A Nano-Rattle SnO2@carbon Composite Anode Material for High-Energy Li-ion Batteries by Melt Diffusion Impregnation



Abstract
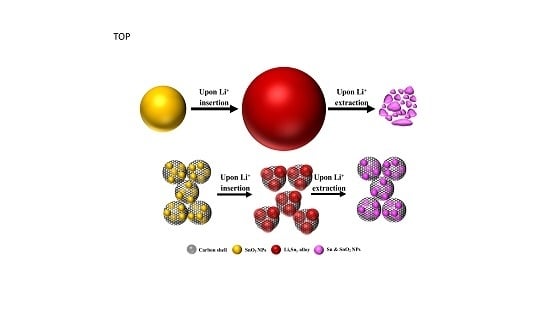
:
1. Introduction
2. Materials and Methods
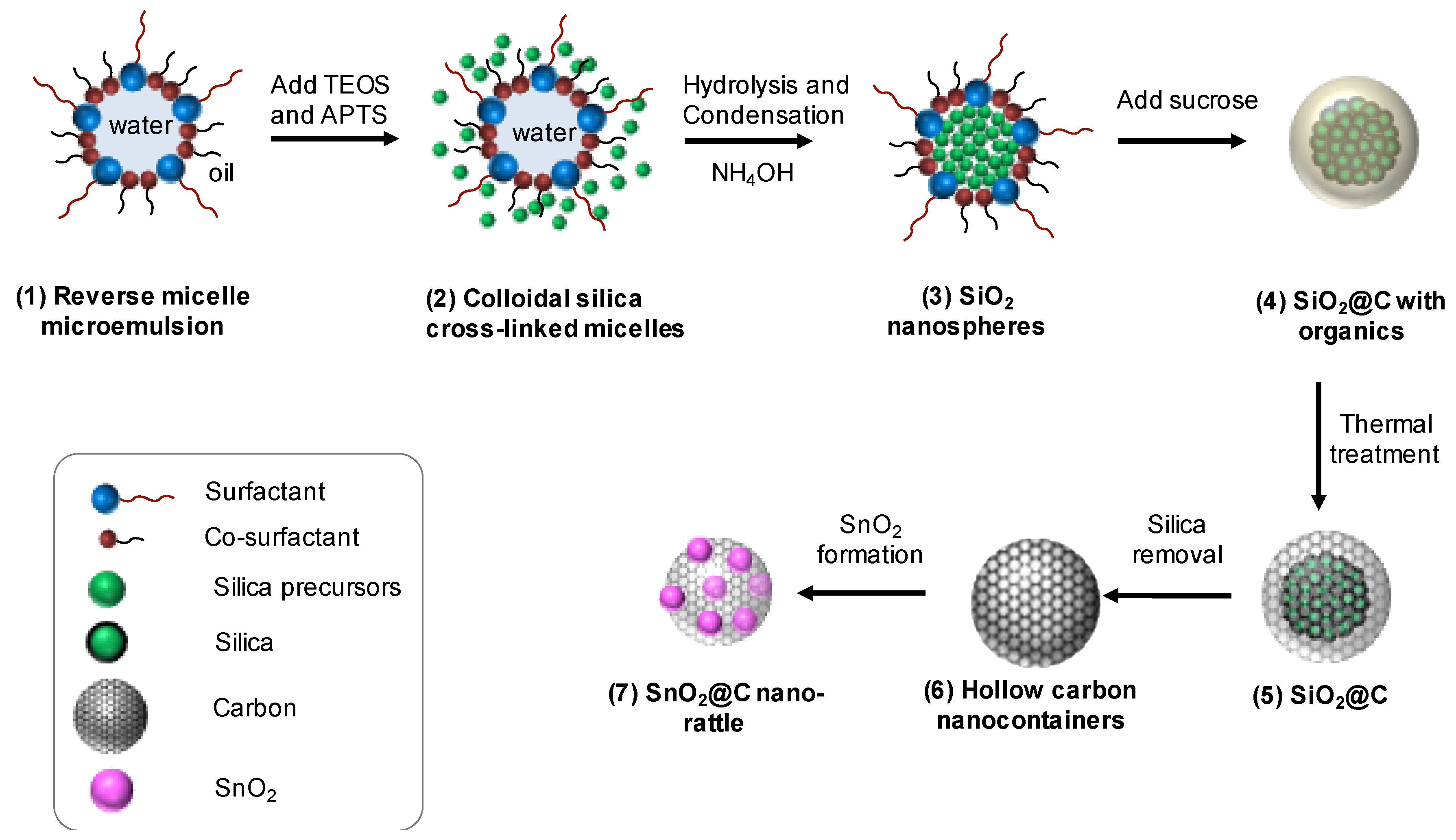
