Nanomedicine and Onco-Immunotherapy: From the Bench to Bedside to Biomarkers

 , ,
, ,  ,
, 
Abstract

1. Introduction
2. Nanomedicine
2.1. Nanotechnology: Brief Description
2.2. Nanomedicine: Concept
2.2.1. Nanodiagnostics
2.2.2. Controlled Drug Release
2.2.3. Regenerative Medicine
2.3. Nanomaterials in Medicine
3. Immuno-Oncology
3.1. Cancer Hallmarks
3.2. Immune Cycle in Cancer
3.3. Cancer Immunotherapy
3.3.1. Cytokines
3.3.2. Monoclonal Antibodies
3.3.3. CAR-T Cells
3.3.4. Therapeutic Onco-Vaccines
- i. Cellular onco-vaccines: Within cell-based vaccines there are two types: (i) autologous or allogeneic whole-cell tumour vaccines and (ii) autologous dendritic cells, pulsed or transfected with tumour antigens (contained in tumour lysates, purified proteins, peptides, DNA or RNA) [108]. Autologous cell-based vaccines are based on patient-derived tumour cells, which are irradiated and combined with an immunostimulatory adjuvant and administered to the same individual from whom the cells were extracted and isolated [109]. These vaccines have been tested in a variety of solid cancers, including lung cancer, colorectal cancer, melanoma, renal cell cancer, and prostate cancer [106], showing potent antitumour immunity in preclinical animal models and, in early human clinical trials, have shown relative safety, as well as the induction of tumour-specific immune responses and evidence of antitumour activity, obtaining clinical benefit, although objective response rates remain low [110,111,112,113]. One of the advantages of this type of vaccine is that it has a high potential to deliver the full spectrum of Tumour-Associated Antigens (TAAs) and, in addition, autologous tumour cells can be modified to acquire more potent immunostimulatory characteristics [106]. However, there are some disadvantages, such as requiring an enough tumour sample and potentially inducing autoimmunity, as tumours also express patient-specific proteins [114]. Allogeneic tumour cell vaccines typically contain two or three human tumour cell lines, and have the advantage that they contain unlimited sources of tumour antigens and can produce standardized, large-scale vaccines [106]. An example is Canvaxin, which contains three melanoma lines combined with Bacillus Calmatte-Guerin (BCG) as an adjuvant [115]. In 2010, the first cell-based vaccine was approved by the FDA, based on dendritic cell vaccine called provenge (sipuleucel-T), which targets Prostatic Acid Phosphatase (PAP) antigen in castration-resistant metastatic prostate cancer. PAP is an TAA, which gives the vaccine some specificity and therefore improves the anti-cancer effect [116]. Other vaccines that use whole tumour cells as antigens are OncoVAX for colon cancer and GVAX for prostate cancer [117,118]. These cells can also be genetically modified to produce immune molecules, as in the case of Lucanix for NSCLC [119]. The disadvantage of cell-based vaccines is that they are expensive and, in the case of autologous vaccines, it is difficult to produce them on a large scale [107].
- ii. Dendritic Cell (DC) Vaccines: These vaccines are based on the main characteristic of DCs, which are professional antigen-presenting cells. DCs act in the peripheral tissues, where they absorb, process and present antigenic peptides of the pathogen or host to the virgin T lymphocytes in the lymphoid organs through the MHC. Therefore, DCs are important for connecting innate and adaptive immunity. Functional characterisation in DCs determine that three signals are necessary for complete activation of DCs: 1. adequate loading of MHC–peptide complexes in DC for priming of T cells; 2. positive regulation of co-stimulatory molecules such as CD40, CD80 and CD86, 3. production of cytokines that polarize the Th1/Tc1 immune response [106]. Ex vivo generated DCs are used as cancer vaccines. For this purpose, human DCs can be generated in culture from CD34+ hematopoietic progenitors or peripheral blood monocytes [120]. DC vaccines are achieved by loading TAAs antigens on autologous DCs from patients, which are then treated with adjuvants (Figure 9) [106]. For example, GM-CSF is essential for ex vivo generation of monocyte-derived DC [121]. These cells required a maturation process, which is associated with morphological and functional changes in the DC, allowing improved expression of MHC-I and -II, co-stimulatory molecules and increased cytokine production [122]. These ex vivo DCs are then administered to patients to induce anti-tumour immunity. Thus, T cell activation is regulated by co-stimulatory molecules expressed in DC, so the potency of the DC vaccine can be improved by modifying the expression levels of these inhibitory or activating molecules. DCs need stimulation of CD40 by active CD4+ T cells, so human DCs expressing high CD40L lead to increased activation of reactive T cells with low immunogenic tumour antigens. The activating molecules expressed in DC are related to the response of pro-inflammatory T cells, while suppressor molecules contribute to the tolerance or suppression of T cells [106]. The first work that laid the foundation for DC vaccine development was carried out by Inaba et al. in 1992. They cultivated mouse DC ex vivo from bone marrow precursors [123]. One of the first trials testing the immunogenicity of DC was performed on metastatic prostate cancer. Patients received autologous pulsed DCs with peptides restricted to HLA-A0201 derived from the prostate-specific membrane antigen (PSMA). Antigen-specific cellular responses and reduced PSA levels were observed in some patients [124]. These vaccines have also been tested in clinical trials for the treatment of prostate cancer, melanoma, renal cell carcinoma, and glioma [125,126,127,128,129,130,131]. The results of these studies are mixed but ultimately indicate that, although studies in mice demonstrate a potent ability of DCs to induce antitumour immunity and autologous DCs generated from peripheral blood in humans are a safe and promising approach, further studies are still needed to demonstrate their clinical efficacy and impact on the survival of patients with these types of cancers. As mentioned above, the DC vaccine Sipuleucel-T (Provenge TM) is the first therapeutic cancer vaccine approved by the FDA and has succeeded in increasing survival with a favourable toxicity profile, opening up new paradigms in cancer treatment [106].iii. Protein or peptide-based vaccines: These vaccines are based on tumour-associated antigens (TAA), cancer germline antigens (CGA), virus-associated antigens or tumour-specific antigens (TSA), along with some adjuvants. Those composed of synthetic peptides generally contain between 20 and 30 amino acids directed at specific epitopes of tumour antigens. Antigens can be modified to bind cytokines, antibodies or immunogenic peptides in these vaccines [107]. In this group of vaccines, a few representative examples are Oncophage, which is used in kidney cancer, melanoma, and brain cancer; and MUC1, which is used in breast cancer and NSCLC [132,133]. These types of vaccines are not very expensive and are also very stable but have the limitation that known peptide epitopes are required to be candidates for use in vaccines. Other disadvantages are immune suppression and the weak immunogenicity of these antigens [134]. Recombinant vaccines based on TAA peptides are classified into different categories: 1. antigens encoded by genes that are normally silenced in adult tissues, but which are transcriptionally reactivated in tumour cells (testicular cancer antigens, such as melanoma associated antigen (MAGE) and SSX-2), 2. Tissue-differentiating antigens, which have a normal tissue origin and appear in both normal and tumour tissue (melanoma, breast carcinomas and prostate cancer, such as gp100, mammaglobin-A and PSA, respectively), 3. Tissue differentiation antigens similar to the above, but which, compared to their normal homologous tissues, are very high in tumour tissues (MUC-1, HER2, p53, hTERT, etc.), 4. tumour-specific antigens, which are normally mutated oncogenes (e.g., Ras, B-Raf) and 5. molecules associated with tumour stem cells or with the epithelium-mesenchyme transition process [106]. This type of vaccine is more cost-effective than individualized vaccines, but also has the disadvantage of targeting only one or a few epitopes of the TAAs. To improve the immunogenicity of an auto-antigen, the peptide sequence of TAAs can be altered by introducing agonist-enhancing epitopes that increase peptide binding to MHC or TCR, enhancing the T cell response against the target [106]. Immuno-stimulatory adjuvants are also used when the TAA display of a weak immunogenic nature. Aluminium salts have been used as adjuvants to promote humoral immunity but are not effective in diseases requiring cellular immunity. To induce the adaptive immune response, activation of innate immunity is necessary, which has led to questions about theories of how adjuvants promote adaptive immunity [106]. Charles Janeway demonstrated that adaptive immune responses are dependent on innate immune receptors activated by microbial components [135]. Pattern-Associated Molecular Pattern Recognition (PAMPs) through pattern recognition receptors (PRRs) involves the coordination of innate and adaptive immunity to microbial pathogens or infected cells. TLR-mediated activation of DC is very important in this process, which is why many vaccines include PAMPs as part of therapeutic immunizations against cancer. That is, these molecules are used as adjuvants, facilitating the development of vaccines. Some examples are the use of BCG to treat bladder carcinoma, by activating TLR2 and TLR4, or LPS, which is a natural ligand of TLR4 [106].
- iv. DNA Vaccines: These are vaccines in the form of genes use either DNA, such as plasmids, or RNA, such as mRNA [107]. Viral DNA vectors can transfuse infiltrated somatic cells or DCs as part of the inflammatory response to vaccination [106]. APCs absorb genetic material and translate peptide and proteins as cancer-specific antigens, stimulating the immune response [107]. Currently, there are some DNA vaccines include mammaglobin-A for breast cancer, PAP for prostate cancer, and gp100 and gp75 DNA for melanoma [136,137,138,139]. Disadvantages may be the method of DNA/RNA delivery and the efficiency of absorption, which may limit transcription and antigenic presentation by APCs [107]. These vaccines have been administered using viral vectors and electroporation, which are effective but difficult to apply in the clinical routine [140,141]. It should also be noted that the administration of live virus may cause side effects and decrease the effectiveness of antiviral antibodies in patients [140].
