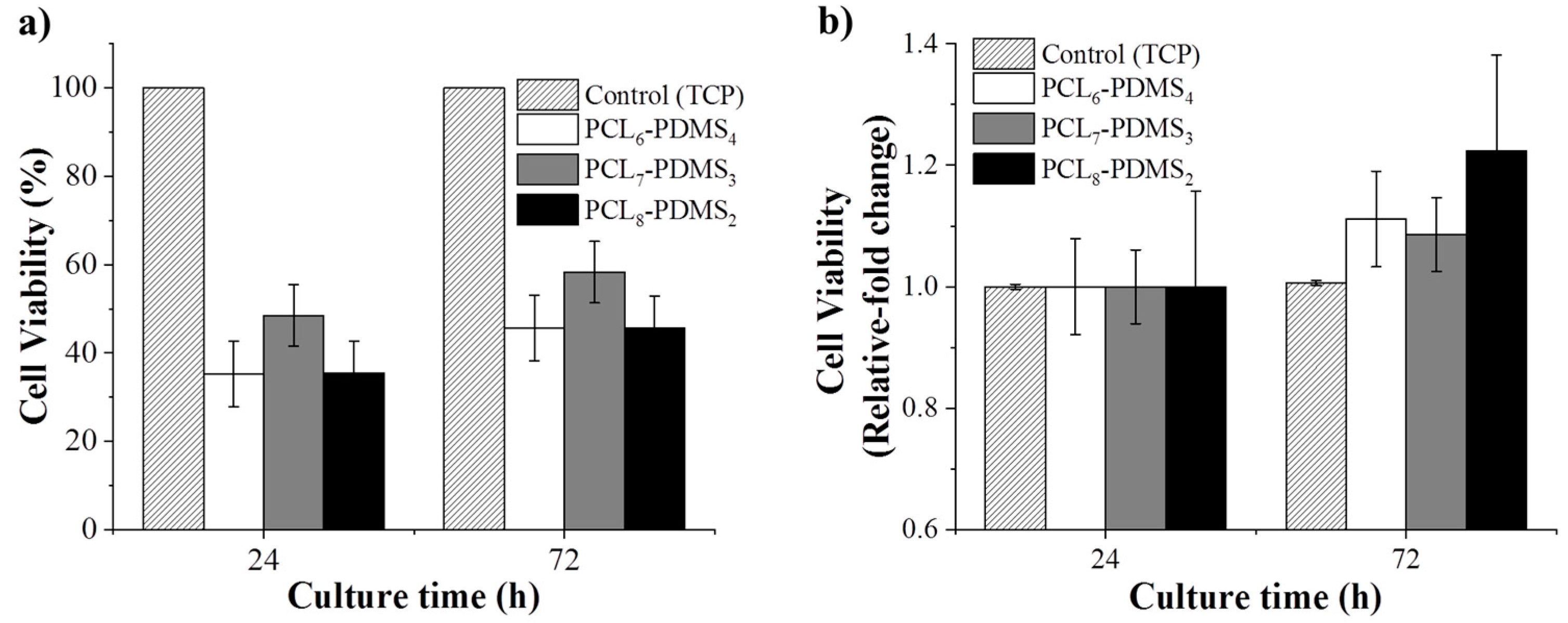
The cytotoxicity of the fabricated nanofibres was statistically evaluated using the CCK-8 assay to validate their application in the biomedical field. Human dermal fibroblast cells were cultured on the fabricated nanofibres and the cytotoxicity effect was observed. The HDF cells cultured on a tissue culture plate (TCP) were regarded as the control group.
Figure 8 shows the CCK-8 result for all samples after 24 h and 72 h. The viability is presented in terms of relative fold change from the control group.
The HDF cell viability is presented in terms of relative fold change from the control group. As expected, since the copolymer samples are based on derivatives of biocompatible PCL and PDMS, no samples showed any toxicity effect to the HDF cells and, as such, this demonstrates compatibility for biomedical application. According to
Figure 8, the cellular viability for all samples increased significantly compared to the control group after 72 h, assuming that the nanofibres can promote cell growth and proliferation. This confirms that the fabricated copolymer nanofibre did not exhibit any toxicity to the cells for 72 h.
Although the fabricated nanofibres showed low cell viability percentage, this shrinkable nanofibre mesh can be applied as an early treatment on acute open wounded area during an emergency situation. Since the fabricated SMP was triggered at near body temperature (37 °C), the pre-stretched fibre mesh can recover to its initial shape upon applying to body. The shrinkage of the fibre during the shape recovery gives pressure to the wound site.
Besides, HDF cells seeded on the nanofibre samples were stained with FDA/DAPI double staining for observable evaluation on the cell morphology. The microscopy images of the cell morphology illustrated in
Figure 9. Survival cells were observed in all samples with spreading cellular extensions.
3.7. Demonstration of the Shape Memory Response
The images in
Figure 10 demonstrate the shape memory responses of the strip made up by electro-spun PCL
6–PDMS
4 nanofibres. To perform a visualized demonstration of shape memory responses, we wrapped a sponge with the PCL–PDMS strip which was programmed as a temporary shape state. Before that, the PCL–PDMS strip with 30 mm in length was, initially, stretched to 50% strain at 40 °C by using the tensile machine under the elongation speed of 20 mm/min. The strip was kept at 45 mm as a temporary shape state on the stage until the environmental temperature reached room temperature. As presented in
Figure 10b, the ambient environment was heated again to 40 °C, and the stretched nanofibres shrunk to its initial length of 30 mm within 10 s as demonstrated in
Figure 10d,e.
Figure 10f,g showed the photos of nanofibre mesh which was cut into the size of 10 mm × 10 mm. Nanofibre mesh had undergone a series of temperature changes to demonstrate its shape memory ability. The nanofibre mesh was stretched to 200% at 40 °C (
Figure 10(f-1)). After that, the nanofibre mesh was cooled to 25 °C until equilibrium was reached. Lastly, we could observe that after reheating nanofibre mesh to 40 °C, the nanofibre mesh recovered to the permanent shape (
Figure 10(g-1)). As presented in the SEM image of
Figure 10(f-2,g-2), we could conclude that the microstructure of nanofibre mesh changed with the shape of nanofibre mesh. The fibres showed the arranged orientation after being stretched (
Figure 10(f-2)). Meanwhile, the energy generated by stretching was stored in the deformed nanofibre mesh. The increase in temperature caused the entropy change which, in turn, leads to changes in the macroscopic appearance. The fibre reverted to random orientation again (
Figure 10(g-2)).

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}