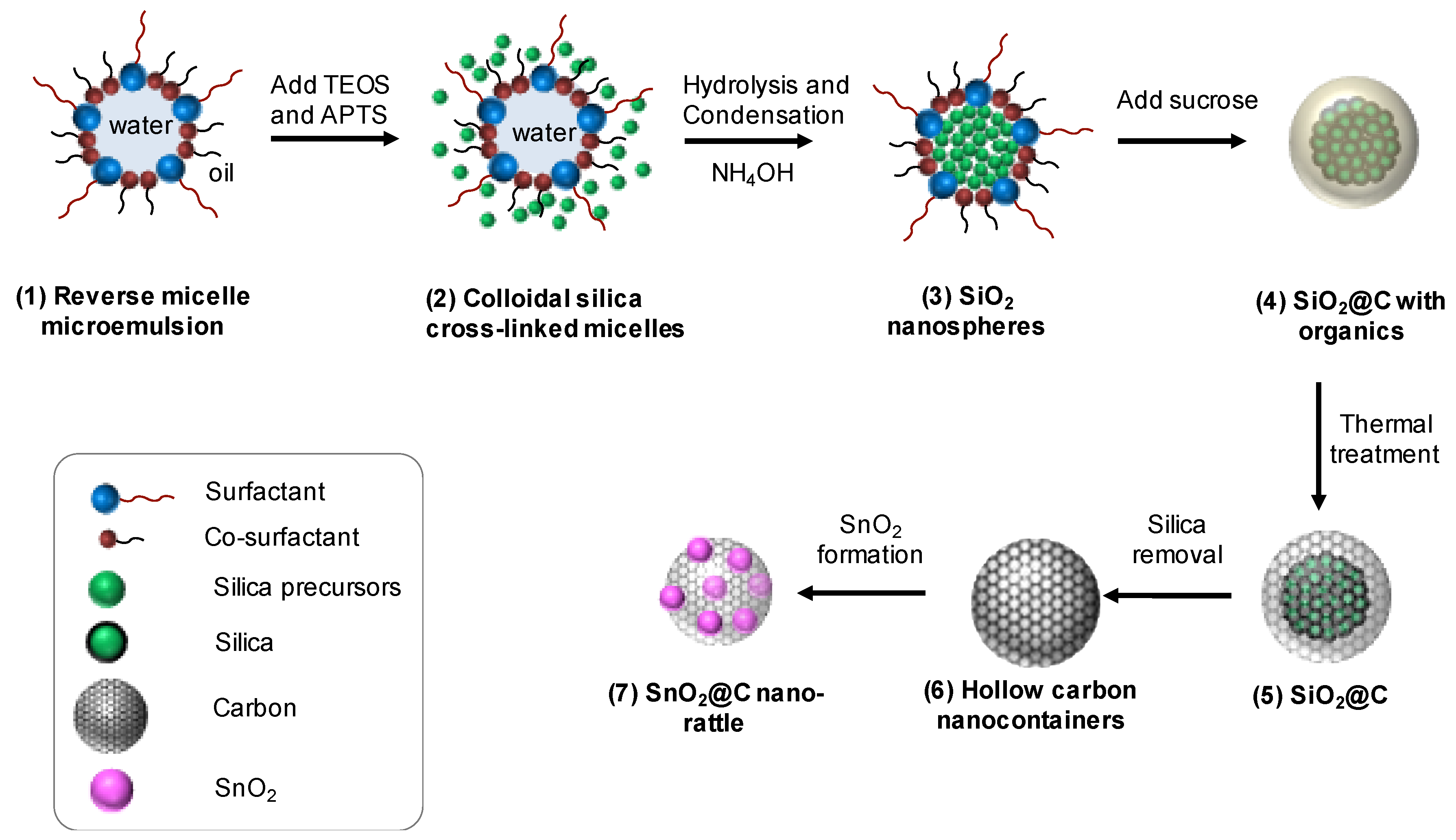
2.1. Synthesis of the Nanospherical Silica Template
2.2. Formation of Hollow Carbon Spheres