- v. Vaccines targeting TAAs: To achieve tumour-specific death, cancer vaccines must target restricted epitopes of MHC-I that activate CD8+ T cells, as these are the most potent cells and when activated recognize TSAs and distinguish normal cells from cancer cells [142]. This involves the following processes: degradation of ubiquitous proteins by the proteasome, interaction of peptides with Hsp90 in the cytosol, which acts as a chaperone, active transport into the endoplasmic reticulum by the TAP transporter, modification of peptides by ERAP to an appropriate length, which are subsequently loaded into the peptide-binding cleft of MHC class I molecules with the help of chaperones such as tapain and transport to the cell surface, and can thus be recognised by the CD8+ T-cell receptor [143]. There are different types of tumour antigens that can be targeted in immunotherapy: (i) tumour-associated antigens (TAA), which are over-expressed on tumour cells and are expressed to a lesser extent on normal cells, (ii) cancer germ-line antigens (CGA), which on normal adult cells are found only in reproductive tissues, but are expressed selectively on several types of tumours, (iii) virus-associated antigens, which arise in tumour cells from oncogenic viral proteins; and (iv) tumour-specific antigens (TSAs), which are the neo-antigens and are only found in tumour cells, as they arise from non-anonymous somatic mutations [107]. Commonly, cancer vaccines should target the broadest possible antigen repertoire, which can be achieved by using autologous tumour lysates, whole-tumour-derived mRNA, irradiated autologous tumour cells, or allogeneic tumour cell lines [144,145]. In addition, effective responses in response to an antigen can result in the immunogenic release of additional endogenous antigens by tumour cell destruction, leading to a broader immune response. This is known as “epitope spread” [146]. Vaccines targeting TAAs have not been very successful so far and are still under development, mainly because many TAAs are also expressed on normal cells, which show central and peripheral tolerance, and the affinity of TCR for these antigens might be very low [147]. In addition, autoimmune toxicities may take place during treatment. Despite this, some AATs are used as targets Despite the weak points on this approach; Currently, several approaches has been quite promising and help to open more studies exploring the full potential, for example: CD19-directed CAR-T therapy in acute lymphoblastic leukemia (ALL), which results in complete remission in a large number of patients [148]. CGAs, such as melanoma associated antigen 3 (MAGE-A3) and NY-ESO-1 antigen, are expressed selectively in some cancers, but when used as a target they result in high toxicities. In particular, severe neurological toxicities and death occur when MAGE-A3 is targeted [149]. On the other hand, virus-coded antigens are only present on tumour cells, not on normal cells, as some cancers are associated with virus infection. Viral oncogenes encode oncoproteins that cause cell transformation. An example is the human papilloma virus (HPV), which is associated with cervical cancer [150]. This method has been effective in treating cancer, but there are also virus-associated antigens with the ability to escape from the immune system [151]. In the approach of these vaccines, the critical and important key aspect is the selection of tumour-specific antigens (TSA), which are the neo-antigens. These are peptides that arise from non-anonymous mutations, alterations in genomic codons, editing, processing and antigen presentation in tumour cells [107]. Among all non-synonymous mutations, a part of them is distributed clonally by the tumour and generates peptides containing mutations (neo-epitopes) that can be recognised by cytotoxic T cells. Deletions and insertions are also highly predictive of response [121]. The use of these mutant derived epitopes is based initially on the responses to checkpoint inhibitors, which are proportional to the mutational load of each tumour [152]. Neoantigens are presented by MHC on the cell surface in order to be recognised by the T lymphocytes of the immune system. TSAs are the best therapeutic targets for cancer vaccines and T-cell-based immunotherapy because they are different from the germline and are not considered proprietary by the immune system. In addition, they are not subject to central or peripheral tolerance, as normal cells do not express them, so they will not cause auto-immunity problems either [107]. To identify immunogenic neo-epitopes in each patient, a combination of genomic sequencing of the tumour, RNA sequencing and bioinformatic tools with algorithms that allow for the prediction of the mutations are required, which will be presented to the T cells based on the processing by the proteasome and the affinity of the molecules for human leukocyte antigen (HLA). The resulting sequences can be synthesized as mRNA or as peptides for use as a vaccine. This methodology has been validated in preclinical trials, demonstrating that mutanome-derived neoantigens can induce an immune response against autologous tumours [153]. There are also phase 1 trials showing the immunogenicity and viability of the vaccine against the neo-antigen in metastatic melanoma [154]. The disadvantage of this customized approach is that it is a lengthy process and is therefore only suitable for certain patients. Neo-antigens have already been identified in different types of cancer such as melanoma, lung cancer, liver and renal cancer [155]. Adoptive cell transfer (ACT) studies of autologous tumour infiltrating lymphocytes (TIL) have shown that an effective antitumour immune response occurs in the presence of tumour specific T cells [156]. Isolated T cell clones or TCR-designed T lymphocytes have demonstrated the epitope patterns of neoantigens that are recognised by T cells [157]. Increasingly, cancer vaccines are being designed based on neo-antigens, targeting immunogenic mutations unique to each patient. Customized RNA mutanome vaccines and peptide-based vaccines have been tested and found to be safe and capable of eliciting T cell responses to neo-epitopes in melanoma patients [154,158]. When neo-epitopes are presented by antigen-presenting cells (APCs), such as dendritic cells and tumour cells themselves, cross presentation—whereby antigen-presenting cells phagocytize exogenous antigens and process them for presentation by MHC-I—plays an important role [159]. For a sufficient response of T cells to a neo-epitope, it is important to consider the affinity of the TCR for its related antigen [142]. Because neo-antigens are small pieces of peptides that contain tumour mutations, immunization with these antigens requires the assistance of other immune-stimulatory agents to produce an efficient immune response. On their own, peptides as vaccines may not be able to stimulate the immune system in a potent way, so they are used in combination with adjuvants [160]. Generally, to activate cytotoxic T cells and obtain a potent immune response, the stimulation of T helper cells is also required [142]. Even peptides with epitopes capable of activating cytotoxic T cells and helper T cells need an adjuvant to obtain an effective vaccine, so containing a potent immune-stimulator is very important to obtain an effective response. Then, CD8+ T cells are induced [161]. The appropriate adjuvant must be able to induce the production of cytokines and co-stimulator molecules from APC and also be able to deliver the optimal amount of antigen, to maintain a balance between antigen persistence, antigen concentration and antigen distribution [162]. In addition, the adjuvant must enhance cell-mediated immunity polarized to type 1 [121]. Adjuvants can function in several ways: gradually releasing the antigen, stimulating pattern recognition receptors in APCs, protecting antigens from rapid degradation, and extending antigen presentation time [142]. Different types of cells with neo-epitopes have also been pressed for immunization, such as B cells, macrophages, splenocytes or dendritic cells, which serve as delivery and adjuvant systems [142]. Since dendritic cells are capable of efficiently capturing, processing and presenting the antigen, initiating the immune response, they are also considered natural adjuvants, but the number of dendritic cells presented in peripheral blood in cancer patients is very low, in addition, this DCs may not be functional due to the effect of TME, so one of the goals is to provide enough functional DCs for each patient. It is also important to determine the DC subtype that works best as an adjuvant, the number of DCs injected, their stage of maturation or the location of the injection [163].
- The identification of neo-epitopes is the most specific approach to cancer treatment, since it allows for a targeted immune response against specific tumour epitopes, but with this approach, no clinically determinant results have been achieved, since these strategies are conditioned by the TME, T-cell depletion, regulation of the immune checkpoint, tumour heterogeneity, etc. For this reason, it is necessary to find an ideal combination of neo-epitope vaccines, chemotherapy, radiotherapy, checkpoint blocking therapies, etc., specific to each patient [142].
3.3.5. Checkpoints Inhibitors
3.4. Limitations of Immunotherapy
4. Nanomedicine and Immunotherapy: Synergy Combination
4.1. Passive Immune Nanomedicine
4.2. Active Immune Nanomedicine
4.2.1. NP Conjugates
4.2.2. Exosomes
4.2.3. Artificial Antigen Presentation Cells (aAPC)
4.2.4. Iron Oxide NPs (IOPNs)
4.3. NP Biosafety: A Critical Aspect in Nanomedicine and Immunotherapy
4.4. Immunogenic Cell Death: A Merge Point of Nanomedicine and Immunotherapy
- Pathogen-induced ICD: It is defined as a defence mechanism against pathogens, such as obligatory intracellular bacteria and viruses. After infection, the cells detect MAMPs through specific PRRs, which will send danger signals to neighbouring cells. Intracellular hazard signalling is generated, which will activate autophagy, and microenvironmental hazard signalling, which will induce the secretion of pro-inflammatory cytokines, including TNF and type I interferons. The adaptive immune response is activated when infected cells die and their bodies are internalized in APC, which will present the various non-self-antigenic epitopes in MHC molecules, thus activating CD8+ and CD4+ T cells [251].
- ICD caused by chemotherapy and/or targeted onco-therapy: Exposure to certain chemotherapeutic agents used in the clinic has been shown to produce ICD in mouse tumour cells [257]. This ICD is based on eIF2A phosphorylation-dependent exposure of endoplasmic reticulum chaperones in the membrane of these tumour cells. Processes such as autophagy-mediated ATP secretion, IFN type I activation, secretion of chemokine ligands such as CXCL10, etc. are also involved. These processes also occur in human tumour cells after chemotherapy. Chemotherapeutic agents that are unable to promote the release of DAMPs do not produce ICD. The degree of antigenicity among tumour cells is very heterogeneous, which may condition the activation of adaptive immunity after ICD. However, the lower level of mutational load associated with oncogenesis has been found to be sufficient to activate this immunogenicity, which is due to the fact that tumour cells express neoantigens that are different from their own and are therefore not subject to tolerance [251,258].
- ICD activated by physical signals: There are three physical interventions that trigger ICDs: irradiation, hypericin-based photodynamic therapy (PDT), and high hydrostatic pressure [251,258]. DCs loaded with irradiated tumour cells have been shown to produce strong immune responses in mice and cancer patients [259]. ICD due to hypericin-based PDT or high hydrostatic pressure shows exposure of ER chaperones on the plasma membrane, ATP secretion, and high-mobility group box 1 (HMGB1) [260,261]. DCs exposed to cells suffering from these two types of ICDs induce positive regulation of DC-activation markers and pro-inflammatory cytokine secretion, resulting in priming of tumour-specific CD8+ T cells [251].
- Necroptotic ICD: This is a form of programmed cell death, initiated by phosphorylation catalyzed by the serine/threonine kinase 3 (RIPK3) protein, which activates the pseudokinase mixed lineage kinase domain-like (MLKL) receptor, which forms oligomers that produce irreversible plasma membrane permeation [262]. Necroptosis is highly pro-inflammatory and is also capable of activating the adaptive immune system, generating a specific antigen response [251]. This has been demonstrated in studies with the mouse cell lines TC-1 and EL4 of lung carcinoma and CT26 of colorectal carcinoma, which were exposed to necroptosis inducers [263,264].
5. Biomarkers in Onco-Immunotherapy
5.1. Critical Role of Predictive Biomarkers in Oncology
5.2. Potential Biomarkers in Immuno-Oncology
5.2.1. Serum-Soluble Biomarkers
5.2.2. Cellular Biomarkers
5.2.3. Specific Tumour Antibodies
5.2.4. Tumoral Microenvironment as Biomarker
5.2.5. Immunocheckpoints (ICs) as Biomarkers
5.2.6. Tumoral Genomics Biomarkers
5.3. Tumour Mutational Burden as Biomarkers
5.4. TCR diversity and MHC Molecules
5.5. Neoantigens as Biomarkers
5.6. Microsatellite-Unstable Tumours
5.7. Immunopeptidome as Biomarkers
5.8. TAAs as Biomarkers
5.9. Techniques for Biomarkers Discovery, Verification and Validation
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lechuga, L. Nanomedicina: Aplicación de la Nanotecnología en la Salud; Digital.csic.es.: Madrid, Spain, 2011. [Google Scholar]
- Auría-Soro, C.; Nesma, T.; Juanes-Velasco, P.; Landeira-Viñuela, A.; Fidalgo-Gomez, H.; Acebes-Fernandez, V.; Gongora, R.; Parra, M.J.A.; Manzano-Roman, R.; Fuentes, M. Interactions of nanoparticles and biosystems: Microenvironment of nanoparticles and biomolecules in nanomedicine. Nanomaterials 2019, 9, 1365. [Google Scholar] [CrossRef]
- Arab, D.; Pourafshary, P. Nanoparticles-assisted surface charge modification of the porous medium to treat colloidal particles migration induced by low salinity water flooding. Colloids Surf. A Physicochem. Eng. Asp. 2013, 436, 803–814. [Google Scholar] [CrossRef]
- Hashemi, R.; Nassar, N.N.; Pereira Almao, P. Nanoparticle technology for heavy oil in-situ upgrading and recovery enhancement: Opportunities and challenges. Appl. Energy 2014, 133, 374–387. [Google Scholar] [CrossRef]
- Mohammadizadeh, M.; Pourabbas, B.; Mahmoodian, M.; Foroutani, K.; Fallahian, M. Facile and rapid production of conductive flexible films by deposition of polythiophene nanoparticles on transparent poly(ethyleneterephthalate): Electrical and morphological properties. Mater. Sci. Semicond. Process. 2014, 20, 74–83. [Google Scholar] [CrossRef]
- Doyle, D.J. Nanotechnology in Anesthesia and Medicine. Adv. Anesth. 2013, 31, 181–200. [Google Scholar] [CrossRef]
- Pelaz, B.; Alexiou, C.; Alvarez-Puebla, R.A.; Alves, F.; Andrews, A.M.; Ashraf, S.; Balogh, L.P.; Ballerini, L.; Bestetti, A.; Brendel, C.; et al. Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef]
- Zhang, Q.; Mochalin, V.N.; Neitzel, I.; Hazeli, K.; Niu, J.; Kontsos, A.; Zhou, J.G.; Lelkes, P.I.; Gogotsi, Y. Mechanical properties and biomineralization of multifunctional nanodiamond-PLLA composites for bone tissue engineering. Biomaterials 2012, 33, 5067–5075. [Google Scholar] [CrossRef]
- Bao, C.; Conde, J.; Polo, E.; del Pino, P.; Moros, M.; Baptista, P.V.; Grazu, V.; Cui, D.; de la Fuente, J.M. A promising road with challenges: Where are gold nanoparticles in translational research? Nanomedicine 2014, 9, 2353–2370. [Google Scholar] [CrossRef]
- Zhou, S.-A.; Brahme, A. Development of phase-contrast X-ray imaging techniques and potential medical applications. Phys. Med. 2008, 24, 129–148. [Google Scholar] [CrossRef]
- Leuvering, J.H.W.; Thal, P.J.H.M.; Van der Waart, M.; Schuurs, A.H.W.M. A sol particle agglutination assay for human chorionic gonadotrophin. J. Immunol. Methods 1981, 45, 183–194. [Google Scholar] [CrossRef]
- Xu, X.; Daniel, W.L.; Wei, W.; Mirkin, C.A. Colorimetric Cu 2+ Detection Using DNA-Modified Gold-Nanoparticle Aggregates as Probes and Click Chemistry. Small 2010, 6, 623–626. [Google Scholar] [CrossRef]
- Ambrosi, A.; Airò, F.; Merkoçi, A. Enhanced Gold Nanoparticle Based ELISA for a Breast Cancer Biomarker. Anal. Chem. 2010, 82, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.; Avadi, M.R.; Attar, F.; Dashtestani, F.; Ghorchian, H.; Rezayat, S.M.; Saboury, A.A.; Falahati, M. Cancer diagnosis using nanomaterials based electrochemical nanobiosensors. Biosens. Bioelectron. 2019, 126, 773–784. [Google Scholar] [CrossRef]
- Meng, Z.; Song, R.; Chen, Y.; Zhu, Y.; Tian, Y.; Li, D.; Cui, D. Rapid screening and identification of dominant B cell epitopes of HBV surface antigen by quantum dot-based fluorescence polarization assay. Nanoscale Res. Lett. 2013, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Koo, Y.-E.L.; Cao, Y.; Kopelman, R.; Koo, S.M.; Brasuel, M.; Philbert, M.A. Real-Time Measurements of Dissolved Oxygen Inside Live Cells by Organically Modified Silicate Fluorescent Nanosensors. Anal. Chem. 2004, 76, 2498–2505. [Google Scholar] [CrossRef]
- Fernandes, C.; Suares, D.; Yergeri, M.C. Tumor Microenvironment Targeted Nanotherapy. Front. Pharmacol. 2018, 9, 1230. [Google Scholar] [CrossRef]
- Tay, C.Y.; Cai, P.; Setyawati, M.I.; Fang, W.; Tan, L.P.; Hong, C.H.L.; Chen, X.; Leong, D.T. Nanoparticles Strengthen Intracellular Tension and Retard Cellular Migration. Nano Lett. 2014, 14, 83–88. [Google Scholar] [CrossRef]
- Stirland, D.L.; Nichols, J.W.; Miura, S.; Bae, Y.H. Mind the gap: A survey of how cancer drug carriers are susceptible to the gap between research and practice. J. Control. Release 2013, 172, 1045–1064. [Google Scholar] [CrossRef]
- MaHam, A.; Tang, Z.; Wu, H.; Wang, J.; Lin, Y. Protein-Based Nanomedicine Platforms for Drug Delivery. Small 2009, 5, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Díez, P.; González-Muñoz, M.; González-González, M.; Dégano, R.M.; Jara-Acevedo, R.; Sánchez-Paradinas, S.; Piñol, R.; Murillo, J.L.; Lou, G.; Palacio, F.; et al. Functional insights into the cellular response triggered by a bile-acid platinum compound conjugated to biocompatible ferric nanoparticles using quantitative proteomic approaches. Nanoscale 2017, 9, 9960–9972. [Google Scholar] [CrossRef] [PubMed]
- Maleki Dizaj, S.; Barzegar-Jalali, M.; Zarrintan, M.H.; Adibkia, K.; Lotfipour, F. Calcium carbonate nanoparticles as cancer drug delivery system. Expert Opin. Drug Deliv. 2015, 12, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Magherini, F.; Fiaschi, T.; Valocchia, E.; Becatti, M.; Pratesi, A.; Marzo, T.; Massai, L.; Gabbiani, C.; Landini, I.; Nobili, S.; et al. Antiproliferative effects of two gold(I)-N-heterocyclic carbene complexes in A2780 human ovarian cancer cells: A comparative proteomic study. Oncotarget 2018, 9, 28042–28068. [Google Scholar] [CrossRef]
- Centi, S.; Tatini, F.; Ratto, F.; Gnerucci, A.; Mercatelli, R.; Romano, G.; Landini, I.; Nobili, S.; Ravalli, A.; Marrazza, G.; et al. In vitro assessment of antibody-conjugated gold nanorods for systemic injections. J. Nanobiotechnol. 2014, 12, 1–10. [Google Scholar] [CrossRef]
- Marzo, T.; Massai, L.; Pratesi, A.; Stefanini, M.; Cirri, D.; Magherini, F.; Becatti, M.; Landini, I.; Nobili, S.; Mini, E.; et al. Replacement of the Thiosugar of Auranofin with Iodide Enhances the Anticancer Potency in a Mouse Model of Ovarian Cancer. ACS Med. Chem. Lett. 2019, 10, 656–660. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, L.; Wang, J.; Feng, Q.; Liu, D.; Yin, Q.; Xu, D.; Wei, Y.; Ding, B.; Shi, X.; et al. Tunable Rigidity of (Polymeric Core)-(Lipid Shell) Nanoparticles for Regulated Cellular Uptake. Adv. Mater. 2015, 27, 1402–1407. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 80 2010, 328, 1031–1035. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; Li, H.; Huang, N.; Jin, Q.; Ren, K.; Ji, J. Enhanced Retention and Cellular Uptake of Nanoparticles in Tumors by Controlling Their Aggregation Behavior. ACS Nano 2013, 7, 6244–6257. [Google Scholar] [CrossRef]
- Dai, W.; Yang, T.; Wang, Y.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Peptide PHSCNK as an integrin α5β1 antagonist targets stealth liposomes to integrin-overexpressing melanoma. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Skirtach, A.G.; Muñoz Javier, A.; Kreft, O.; Köhler, K.; Piera Alberola, A.; Möhwald, H.; Parak, W.J.; Sukhorukov, G.B. Laser-Induced Release of Encapsulated Materials inside Living Cells. Angew. Chem. Int. Ed. 2006, 45, 4612–4617. [Google Scholar] [CrossRef] [PubMed]
- Ochs, M.; Carregal-Romero, S.; Rejman, J.; Braeckmans, K.; De Smedt, S.C.; Parak, W.J. Light-Addressable Capsules as Caged Compound Matrix for Controlled Triggering of Cytosolic Reactions. Angew. Chem. Int. Ed. 2013, 52, 695–699. [Google Scholar] [CrossRef]
- Kim, Y.; Zhu, J.; Yeom, B.; Di Prima, M.; Su, X.; Kim, J.-G.; Yoo, S.J.; Uher, C.; Kotov, N.A. Stretchable nanoparticle conductors with self-organized conductive pathways. Nature 2013, 500, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Lovat, V.; Pantarotto, D.; Lagostena, L.; Cacciari, B.; Grandolfo, M.; Righi, M.; Spalluto, G.; Prato, M.; Ballerini, L. Carbon Nanotube Substrates Boost Neuronal Electrical Signaling. Nano Lett. 2005, 5, 1107–1110. [Google Scholar] [CrossRef]
- Mattson, M.P.; Haddon, R.C.; Rao, A.M. Molecular Functionalization of Carbon Nanotubes and Use as Substrates for Neuronal Growth. J. Mol. Neurosci. 2000, 14, 175–182. [Google Scholar] [CrossRef]
- Shin, S.R.; Jung, S.M.; Zalabany, M.; Kim, K.; Zorlutuna, P.; Kim, S.; Nikkhah, M.; Khabiry, M.; Azize, M.; Kong, J.; et al. Carbon-Nanotube-Embedded Hydrogel Sheets for Engineering Cardiac Constructs and Bioactuators. ACS Nano 2013, 7, 2369–2380. [Google Scholar] [CrossRef]
- Lee, J.; Kotov, N.A. Notch Ligand Presenting Acellular 3D Microenvironments for ex vivo Human Hematopoietic Stem-Cell Culture made by Layer-By-Layer Assembly. Small 2009, 5, 1008–1013. [Google Scholar] [CrossRef]
- Torchilin, V.P. Micellar Nanocarriers: Pharmaceutical Perspectives. Pharm. Res. 2006, 24, 1–16. [Google Scholar] [CrossRef]
- Lee, Y.; Fukushima, S.; Bae, Y.; Hiki, S.; Ishii, T.; Kataoka, K. A Protein Nanocarrier from Charge-Conversion Polymer in Response to Endosomal pH. J. Am. Chem. Soc. 2007, 129, 5362–5363. [Google Scholar] [CrossRef]
- Colombo, M.; Carregal-Romero, S.; Casula, M.F.; Gutiérrez, L.; Morales, M.P.; Böhm, I.B.; Heverhagen, J.T.; Prosperi, D.; Parak, W.J. Biological applications of magnetic nanoparticles. Chem. Soc. Rev. 2012, 41, 4306. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.H. Carbon Nanotubes--the Route Toward Applications. Science 80 2002, 297, 787–792. [Google Scholar] [CrossRef]
- Yu, S.-J.; Kang, M.-W.; Chang, H.-C.; Chen, K.-M.; Yu, Y.-C. Bright Fluorescent Nanodiamonds: No Photobleaching and Low Cytotoxicity. J. Am. Chem. Soc. 2005, 127, 17604–17605. [Google Scholar] [CrossRef] [PubMed]
- Guo, P. The emerging field of RNA nanotechnology. Nat. Nanotechnol. 2010, 5, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.E.; Tyner, K.M.; Dair, B.J.; Deschamps, J.R.; Medintz, I.L. Analyzing Nanomaterial Bioconjugates: A Review of Current and Emerging Purification and Characterization Techniques. Anal. Chem. 2011, 83, 4453–4488. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yang, D.; Fang, Y.; Lin, X.; Jin, X.; Wang, Q.; Wang, X.; Ke, L.; Shi, K. Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics 2019, 9, 126–151. [Google Scholar] [CrossRef]
- Decuzzi, P.; Godin, B.; Tanaka, T.; Lee, S.Y.; Chiappini, C.; Liu, X.; Ferrari, M. Size and shape effects in the biodistribution of intravascularly injected particles. J. Control. Release 2010, 141, 320–327. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Heo, M.B.; Lim, Y.T. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials 2014, 35, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Xu, J.; Xu, L.; Sun, X.; Chen, Q.; Zhao, Y.; Peng, R.; Liu, Z. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano 2018, 12, 5121–5129. [Google Scholar] [CrossRef]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific t cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Chan, C.; Guo, N.; Han, W.; Weichselbaum, R.R.; Lin, W. Photodynamic Therapy Mediated by Nontoxic Core-Shell Nanoparticles Synergizes with Immune Checkpoint Blockade To Elicit Antitumor Immunity and Antimetastatic Effect on Breast Cancer. J. Am. Chem. Soc. 2016, 138, 16686–16695. [Google Scholar] [CrossRef]