2.3. SnO2@C Nano-Rattles via Melt Diffusion
2.4. Sn@C Nano-Rattles via Wet Impregnation
2.5. Characterization
2.6. Preparation of the Electrodes
2.7. Electrochemical Characterizations of Electrodes
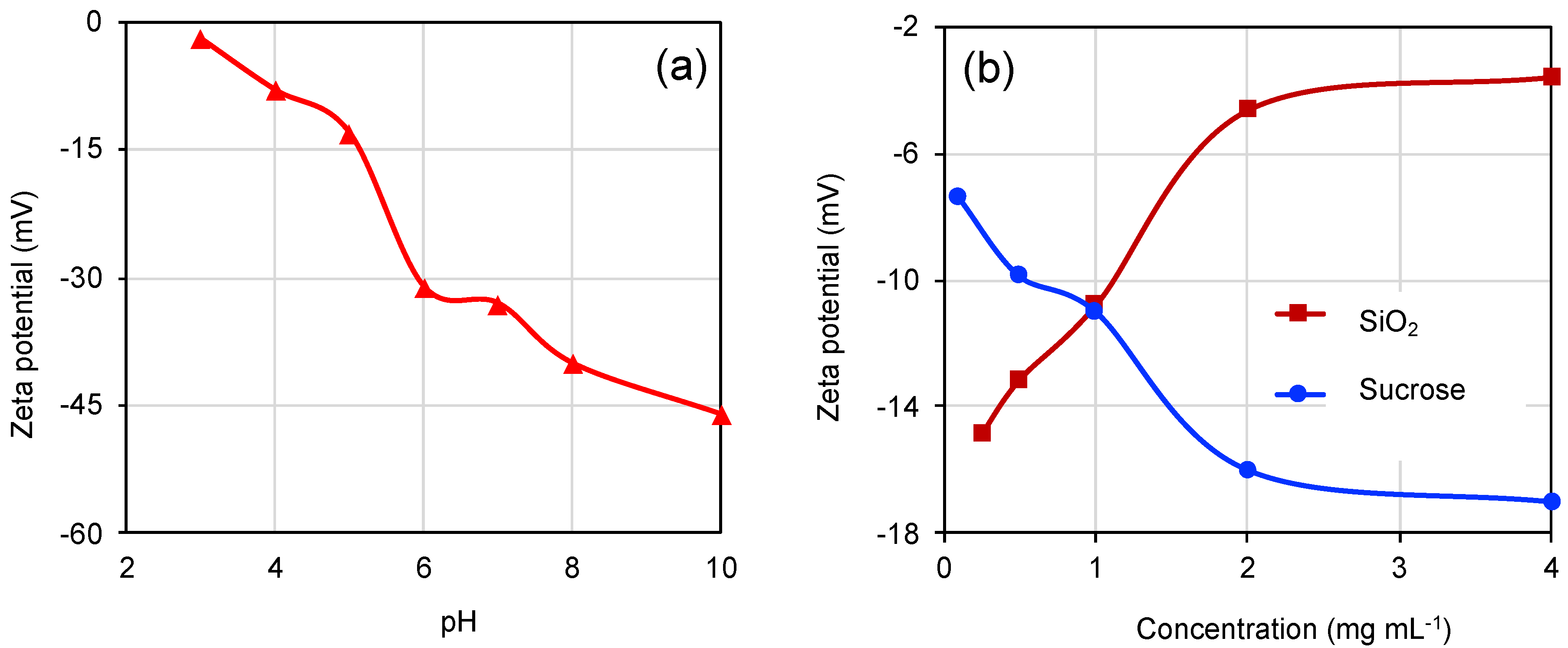