- Liu, X.; Gao, X.; Zheng, S.; Wang, B.; Li, Y.; Zhao, C.; Muftuoglu, Y.; Chen, S.; Li, Y.; Yao, H.; et al. Modified nanoparticle mediated IL-12 immunogene therapy for colon cancer. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhao, X.; Du, B.; Li, X.; Liu, S.; Yang, X.Y.; Ding, H.; Yang, W.; Pan, F.; Wu, X.; et al. Gold Nanoparticle-Mediated Targeted Delivery of Recombinant Human Endostatin Normalizes Tumour Vasculature and Improves Cancer Therapy. Sci. Rep. 2016, 6, 30619. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Holay, M.; Park, J.H.; Fang, R.H.; Zhang, J.; Zhang, L. Nanoparticle delivery of immunostimulatory agents for cancer immunotherapy. Theranostics 2019, 9, 7826–7848. [Google Scholar] [CrossRef]
- Chen, W.L.; Liu, S.J.; Leng, C.H.; Chen, H.W.; Chong, P.; Huang, M.H. Disintegration and cancer immunotherapy efficacy of a squalane-in-water delivery system emulsified by bioresorbable poly(ethylene glycol)-block-polylactide. Biomaterials 2014, 35, 1686–1695. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Li, N.; Suh, H.; Irvine, D.J. Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Rahimian, S.; Fransen, M.F.; Kleinovink, J.W.; Amidi, M.; Ossendorp, F.; Hennink, W.E. Polymeric microparticles for sustained and local delivery of antiCD40 and antiCTLA-4 in immunotherapy of cancer. Biomaterials 2015, 61, 33–40. [Google Scholar] [CrossRef]
- Akiyoshi, K.; Deguchi, S.; Moriguchi, N.; Yamaguchi, S.; Sunamoto, J. Self-Aggregates of Hydrophobized Polysaccharides in Water. Formation and Characteristics of Nanoparticles. Macromolecules 1993, 26, 3062–3068. [Google Scholar] [CrossRef]
- Muraoka, D.; Harada, N.; Hayashi, T.; Tahara, Y.; Momose, F.; Sawada, S.I.; Mukai, S.A.; Akiyoshi, K.; Shiku, H. Nanogel-based immunologically stealth vaccine targets macrophages in the medulla of lymph node and induces potent antitumor immunity. ACS Nano 2014, 8, 9209–9218. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Luo, Z.; Liu, P.; Gao, N.; Zhang, Y.; Pan, H.; Liu, L.; Wang, C.; Cai, L.; Ma, Y. Bioreducible alginate-poly(ethylenimine) nanogels as an antigen-delivery system robustly enhance vaccine-elicited humoral and cellular immune responses. J. Control. Release 2013, 168, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kishida, T.; Hasegawa, U.; Ueda, Y.; Imanishi, J.; Yamagishi, H.; Akiyoshi, K.; Otsuji, E.; Mazda, O. Nanogel DDS enables sustained release of IL-12 for tumor immunotherapy. Biochem. Biophys. Res. Commun. 2008, 367, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Curnis, F.; Fiocchi, M.; Sacchi, A.; Gori, A.; Gasparri, A.; Corti, A. NGR-tagged nano-gold: A new CD13-selective carrier for cytokine delivery to tumors. Nano Res. 2016, 9, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Meir, R.; Shamalov, K.; Sadan, T.; Motiei, M.; Yaari, G.; Cohen, C.J.; Popovtzer, R. Fast Image-Guided Stratification Using Anti-Programmed Death Ligand 1 Gold Nanoparticles for Cancer Immunotherapy. ACS Nano 2017, 11, 11127–11134. [Google Scholar] [CrossRef]
- Lu, J.; Liu, X.; Liao, Y.P.; Salazar, F.; Sun, B.; Jiang, W.; Chang, C.H.; Jiang, J.; Wang, X.; Wu, A.M.; et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Ding, B.; Shao, S.; Yu, C.; Teng, B.; Wang, M.; Cheng, Z.; Wong, K.L.; Ma, P.; Lin, J. Large-Pore Mesoporous-Silica-Coated Upconversion Nanoparticles as Multifunctional Immunoadjuvants with Ultrahigh Photosensitizer and Antigen Loading Efficiency for Improved Cancer Photodynamic Immunotherapy. Adv. Mater. 2018, 30, 1802479. [Google Scholar] [CrossRef]
- Irache, J.M.; Salman, H.H.; Gamazo, C.; Espuelas, S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin. Drug Deliv. 2008, 5, 703–724. [Google Scholar] [CrossRef]
- Wang, C.; Li, P.; Liu, L.; Pan, H.; Li, H.; Cai, L.; Ma, Y. Self-adjuvanted nanovaccine for cancer immunotherapy: Role of lysosomal rupture-induced ROS in MHC class I antigen presentation. Biomaterials 2016, 79, 88–100. [Google Scholar] [CrossRef]
- Shukla, S.; Jandzinski, M.; Wang, C.; Gong, X.; Bonk, K.W.; Keri, R.A.; Steinmetz, N.F. A Viral Nanoparticle Cancer Vaccine Delays Tumor Progression and Prolongs Survival in a HER2 + Tumor Mouse Model. Adv. Ther. 2019, 2, 1800139. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Ahn, B.; Oehlke, J.; Kivett, V.; Fedoroff, A.; Butowski, N.; Chang, S.M.; Clarke, J.; Berger, M.S.; et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res. 2013, 19, 205–214. [Google Scholar] [CrossRef]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–498. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Christian, D.A.; Hunter, C.A. Particle-mediated delivery of cytokines for immunotherapy. Immunotherapy 2012, 4, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Cytokines in cancer immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.M.; Patterson, A.; Johnson, C.B.; Neitzke, D.J.; Mehrotra, S.; Denlinger, C.E.; Paulos, C.M.; Li, Z.; Cole, D.J.; Rubinstein, M.P. IL-2 and beyond in Cancer Immunotherapy. J. Interf. Cytokine Res. 2018, 38, 45–68. [Google Scholar] [CrossRef]
- Ratain, M.J.; Golomb, H.M.; Vardiman, J.W.; Vokes, E.E.; Jacobs, R.H.; Daly, K. Treatment of Hairy Cell Leukemia With Recombinant Alpha2 Interferon. Blood 1985, 65, 644–648. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interf. Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Meeker, T.C.; Lowder, J.; Maloney, D.G.; Miller, R.A.; Thielemans, K.; Warnke, R.; Levy, R. A clinical trial of anti-idiotype therapy for B cell malignancy. Blood 1985, 65, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.L.; French, R.R.; Illidge, T.M.; Honeychurch, J.; McBride, H.M.; Penfold, C.A.; Fearon, D.T.; Parkhouse, R.M.E.; Klaus, G.G.B.; Glennie, M.J. Monoclonal antibody therapy of B cell lymphoma: Signaling activity on tumor cells appears more important than recruitment of effectors. J. Immunol. 1998, 161, 3176–3185. [Google Scholar]
- Clynes, R.; Takechi, Y.; Moroi, Y.; Houghton, A.; Ravetch, J.V. Fc receptors are required in passive and active immunity to melanoma. Proc. Natl. Acad. Sci. USA 1998, 95, 652–656. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; De Vos, A.M.; Sliwkowski, M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Beurskens, F.J.; Beum, P.V.; Lindorfer, M.A.; van de Winkel, J.G.J.; Parren, P.W.H.I.; Taylor, R.P. Binding of Submaximal C1q Promotes Complement-Dependent Cytotoxicity (CDC) of B Cells Opsonized with Anti-CD20 mAbs Ofatumumab (OFA) or Rituximab (RTX): Considerably Higher Levels of CDC Are Induced by OFA than by RTX. J. Immunol. 2009, 183, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, M.L.; Krause, S.W.; Salcedo, M.; Nardin, A. Ex vivo-activated human macrophages kill chronic lymphocytic leukemia cells in the presence of rituximab: Mechanism of antibody-dependent cellular cytotoxicity and impact of human serum. J. Immunother. 2006, 29, 388–397. [Google Scholar] [CrossRef]
- Dall’ozzo, S.; Tartas, S.; Paintaud, G.; Cartron, G.; Colombat, P.; Bardos, P.; Watier, H.; Thibault, G. Rituximab-Dependent Cytotoxicity by Natural Killer Cells: Influence of FCGR3A Polymorphism on the Concentration-Effect Relationship. Cancer Res. 2004, 64, 4664–4669. [Google Scholar]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; McDonald, D.M. Controlling escape from angiogenesis inhibitors. Nat. Rev. Cancer 2012, 12, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.A.; Assadipour, Y.; Kriley, I.; Goff, S.L.; Rosenberg, S.A. Adoptive Cell Therapy—Tumor-Infiltrating Lymphocytes, T-Cell Receptors, and Chimeric Antigen Receptors. Semin. Oncol. 2015, 42, 626–639. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 80 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.T.; Carpenter, R.O.; Maryalice, S.S.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Doan, A.; Pulsipher, M.A. Hypogammaglobulinemia due to CAR T-cell therapy. Pediatr. Blood Cancer 2018, 65, e26914. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines. Past, present, and future. In Advances in Cancer Research; Academic Press Inc.: Cambridge, MA, USA, 2013; Volume 119, pp. 421–475. [Google Scholar]
- Pan, R.-Y.; Chung, W.-H.; Chu, M.-T.; Chen, S.-J.; Chen, H.-C.; Zheng, L.; Hung, S.-I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.M.; Buqué, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef]
- Berger, M.; Kreutz, F.T.; Horst, J.L.; Baldi, A.C.; Koff, W.J. Phase I Study with an Autologous Tumor Cell Vaccine for Locally Advanced or Metastatic Prostate Cancer. J. Pharm. Pharm. Sci. 2007, 10, 144–152. [Google Scholar]
- Hanna, M.G.; Hoover, H.C.; Vermorken, J.B.; Harris, J.E.; Pinedo, H.M. Adjuvant active specific immunotherapy of stage II and stage III colon cancer with an autologous tumor cell vaccine: First randomized phase III trials show promise. In Proceedings of the Vaccine; Elsevier: Amsterdam, The Netherlands, 2001; Volume 19, pp. 2576–2582. [Google Scholar]
- Rüttinger, D.; van den Engel, N.K.; Winter, H.; Schlemmer, M.; Pohla, H.; Grützner, S.; Wagner, B.; Schendel, D.J.; Fox, B.A.; Jauch, K.W.; et al. Adjuvant therapeutic vaccination in patients with non-small cell lung cancer made lymphopenic and reconstituted with autologous PBMC: First clinical experience and evidence of an immune response. J. Transl. Med. 2007, 5, 43. [Google Scholar] [CrossRef]
- Berd, D.; Maguire, H.C.; McCue, P.; Mastrangelo, M.J. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: Clinical and immunologic results in 64 patients. J. Clin. Oncol. 1990, 8, 1858–1867. [Google Scholar] [CrossRef]
- Antonia, S.J.; Seigne, J.; Diaz, J.; Muro-Cacho, C.; Extermann, M.; Farmelo, M.J.; Friberg, M.; Alsarraj, M.; Mahany, J.J.; Pow-Sang, J.; et al. Phase I trial of a B7-1 (CD80) gene modified autologous tumor cell vaccine in combination with systemic interleukin-2 in patients with metastatic renal cell carcinoma. J. Urol. 2002, 167, 1995–2000. [Google Scholar] [CrossRef]
- Butterfield, L.H. Cancer vaccines. BMJ 2015, 350, h988. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.L.; Foshag, L.J.; Hoon, D.S.B.; Nizze, J.A.; Wanek, L.A.; Chang, C.; Davtyan, D.G.; Gupta, R.K.; Elashoff, R.; Irie, R.F.; et al. Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma vaccine. In Proceedings of the Annals of Surgery; Lippincott, Williams, and Wilkins: Philadelphia, PA, USA, 1992; Volume 216, pp. 463–482. [Google Scholar]
- Cheever, M.A.; Higano, C.S. PROVENGE (sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G. Immunotherapy with autologous tumor cell vaccines for treatment of occult disease in early stage colon cancer. Hum. Vaccines Immunother. 2012, 8, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Geary, S.M.; Salem, A.K. Prostate cancer vaccines update on clinical development. Oncoimmunology 2013, 2, e24523. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [Google Scholar] [CrossRef]
- Vermaelen, K. Vaccine strategies to improve anticancer cellular immune responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef]
- Van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; De Vries, I.J.M.; Bol, K.F. Dendritic cell cancer therapy: Vaccinating the right patient at the right time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef]
- Inba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef]
- Murphy, G.P.; Tjoa, B.; Ragde, H.; Kenny, G.; Boynton, A. Phase I clinical trial: T-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate 1996, 29, 371–380. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with Sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef]
- Romano, E.; Rossi, M.; Ratzinger, G.; De Cos, M.A.; Chung, D.J.; Panageas, K.S.; Wolchock, J.D.; Houghton, A.N.; Chapman, P.B.; Heller, G.; et al. Peptide-loaded langerhans cells, despite increased IL15 secretion and T-cell activation in vitro, elicit antitumor T-cell responses comparable to peptide-loaded monocyte-derived dendritic cells in vivo. Clin. Cancer Res. 2011, 17, 1984–1997. [Google Scholar] [CrossRef]
- Thurner, B.; Haendle, I.; Röder, C.; Dieckmann, D.; Keikavoussi, P.; Jonuleit, H.; Bender, A.; Maczek, C.; Schreiner, D.; Von Den Driesch, P.; et al. Vaccination with Mage-3A1 peptide-pulsed nature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J. Exp. Med. 1999, 190, 1669–1678. [Google Scholar] [CrossRef]
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004, 64, 4973–4979. [Google Scholar] [CrossRef]
- Höltl, L.; Rieser, C.; Papesh, C.; Ramoner, R.; Herold, M.; Klocker, H.; Radmayr, C.; Stenzl, A.; Bartsch, G.; Thurnher, M. Cellular and humoral immune responses in patients with metastatic renal cell carcinoma after vaccination with antigen pulsed dendritic cells. J. Urol. 1999, 161, 777–782. [Google Scholar] [CrossRef]
- di Pietro, A.; Tosti, G.; Ferrucci, P.F.; Testori, A. Oncophage: Step to the future for vaccine therapy in melanoma. Expert Opin. Biol. Ther. 2008, 8, 1973–1984. [Google Scholar] [CrossRef]
- Xia, W.; Wang, J.; Xu, Y.; Jiang, F.; Xu, L. L-BLP25 as a peptide vaccine therapy in non-small cell lung cancer: A review. J. Thorac. Dis. 2014, 6, 1513–1520. [Google Scholar]
- Mocellin, S.; Pilati, P.; Nitti, D. Peptide-Based Anticancer Vaccines: Recent Advances and Future Perspectives. Curr. Med. Chem. 2009, 16, 4779–4796. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today 1992, 13, 11–16. [Google Scholar] [CrossRef]
- FLEMING, T.P.; WATSON, M.A. Mammaglobin, a Breast-Specific Gene, and Its Utility as a Marker for Breast Cancer. Ann. N. Y. Acad. Sci. 2006, 923, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.E.; Brockstedt, D.; Leong, M.; Lauer, P.; Theisen, E.; Sauer, J.D.; McNeel, D.G. Heterologous vaccination targeting prostatic acid phosphatase (PAP) using DNA and Listeria vaccines elicits superior anti-tumor immunity dependent on CD4+ T cells elicited by DNA priming. Oncoimmunology 2018, 7, e1456603. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef]
- Ghanem, G.; Fabrice, J. Tyrosinase related protein 1 (TYRP1/gp75) in human cutaneous melanoma. Mol. Oncol. 2011, 5, 150–155. [Google Scholar] [CrossRef]
- Osada, T.; Morse, M.A.; Hobeika, A.; Lyerly Kim, H. Novel recombinant alphaviral and adenoviral vectors for cancer immunotherapy. Semin. Oncol. 2012, 39, 305–310. [Google Scholar] [CrossRef]
- Lee, S.H.; Danishmalik, S.N.; Sin, J.I. DNA vaccines, electroporation and their applications in cancer treatment. Hum. Vaccines Immunother. 2015, 11, 1889–1900. [Google Scholar] [CrossRef]
- Brennick, C.A.; George, M.M.; Corwin, W.L.; Srivastava, P.K.; Ebrahimi-Nik, H. Neoepitopes as cancer immunotherapy targets: Key challenges and opportunities. Immunotherapy 2017, 9, 361–371. [Google Scholar] [CrossRef]
- Srivastava, P.K. Peptide-binding heat shock proteins in the endoplasmic reticulum: Role in immune response to cancer and in antigen presentation. Adv. Cancer Res. 1993, 62, 153–177. [Google Scholar] [CrossRef]
- Keenan, B.P.; Jaffee, E.M. Whole cell vaccines—Past progress and future strategies. Semin. Oncol. 2012, 39, 276–286. [Google Scholar] [CrossRef]
- Chiang, C.L.L.; Coukos, G.; Kandalaft, L.E. Whole tumor antigen vaccines: Where are we? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.; Madan, R.; Pachynski, R.; Mulders, P.; Sheikh, N.; Trager, J.; CG, D. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. JNCI J. Natl. Cancer Inst. 2017, 109, djw261. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Harris, D.T.; Kranz, D.M. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: Impact on efficacy and toxicity. Curr. Opin. Immunol. 2015, 33, 16–22. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first CAR T therapy. Nat. Rev. Drug Discov. 2017, 16, 669. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a Causal Association Between Human Papillomavirus and a Subset of Head and Neck Cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.G.; Revskaya, E.; Bryan, R.A.; Strickler, H.D.; Burk, R.D.; Casadevall, A.; Dadachova, E. Treating Cancer as an Infectious Disease—Viral Antigens as Novel Targets for Treatment and Potential Prevention of Tumors of Viral Etiology. PLoS ONE 2007, 2, e1114. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 80 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Kreiter, S.; Castle, J.C.; Türeci, Ö.; Sahin, U. Targeting the tumor mutanome for personalized vaccination therapy. Oncoimmunology 2012, 1, 768–769. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 80 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Strønen, E.; Toebes, M.; Kelderman, S.; Van Buuren, M.M.; Yang, W.; Van Rooij, N.; Donia, M.; Böschen, M.L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 80 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Kovacsovics-Bankowski, M.; Rock, K.L. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 80 1995, 267, 243–246. [Google Scholar] [CrossRef]
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. In Proceedings of the Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989; Volume 54, pp. 1–13. [Google Scholar]
- Apetoh, L.; Smyth, M.J.; Drake, C.G.; Abastado, J.P.; Apte, R.N.; Ayyoub, M.; Blay, J.Y.; Bonneville, M.; Butterfield, L.H.; Caignard, A.; et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology 2015, 4, e998538. [Google Scholar] [CrossRef]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef]
- Sabado, R.L.; Bhardwaj, N. Cancer immunotherapy: Dendritic-cell vaccines on the move. Nature 2015, 519, 300–301. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Duggan, S. Cemiplimab: First Global Approval. Drugs 2018, 78, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular interactions of antibody drugs targeting PD-1, PD-L1, and CTLA-4 in immuno-oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef]
- Carlino, M.S.; Long, G.V. Ipilimumab combined with nivolumab: A standard of care for the treatment of advanced melanoma? Clin. Cancer Res. 2016, 22, 3992–3998. [Google Scholar] [CrossRef]
- Song, M.; Chen, X.; Wang, L.; Zhang, Y. Future of anti-PD-1/PD-L1 applications: Combinations with other therapeutic regimens. Chin. J. Cancer Res. 2018, 30, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Zamarin, D. Combination Immune Checkpoint Blockade Strategies to Maximize Immune Response in Gynecological Cancers. Curr. Oncol. Rep. 2018, 20, 94. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Cytokines in Cancer Immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Jeang, J.; Yang, A.; Wu, T.C.; Hung, C.F. DNA vaccine for cancer immunotherapy. Hum. Vaccines Immunother. 2014, 10, 3153–3164. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Webber, M.J.; Anderson, D.G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. [Google Scholar] [CrossRef]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef]
- Byun, D.J.; Wolchok, J.D.; Rosenberg, L.M.; Girotra, M. Cancer immunotherapy-immune checkpoint blockade and associated endocrinopathies. Nat. Rev. Endocrinol. 2017, 13, 195–207. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Milling, L.; Zhang, Y.; Irvine, D.J. Delivering safer immunotherapies for cancer. Adv. Drug Deliv. Rev. 2017, 114, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V.P.; Jiang, W.; Popovíc, Z.; Jain, R.K.; Bawendi, M.G.; Fukumura, D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 2426–2431. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhang, F.; Ni, Q.; Niu, G.; Chen, X. Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano 2017, 11, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bertoni, H.; Kozielski, K.L.; Rui, Y.; Lal, B.; Vaughan, H.; Wilson, D.R.; Mihelson, N.; Eberhart, C.G.; Laterra, J.; Green, J.J. Bioreducible Polymeric Nanoparticles Containing Multiplexed Cancer Stem Cell Regulating miRNAs Inhibit Glioblastoma Growth and Prolong Survival. Nano Lett. 2018, 18, 4086–4094. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, W.; Nam, J.; Moon, J.J.; Kim, B.Y.S. Immunomodulating Nanomedicine for Cancer Therapy. Nano Lett. 2018, 18, 6655–6659. [Google Scholar] [CrossRef]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Lambert, L.H.; Goebrecht, G.K.E.; De Leo, S.E.; O’Connor, R.S.; Nunez-Cruz, S.; De Li, T.; Yuan, J.; Milone, M.C.; Kam, L.C. Improving T Cell Expansion with a Soft Touch. Nano Lett. 2017, 17, 821–826. [Google Scholar] [CrossRef]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Wang, C.; Sun, W.; Ye, Y.; Hu, Q.; Bomba, H.N.; Gu, Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat. Biomed. Eng. 2017, 1, 1–10. [Google Scholar] [CrossRef]
- Hickey, J.W.; Vicente, F.P.; Howard, G.P.; Mao, H.Q.; Schneck, J.P. Biologically Inspired Design of Nanoparticle Artificial Antigen-Presenting Cells for Immunomodulation. Nano Lett. 2017, 17, 7045–7054. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.S.; Lin, Y.J.; Lee, R.; Lai, Y.H.; Cheng, H.W.; Hsieh, C.H.; Shyu, W.C.; Chen, S.Y. Combination of fucoidan-based magnetic nanoparticles and immunomodulators enhances tumour-localized immunotherapy. Nat. Nanotechnol. 2018, 13, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Jiang, W.; Von Roemeling, C.A.; Qie, Y.; Liu, X.; Chen, Y.; Wang, Y.; Wharen, R.E.; Yun, K.; Bu, G.; et al. Multivalent bi-specific nanobioconjugate engager for targeted cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L. Post isolation modification of exosomes for nanomedicine applications. Nanomedicine 2016, 11, 1745–1756. [Google Scholar] [CrossRef]
- Yu, B.; Zhang, X.; Li, X. Exosomes derived from mesenchymal stem cells. Int. J. Mol. Sci. 2014, 15, 4142–4157. [Google Scholar] [CrossRef]
- Zöller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; Wickline, S.A. A systematic approach to exosome-based translational nanomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Munich, S.; Sobo-Vujanovic, A.; Buchser, W.J.; Beer-Stolz, D.; Vujanovic, N.L. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology 2012, 1, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Saunderson, S.C.; Schuberth, P.C.; Dunn, A.C.; Miller, L.; Hock, B.D.; MacKay, P.A.; Koch, N.; Jack, R.W.; McLellan, A.D. Induction of Exosome Release in Primary B Cells Stimulated via CD40 and the IL-4 Receptor. J. Immunol. 2008, 180, 8146–8152. [Google Scholar] [CrossRef]
- Raimondo, F.; Morosi, L.; Chinello, C.; Magni, F.; Pitto, M. Advances in membranous vesicle and exosome proteomics improving biological understanding and biomarker discovery. Proteomics 2011, 11, 709–720. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef]
- Bunggulawa, E.J.; Wang, W.; Yin, T.; Wang, N.; Durkan, C.; Wang, Y.; Wang, G. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnol. 2018, 16, 1–13. [Google Scholar] [CrossRef]
- Mokarizadeh, A.; Delirezh, N.; Morshedi, A.; Mosayebi, G.; Farshid, A.-A.; Dalir-Naghadeh, B. Phenotypic modulation of auto-reactive cells by insertion of tolerogenic molecules via MSC-derived exosomes. Vet. Res. Forum Int. Q. J. 2012, 3, 257–25761. [Google Scholar]
- Yeo, R.W.Y.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef]
- Mokarizadeh, A.; Delirezh, N.; Morshedi, A.; Mosayebi, G.; Farshid, A.A.; Mardani, K. Microvesicles derived from mesenchymal stem cells: Potent organelles for induction of tolerogenic signaling. Immunol. Lett. 2012, 147, 47–54. [Google Scholar] [CrossRef]
- Zhang, B.; Yin, Y.; Lai, R.C.; Tan, S.S.; Choo, A.B.H.; Lim, S.K. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 2014, 23, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Huang, L.; Li, Y.; Zhang, X.; Gu, J.; Yan, Y.; Xu, X.; Wang, M.; Qian, H.; Xu, W. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012, 315, 28–37. [Google Scholar] [CrossRef]
- Lee, J.K.; Park, S.R.; Jung, B.K.; Jeon, Y.K.; Lee, Y.S.; Kim, M.K.; Kim, Y.G.; Jang, J.Y.; Kim, C.W. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS ONE 2013, 8, e84256. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Wahlgren, J.; Karlson, T.D.L.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Bala, S.; Bukong, T.; Szabo, G. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Leijendekker, R.; Harding, C.V.; Melief, C.J.M.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Conlan, R.S.; Pisano, S.; Oliveira, M.I.; Ferrari, M.; Mendes Pinto, I. Exosomes as Reconfigurable Therapeutic Systems. Trends Mol. Med. 2017, 23, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Perica, K.; De León Medero, A.; Durai, M.; Chiu, Y.L.; Bieler, J.G.; Sibener, L.; Niemöller, M.; Assenmacher, M.; Richter, A.; Edidin, M.; et al. Nanoscale artificial antigen presenting cells for T cell immunotherapy. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 119–129. [Google Scholar] [CrossRef]
- Zhang, N.; Bevan, M.J. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Perica, K.; Bieler, J.G.; Schütz, C.; Varela, J.C.; Douglass, J.; Skora, A.; Chiu, Y.L.; Oelke, M.; Kinzler, K.; Zhou, S.; et al. Enrichment and Expansion with Nanoscale Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano 2015, 9, 6861–6871. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies that are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Butler, M.O.; Hirano, N. Human cell-based artificial antigen-presenting cells for cancer immunotherapy. Immunol. Rev. 2014, 257, 191–209. [Google Scholar] [CrossRef]
- Rhodes, K.R.; Green, J.J. Nanoscale artificial antigen presenting cells for cancer immunotherapy. Mol. Immunol. 2018, 98, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Z.; Mao, H.; Yang, L. Magnetic nanoparticles for precision oncology: Theranostic magnetic iron oxide nanoparticles for image-guided and targeted cancer therapy. Nanomedicine 2017, 12, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Sant, S.; Wang, B.; Laurent, S.; Sen, T. Superparamagnetic iron oxide nanoparticles (SPIONs): Development, surface modification and applications in chemotherapy. Adv. Drug Deliv. Rev. 2011, 63, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Petros, R.A.; Desimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Bonnemain, B. Superparamagnetic agents in magnetic resonance imaging: Physicochemical characteristics and clinical applications. A review. J. Drug Target. 1998, 6, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.K.; Park, J.; Jon, S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics 2012, 2, 3–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, X.; Cheng, L.; Yin, S.; Yang, G.; Li, Y.; Liu, Z. Multifunctional theranostic red blood cells for magnetic-field-enhanced in vivo combination therapy of cancer. Adv. Mater. 2014, 26, 4794–4802. [Google Scholar] [CrossRef]
- Williams, D.F. On the mechanisms of biocompatibility. Biomaterials 2008, 29, 2941–2953. [Google Scholar] [CrossRef]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of engineered nanoparticles for drug delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef]
- Guerrero, S.; Herance, J.R.; Rojas, S.; Mena, J.F.; Gispert, J.D.; Acosta, G.A.; Albericio, F.; Kogan, M.J. Synthesis and in vivo evaluation of the biodistribution of a 18F-labeled conjugate gold-nanoparticle-peptide with potential biomedical application. Bioconjug. Chem. 2012, 23, 399–408. [Google Scholar] [CrossRef]
- Jiang, W.; Gupta, R.K.; Deshpande, M.C.; Schwendeman, S.P. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv. Drug Deliv. Rev. 2005, 57, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Zyuzin, M.V.; Díez, P.; Goldsmith, M.; Carregal-Romero, S.; Teodosio, C.; Rejman, J.; Feliu, N.; Escudero, A.; Almendral, M.J.; Linne, U.; et al. Comprehensive and Systematic Analysis of the Immunocompatibility of Polyelectrolyte Capsules. Bioconjug. Chem. 2017, 28, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Zhang, S.; Yang, Q.; Zhang, T.; Wei, X.Q.; Jiang, L.; Zhang, C.L.; Chen, Q.M.; Zhang, Z.R.; Lin, Y.F. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials 2013, 34, 8521–8530. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, S.L.; McKenzie, D.R.; Nosworthy, N.J.; Denman, J.A.; Sezerman, O.U.; Bilek, M.M.M. The Vroman effect: Competitive protein exchange with dynamic multilayer protein aggregates. Colloids Surf. B Biointerfaces 2013, 103, 395–404. [Google Scholar] [CrossRef]
- Liu, W.; Rose, J.; Auffan, M.; Bottero, J.-Y. Protein corona formation for nanomaterials and proteins of a similar size: Hard or soft corona? View project. Nanoscale 2014, 5, 1658–1665. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Van Kempen, T.S.; Wenink, M.H.; Leijten, E.F.A.; Radstake, T.R.D.J.; Boes, M. Perception of self: Distinguishing autoimmunity from autoinflammation. Nat. Rev. Rheumatol. 2015, 11, 483–492. [Google Scholar] [CrossRef]
- Serrano-del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic cell death and immunotherapy of multiple myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Li, X. The inducers of immunogenic cell death for tumor immunotherapy. Tumori J. 2018, 104, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, T.; Oelke, M.; Mackensen, A. Induction and clonal expansion of tumor-specific cytotoxic T lymphocytes from renal cell carcinoma patients after stimulation with autologous dendritic cells loaded with tumor cells. Int. J. Cancer 2001, 91, 749–756. [Google Scholar] [CrossRef]
- Fucikova, J.; Moserova, I.; Truxova, I.; Hermanova, I.; Vancurova, I.; Partlova, S.; Fialova, A.; Sojka, L.; Cartron, P.F.; Houska, M.; et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int. J. Cancer 2014, 135, 1165–1177. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Zappasodi, R.; Pupa, S.M.; Ghedini, G.C.; Bongarzone, I.; Magni, M.; Cabras, A.D.; Colombo, M.P.; Carlo-Stella, C.; Gianni, A.M.; Di Nicola, M. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010, 70, 9062–9072. [Google Scholar] [CrossRef] [PubMed]
- Wemeau, M.; Kepp, O.; Tesnière, A.; Panaretakis, T.; Flament, C.; De Botton, S.; Zitvogel, L.; Kroemer, G.; Chaput, N. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis. 2010, 1, e104. [Google Scholar] [CrossRef]
- Peng, R.Q.; Chen, Y.B.; Ding, Y.; Zhang, R.; Zhang, X.; Yu, X.J.; Zhou, Z.W.; Zeng, Y.X.; Zhang, X.S. Expression of calreticulin is associated with infiltration of T-cells in stage III B colon cancer. World J. Gastroenterol. 2010, 16, 2428–2434. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 80 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Andersson, U.; Tracey, K.J. HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Ma, Y.; Aymeric, L.; Locher, C.; Mattarollo, S.R.; Delahaye, N.F.; Pereira, P.; Boucontet, L.; Apetoh, L.; Ghiringhelli, F.; Casares, N.; et al. Contribution of IL-17-producing γδ T cells to the efficacy of anticancer chemotherapy. J. Exp. Med. 2011, 208, 869. [Google Scholar] [CrossRef]
- Fan, Y.; Kuai, R.; Xu, Y.; Ochyl, L.J.; Irvine, D.J.; Moon, J.J. Immunogenic Cell Death Amplified by Co-localized Adjuvant Delivery for Cancer Immunotherapy. Nano Lett. 2017, 17, 7387–7393. [Google Scholar] [CrossRef]
- Kuai, R.; Yuan, W.; Son, S.; Nam, J.; Xu, Y.; Fan, Y.; Schwendeman, A.; Moon, J.J. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv. 2018, 4, eaao1736. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Kennedy, L.C.; Young, J.K.; Perna, S.K.; Mattos Almeida, J.P.; Lin, A.Y.; Eckels, P.C.; Drezek, R.A.; Foster, A.E. Elimination of Metastatic Melanoma Using Gold Nanoshell-Enabled Photothermal Therapy and Adoptive T Cell Transfer. PLoS ONE 2013, 8, e69073. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Roche, K.C.; Tian, S.; Eblan, M.J.; McKinnon, K.P.; Caster, J.M.; Chai, S.; Herring, L.E.; Zhang, L.; Zhang, T.; et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 2012, 4, 143ra99. [Google Scholar] [CrossRef]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef]
- Tray, N.; Weber, J.S.; Adams, S. Predictive biomarkers for checkpoint immunotherapy: Current status and challenges for clinical application. Cancer Immunol. Res. 2018, 6, 1122–1128. [Google Scholar] [CrossRef]
- Spencer, K.R.; Wang, J.; Silk, A.W.; Ganesan, S.; Kaufman, H.L.; Mehnert, J.M. Biomarkers for Immunotherapy: Current Developments and Challenges. Am. Soc. Clin. Oncol. Educ. B. 2016, e493–e503. [Google Scholar] [CrossRef]
- Nelson, D.; Fisher, S.; Robinson, B. The “Trojan Horse” Approach to Tumor Immunotherapy: Targeting the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 789069. [Google Scholar] [CrossRef]
- Masucci, G.V.; Cesano, A.; Hawtin, R.; Janetzki, S.; Zhang, J.; Kirsch, I.; Dobbin, K.K.; Alvarez, J.; Robbins, P.B.; Selvan, S.R.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume I-pre-analytical and analytical validation. J. Immunother. Cancer 2016, 4, 76. [Google Scholar] [CrossRef]
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and Other Tools); Food and Drug Administration (US): New York, NY, USA, 2017; Volume 55. [CrossRef]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- May, J.Y.; Negrier, S.; Combaret, V.; Attali, S.; Goillot, E.; Mercatello, A.; Ravault, A.; Tourani, J.M.; Moskovtchenko, J.F.; Philip, T.; et al. Serum Level of Interleukin 6 as a Prognosis Factor in Metastatic Renal Cell Carcinoma. Cancer Res. 1992, 52, 3317–3322. [Google Scholar]
- Tartour, E.; Blay, J.Y.; Dorval, T.; Escudier, B.; Mosseri, V.; Douillard, J.Y.; Deneux, L.; Gorin, I.; Negrier, S.; Mathiot, C.; et al. Predictors of clinical response to interleukin-2-based immunotherapy in melanoma patients: A French multiinstitutional study. J. Clin. Oncol. 1996, 14, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, M.; Kim-Schulze, S.; Panelli, M.C.; Stroncek, D.; Wang, E.; Taback, B.; Kim, D.W.; DeRaffele, G.; Pos, Z.; Marincola, F.M.; et al. Serum vascular endothelial growth factor and fibronectin predict clinical response to high-dose interleukin-2 therapy. J. Clin. Oncol. 2009, 27, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Simeone, E.; Gentilcore, G.; Giannarelli, D.; Grimaldi, A.M.; Caracò, C.; Curvietto, M.; Esposito, A.; Paone, M.; Palla, M.; Cavalcanti, E.; et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol. Immunother. 2014, 63, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Hannani, D.; Vétizou, M.; Enot, D.; Rusakiewicz, S.; Chaput, N.; Klatzmann, D.; Desbois, M.; Jacquelot, N.; Vimond, N.; Chouaib, S.; et al. Anticancer immunotherapy by CTLA-4 blockade: Obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. 2015, 25, 208–224. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef]
- Gnjatic, S.; Bronte, V.; Brunet, L.R.; Butler, M.O.; Disis, M.L.; Galon, J.; Hakansson, L.G.; Hanks, B.A.; Karanikas, V.; Khleif, S.N.; et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J. Immunother. Cancer 2017, 5, 1–18. [Google Scholar] [CrossRef]
- Schmidt, H.; Bastholt, L.; Geertsen, P.; Christensen, I.J.; Larsen, S.; Gehl, J.; Von Der Maase, H. Elevated neutrophil and monocyte counts in peripheral blood are associated with poor survival in patients with metastatic melanoma: A prognostic model. Br. J. Cancer 2005, 93, 273–278. [Google Scholar] [CrossRef]
- Powell, D.J.; Dudley, M.E.; Robbins, P.F.; Rosenberg, S.A. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood 2005, 105, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Ochsenbein, A.F.; Riddell, S.R.; Brown, M.; Corey, L.; Baerlocher, G.M.; Lansdorp, P.M.; Greenberg, P.D. CD27 Expression Promotes Long-Term Survival of Functional Effector–Memory CD8+Cytotoxic T Lymphocytes in HIV-infected Patients. J. Exp. Med. 2004, 200, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, A.W.; Jillab, M.; Plimack, E.R.; Hudes, G.R.; Uzzo, R.G.; Litwin, S.; Dulaimi, E.; Al-Saleem, T.; Campbell, K.S. PD-1 Expression on Peripheral Blood Cells Increases with Stage in Renal Cell Carcinoma Patients and Is Rapidly Reduced after Surgical Tumor Resection. Cancer Immunol. Res. 2014, 2, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Adamow, M.; Ginsberg, B.A.; Rasalan, T.S.; Ritter, E.; Gallardo, H.F.; Xu, Y.; Pogoriler, E.; Terzulli, S.L.; Kuk, D.; et al. Integrated NY-ESO-1 antibody and CD8 + T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc. Natl. Acad. Sci. USA 2011, 108, 16723–16728. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Looi, K.S.; Tan, E.M. Identification of tumor-associated antigens as diagnostic and predictive biomarkers in cancer. Methods Mol. Biol. 2009, 520, 1–10. [Google Scholar] [CrossRef]
- Lu, M.; Nakamura, R.M.; Dent, E.D.B.; Zhang, J.Y.; Nielsen, F.C.; Christiansen, J.; Chan, E.K.L.; Tan, E.M. Aberrant expression of fetal RNA-binding protein p62 in liver cancer and liver cirrhosis. Am. J. Pathol. 2001, 159, 945–953. [Google Scholar] [CrossRef]
- Hoo, L.S.; Zhang, J.Y.; Chan, E.K.L. Cloning and characterization of a novel 90 kDa “companion” auto-antigen of p62 overexpressed in cancer. Oncogene 2002, 21, 5006–5015. [Google Scholar] [CrossRef]
- Noguchi, T.; Kato, T.; Wang, L.; Maeda, Y.; Ikeda, H.; Sato, E.; Knuth, A.; Gnjatic, S.; Ritter, G.; Sakaguchi, S.; et al. Intracellular Tumor-Associated Antigens Represent Effective Targets for Passive Immunotherapy. AACR 2012, 72, 1672–1682. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Dieu-Nosjean, M.-C. Tertiary Lymphoid Structure-Associated B Cells are Key Players in Anti-Tumor Immunity. Front. Immunol. 2015, 6, 67. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Erémyer´erémy Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B Cells in Tertiary Lymphoid Structures Is Associated with a Protective Immunity in Patients with Lung Cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Videssa Breast | Detección de Cáncer de Mama a Base de Sangre. Available online: https://www.provistadx.com/videssa-breast (accessed on 16 May 2020).
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- De Vries, I.J.M.; Bernsen, M.R.; van Geloof, W.L.; Scharenborg, N.M.; Lesterhuis, W.J.; Rombout, P.D.M.; Van Muijen, G.N.P.; Figdor, C.G.; Punt, C.J.A.; Ruiter, D.J.; et al. In situ detection of antigen-specific T cells in cryo-sections using MHC class I tetramers after dendritic cell vaccination of melanoma patients. Cancer Immunol. Immunother. 2007, 56, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Robins, H.S.; Campregher, P.V.; Srivastava, S.K.; Wacher, A.; Turtle, C.J.; Kahsai, O.; Riddell, S.R.; Warren, E.H.; Carlson, C.S. Comprehensive assessment of T-cell receptor β-chain diversity in αβ T cells. Blood 2009, 114, 4099–4107. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Meng, X.; Huang, Z.; Teng, F.; Xing, L.; Yu, J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat. Rev. 2015, 41, 868–876. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 80 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Chan, T.A.; Wolchok, J.D.; Snyder, A. Correction: Genetic basis for clinical response to CTLA-4 blockade in melanoma: To the editor. N. Engl. J. Med. 2015, 373, 1984. [Google Scholar] [CrossRef]
- Cirenajwis, H.; Ekedahl, H.; Lauss, M.; Harbst, K.; Carneiro, A.; Enoksson, J.; Rosengren, F.; Werner-Hartman, L.; Törngren, T.; Kvist, A.; et al. Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget 2015, 6, 12297–12309. [Google Scholar] [CrossRef]
- Stoll, G.; Enot, D.; Mlecnik, B.; Galon, J.; Zitvogel, L.; Kroemer, G. Immune-related gene signatures predict the outcome of neoadjuvant chemotherapy. Oncoimmunology 2014, 3, e27884. [Google Scholar] [CrossRef] [PubMed]
- Stenzinger, A.; Allen, J.D.; Maas, J.; Stewart, M.D.; Merino, D.M.; Wempe, M.M.; Dietel, M. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosom. Cancer 2019, 58, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and search for new cancer genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Health, N.; Bank, B.B.; Nahmani, A.; Cancer, P.; Trust, R.T.H.; Foundation, P.; Ministry, S. Signatures of mutational processes in human cancer. Nature 2013, 500, 1–108. [Google Scholar] [CrossRef]
- Chen, Y.P.; Zhang, Y.; Lv, J.W.; Li, Y.Q.; Wang, Y.Q.; He, Q.M.; Yang, X.J.; Sun, Y.; Mao, Y.P.; Yun, J.P.; et al. Genomic Analysis of Tumor Microenvironment Immune Types across 14 Solid Cancer Types: Immunotherapeutic Implications. Theranostics 2017, 7, 3585. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 80 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Vigneron, N.; Stroobant, V.; Chapiro, J.; Ooms, A.; Degiovanni, G.; Morel, S.; Van Der Bruggen, P.; Boon, T.; Van Den Eynde, B.J. An Antigenic Peptide Produced by Peptide Splicing in the Proteasome. Science 80 2004, 304, 587–590. [Google Scholar] [CrossRef]
- Madsen, C.B.; Petersen, C.; Lavrsen, K.; Harndahl, M.; Buus, S.; Clausen, H.; Pedersen, A.E.; Wandall, H.H. Cancer Associated Aberrant Protein O-Glycosylation Can Modify Antigen Processing and Immune Response. PLoS ONE 2012, 7, e50139. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, M.; De La Pena, H.; Norris, A.; Polefrone, J.M.; Qian, J.; English, A.M.; Cummings, K.L.; Penny, S.; Turner, J.E.; Cottine, J.; et al. MHC Class I-Associated Phosphopeptides Are the Targets of Memory-like Immunity in Leukemia. Sci. Transl. Med. 2013, 5, 203ra125. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, B.; Van Campenhout, C.; Rorive, S.; Remmelink, M.; Salmon, I.; D’Haene, N. Methods of measurement for tumor mutational burden in tumor tissue. Transl. Lung Cancer Res. 2018, 7, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Ock, C.Y.; Keam, B.; Kim, S.; Lee, J.S.; Kim, M.; Kim, T.M.; Jeon, Y.K.; Kim, D.W.; Chung, D.H.; Heo, D.S. Pan-Cancer Immunogenomic Perspective on the Tumor Microenvironment Based on PD-L1 and CD8 T-Cell Infiltration. Clin. Cancer Res. 2016, 22, 2261–2270. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res 2015, 75, 2139–2145. [Google Scholar] [CrossRef]
- Bald, T.; Landsberg, J.; Lopez-Ramos, D.; Renn, M.; Glodde, N.; Jansen, P.; Gaffal, E.; Steitz, J.; Tolba, R.; Kalinke, U.; et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014, 4, 674–687. [Google Scholar] [CrossRef]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Investig. 2013, 123, 2756–2763. [Google Scholar] [CrossRef]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 80 2018, 359, 582–587. [Google Scholar] [CrossRef]
- Robert, L.; Harview, C.; Emerson, R.; Wang, X.; Mok, S.; Homet, B.; Comin-Anduix, B.; Koya, R.C.; Robins, H.; Tumeh, P.C.; et al. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology 2014, 3, e29244. [Google Scholar] [CrossRef] [PubMed]
- Bobisse, S.; Foukas, P.G.; Coukos, G.; Harari, A. Neoantigen-based cancer immunotherapy. Ann. Transl. Med. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; L€ Ower, M.; Van De Roemer, N.; De Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. AACR 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- van Rooij, N.; van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; van Dijk, L.J.A.; Behjati, S.; Hilkmann, H.; el Atmioui, D.; et al. Tumor Exome Analysis Reveals Neoantigen-Specific T-Cell Reactivity in an Ipilimumab-Responsive Melanoma. J. Clin. Oncol. 2013, 31, e439–e442. [Google Scholar] [CrossRef]
- Wick, D.A.; Webb, J.R.; Nielsen, J.S.; Martin, S.D.; Kroeger, D.R.; Milne, K.; Castellarin, M.; Twumasi-Boateng, K.; Watson, P.H.; Holt, R.A.; et al. Surveillance of the Tumor Mutanome by T Cells during Progression from Primary to Recurrent Ovarian Cancer. Hum. Cancer Biol. 2014, 20, 1125–1134. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 80 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef]
- Lundegaard, C.; Lund, O.; Nielsen, M. Prediction of epitopes using neural network based methods. J. Immunol. Methods 2011, 374, 26–34. [Google Scholar] [CrossRef]
- Wirth, T.C.; Kühnel, F. Neoantigen targeting—Dawn of a new era in cancer immunotherapy? Front. Immunol. 2017, 8, 1848. [Google Scholar] [CrossRef]
- Kloor, M.; Michel, S.; Von Knebel Doeberitz, M. Immune evasion of microsatellite unstable colorectal cancers. Int. J. Cancer 2010, 127, 1001–1010. [Google Scholar] [CrossRef]
- Axelrod, M.L.; Cook, R.S.; Johnson, D.B.; Balko, J.M. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 2019, 25, 2392–2402. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, J.; Rakitsch, B.; Hoffgaard, F.; Römer, M.; Schuster, H.; Kowalewski, D.J.; Priemer, M.; Stos-Zweifel, V.; Hörzer, H.; Satelli, A.; et al. Translating Immunopeptidomics to Immunotherapy-Decision-Making for Patient and Personalized Target Selection. Proteomics 2018, 18, 1700284. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.M.; Singh-Jasuja, H.; Flamion, B.; Hoos, A.; Huber, C.; Kallen, K.J.; Khleif, S.N.; Kreiter, S.; Nielsen, M.; Rammensee, H.G.; et al. The regulatory landscape for actively personalized cancer immunotherapies. Nat. Biotechnol. 2013, 31, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Fortier, M.H.; Caron, É.; Hardy, M.P.; Voisin, G.; Lemieux, S.; Perreault, C.; Thibault, P. The MHC class I peptide repertoire is molded by the transcriptome. J. Exp. Med. 2008, 205, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Weinzierl, A.O.; Lemmel, C.; Schoor, O.; Müller, M.; Krüger, T.; Wernet, D.; Hennenlotter, J.; Stenzl, A.; Klingel, K.; Rammensee, H.G.; et al. Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol. Cell. Proteom. 2007, 6, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Barnea, E.; Beer, I.; Avivi, I.; Katz, T.; Admon, A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc. Natl. Acad. Sci. USA 2010, 107, 18769–18776. [Google Scholar] [CrossRef]
- Zavazava, N.; Kronke, M. Soluble HLA class I molecules induce apoptosis in alloreactive cytotoxic T lymphocytes. Nat. Med. 1996, 2, 1267. [Google Scholar] [CrossRef] [PubMed]
- HUPO-Proyecto de Inmunopéptido Humano. Available online: https://hupo.org/human-immuno-peptidome-project/ (accessed on 18 May 2020).
- Shao, W.; Pedrioli, P.G.A.; Wolski, W.; Scurtescu, C.; Schmid, E.; Vizcaíno, J.A.; Courcelles, M.; Schuster, H.; Kowalewski, D.; Marino, F.; et al. The SysteMHC Atlas project. Nucleic Acids Res. 2018, 46, D1237–D1247. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Karosiene, E.; Rasmussen, M.; Stryhn, A.; Buus, S.; Nielsen, M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015, 67, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 2009, 10, 296. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef]
- Tan, E.M. Antinuclear Antibodies: Diagnostic Markers for Autoimmune Diseases and Probes for Cell Biology. Adv. Immunol. 1989, 44, 93–151. [Google Scholar] [CrossRef]
- Crawford, L.V.; Pim, D.C.; Bulbrook, R.D. Detection of antibodies against the cellular protein p53 in sera from patients with breast cancer. Int. J. Cancer 1982, 30, 403–408. [Google Scholar] [CrossRef]
- Tan, E.M.; Zhang, J. Autoantibodies to tumor-associated antigens: Reporters from the immune system. Immunol. Rev. 2008, 222, 328–340. [Google Scholar] [CrossRef]
- Tan, E.M.; Shi, F.D. Relative paradigms between autoantibodies in lupus and autoantibodies in cancer. Clin. Exp. Immunol. 2003, 134, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Lubin, R.; Zalcman, G.; Bouchet, L.; Tr É Daniel, J.; Legros, Y.; Cazals, D.; Hirsch, A.; Soussi, T. Serum p53 antibodies as early markers of lung cancer. Nat. Med. 1995, 1, 701–702. [Google Scholar] [CrossRef]
- Trivers, G.E.; De Benedetti, V.M.G.; Cawley, H.L.; Caron, G.; Harrington, A.M.; Bennett, W.P.; Jett, J.R.; Colby, T.V.; Tazelaar, H.; Pairolero, P.; et al. Anti-p53 Antibodies in Sera from Patients with Chronic Obstructive Pulmonary Disease Can Predate a Diagnosis of Cancer. Clin. Cancer Res. 1996, 2, 1767–1775. [Google Scholar]
- Trivers, G.E.; Cawley, H.L.; Debenedetti, V.M.G.; Hollstein, M.; Marion, M.J.; Bennett, W.P.; Hoover, M.L.; Prives, C.C.; Tamburro, C.C.; Harris, C.C. Anti-p53 antibodies in sera of workers occupationally exposed to vinyl chloride. J. Natl. Cancer Inst. 1995, 87, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Nakano, Y.; Kiyosawa, K.; Tan, E.M. Increasing titers and changing specificities of antinuclear antibodies in patients with chronic liver disease who develop hepatocellular carcinoma. Cancer 1993, 71, 26–35. [Google Scholar] [CrossRef]
- Woessner, R.D.; Mattern, M.R.; Mirabelli, C.K.; Johnson, R.K.; Drake, F.H. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991, 2, 209–214. [Google Scholar] [PubMed]
- Winter, S.F.; Minna, J.D.; Johnson, B.E.; Takahashi, T.; Gazdar, A.F.; Carbone, D.P. Development of antibodies against p53 in lung cancer patients appears to be dependent on the type of p53 mutation. Cancer Res. 1992, 52, 4168–4174. [Google Scholar] [PubMed]
- Suzuki, H.; Graziano, D.F.; McKolanis, J.; Finn, O.J. T cell-dependent antibody responses against aberrantly expressed cyclin B1 protein in patients with cancer and premalignant disease. Clin. Cancer Res. 2005, 11, 1521–1526. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Zhang, P.; LaBaer, J.; Hermjakob, H.; Li, D.; Yu, X. AAgAtlas 1.0: A human autoantigen database. Nucleic Acids Res. 2017, 45, D769–D776. [Google Scholar] [CrossRef]
- Soares, M.M.; Mehta, V.; Finn, O.J. Three Different Vaccines Based on the 140-Amino Acid MUC1 Peptide with Seven Tandemly Repeated Tumor-Specific Epitopes Elicit Distinct Immune Effector Mechanisms in Wild-Type Versus MUC1-Transgenic Mice with Different Potential for Tumor Rejection. J. Immunol. 2001, 166, 6555–6563. [Google Scholar] [CrossRef]
- Kao, H.; Marto, J.A.; Hoffmann, T.K.; Shabanowitz, J.; Finkelstein, S.D.; Whiteside, T.L.; Hunt, D.F.; Finn, O.J. Identification of cyclin B1 as a shared human epithelial tumor-associated antigen recognized by T cells. J. Exp. Med. 2001, 194, 1313–1323. [Google Scholar] [CrossRef]
- Wild, D. The Immunoassay Handbook: Theory and Applications of Ligand Binding, ELISA and Related Techniques; Newnes: Oxford, UK, 2013; ISBN 9780080970370. [Google Scholar]
- Vogeser, M.; Seger, C. Mass spectrometry methods in clinical diagnostics—State of the art and perspectives. TrAC-Trends Anal. Chem. 2016, 84, 1–4. [Google Scholar] [CrossRef]
- Pedersen, A.W.; Kopp, K.L.; Andersen, M.H.; Zocca, M.-B. Immunoregulatory antigens—Novel targets for cancer immunotherapy. Chin. Clin. Oncol. 2018, 7, 8. [Google Scholar] [CrossRef]
- Montico, B.; Lapenta, C.; Ravo, M.; Martorelli, D.; Muraro, E.; Zeng, B.; Comaro, E.; Spada, M.; Donati, S.; Santini, S.M.; et al. Exploiting a new strategy to induce immunogenic cell death to improve dendritic cell-based vaccines for lymphoma immunotherapy. Oncoimmunology 2017, 6, e1356964. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta-Rev. Cancer 2010, 1805, 53–71. [Google Scholar] [CrossRef]
- Das, D.K.; Feng, Y.; Mallis, R.J.; Li, X.; Keskin, D.B.; Hussey, R.E.; Brady, S.K.; Wang, J.H.; Wagner, G.; Reinherz, E.L.; et al. Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. USA 2015, 112, 1517–1522. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Reinherz, E.L. αβ TCR-mediated recognition: Relevance to tumor-antigen discovery and cancer immunotherapy. Cancer Immunol. Res. 2015, 3, 305–312. [Google Scholar] [CrossRef]
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. 1994, 153, 5586–5592. [Google Scholar]
















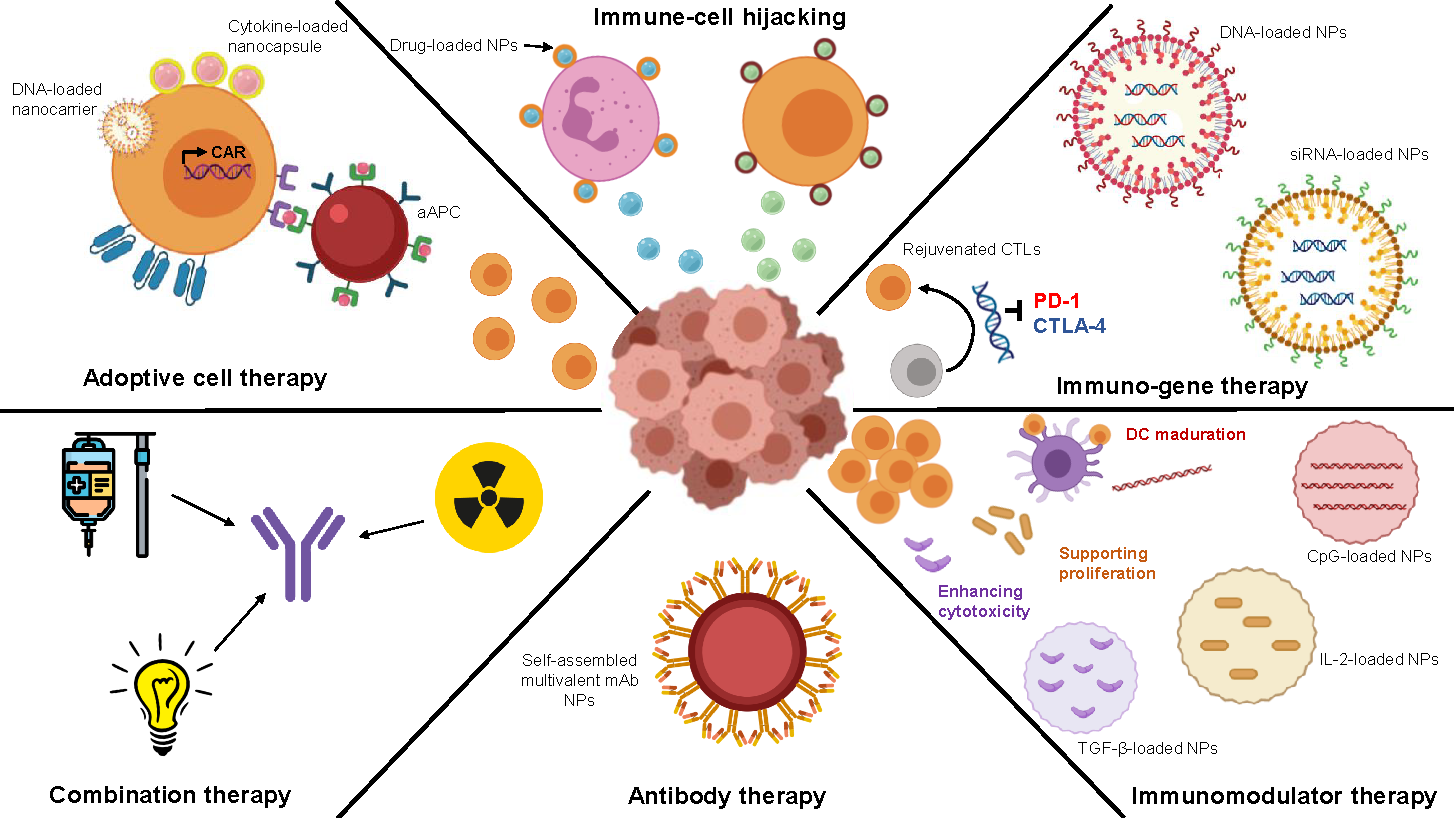{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Danger Signal | PRR | Function |
|---|---|---|
| CALR HSP70 HSP90 | LRP1 | Promotes the uptake of dead cell-asociated antigens. |
| Extracellular ATP | P2RX7/P2RY2 | Favours the recruitment of APCs and their activation. |
| HMGB1 dsRNA Cellular RNA LPS Flagellin ssRNA CpG DNA Viral RNA dsDNA | TLR2/TLR4 TLR3 TLR3 TLR4 TLR5 TLR7 TLR9 RLRs CDSs | Activate the synthesis of pro-inflammatory factors (type I IFNs). |
| Type I IFNs | IFNAR | Promotes CXCL10 secretion by cancer cells and has immune-stimulatory effects. |
| ANXA1 | FPR1 | Guides the final approach of APCs to dying cells. |
| CXCL10 | CXCR3 | Favours T cell recruitment. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acebes-Fernández, V.; Landeira-Viñuela, A.; Juanes-Velasco, P.; Hernández, A.-P.; Otazo-Perez, A.; Manzano-Román, R.; Gongora, R.; Fuentes, M. Nanomedicine and Onco-Immunotherapy: From the Bench to Bedside to Biomarkers. Nanomaterials 2020, 10, 1274. https://doi.org/10.3390/nano10071274
Acebes-Fernández V, Landeira-Viñuela A, Juanes-Velasco P, Hernández A-P, Otazo-Perez A, Manzano-Román R, Gongora R, Fuentes M. Nanomedicine and Onco-Immunotherapy: From the Bench to Bedside to Biomarkers. Nanomaterials. 2020; 10(7):1274. https://doi.org/10.3390/nano10071274
Chicago/Turabian StyleAcebes-Fernández, Vanessa, Alicia Landeira-Viñuela, Pablo Juanes-Velasco, Angela-Patricia Hernández, Andrea Otazo-Perez, Raúl Manzano-Román, Rafael Gongora, and Manuel Fuentes. 2020. "Nanomedicine and Onco-Immunotherapy: From the Bench to Bedside to Biomarkers" Nanomaterials 10, no. 7: 1274. https://doi.org/10.3390/nano10071274
APA StyleAcebes-Fernández, V., Landeira-Viñuela, A., Juanes-Velasco, P., Hernández, A.-P., Otazo-Perez, A., Manzano-Román, R., Gongora, R., & Fuentes, M. (2020). Nanomedicine and Onco-Immunotherapy: From the Bench to Bedside to Biomarkers. Nanomaterials, 10(7), 1274. https://doi.org/10.3390/nano10071274