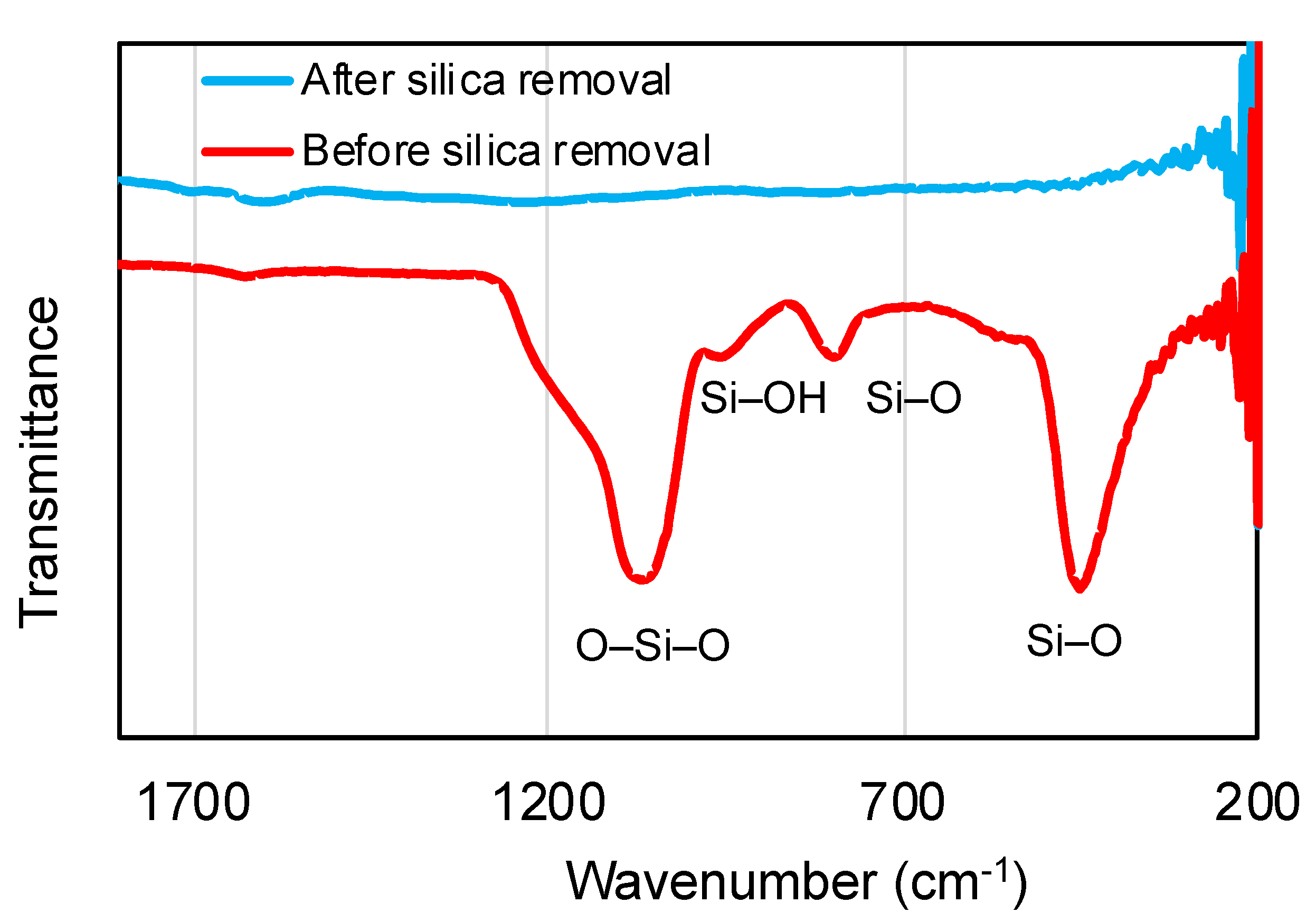
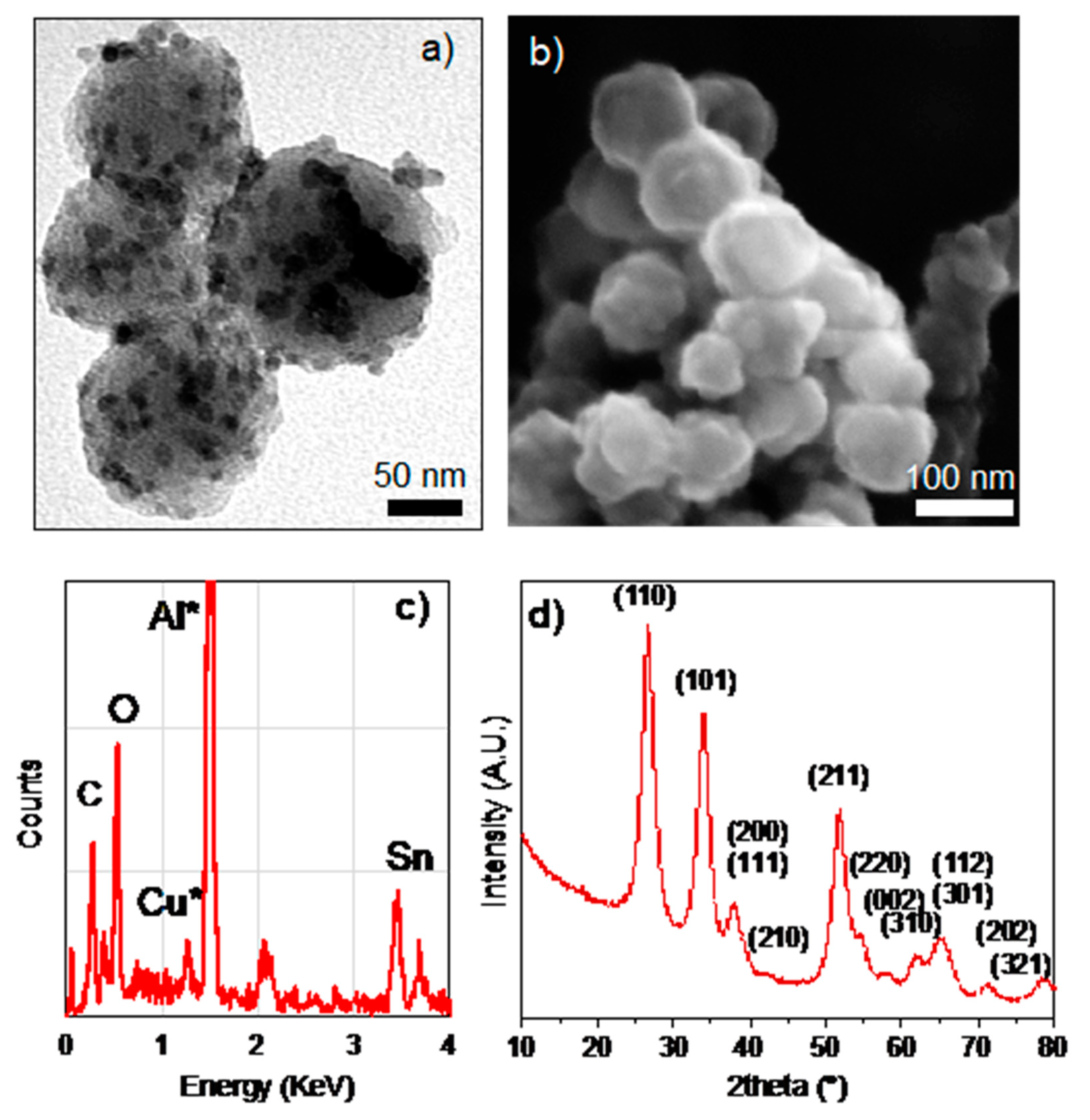
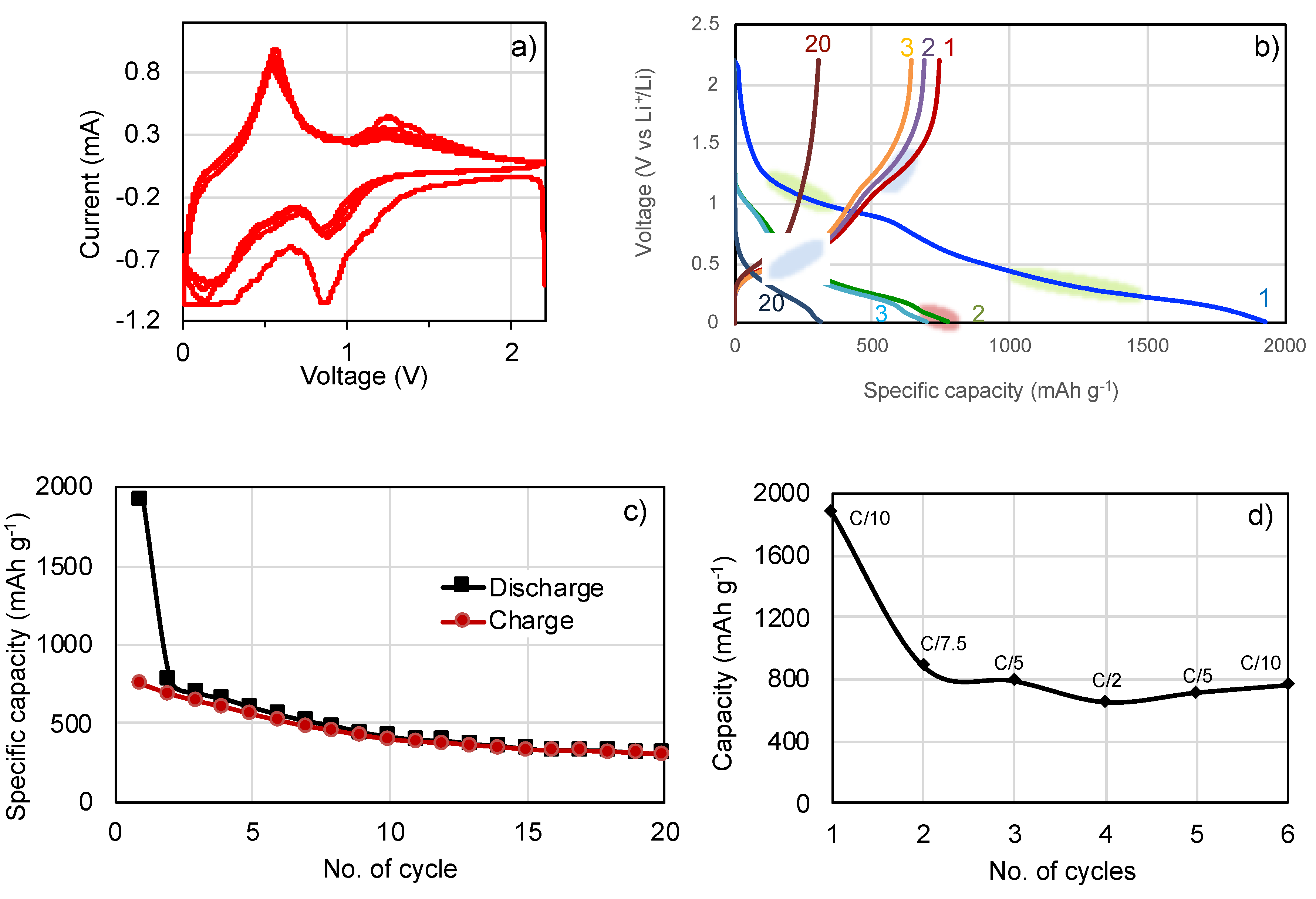
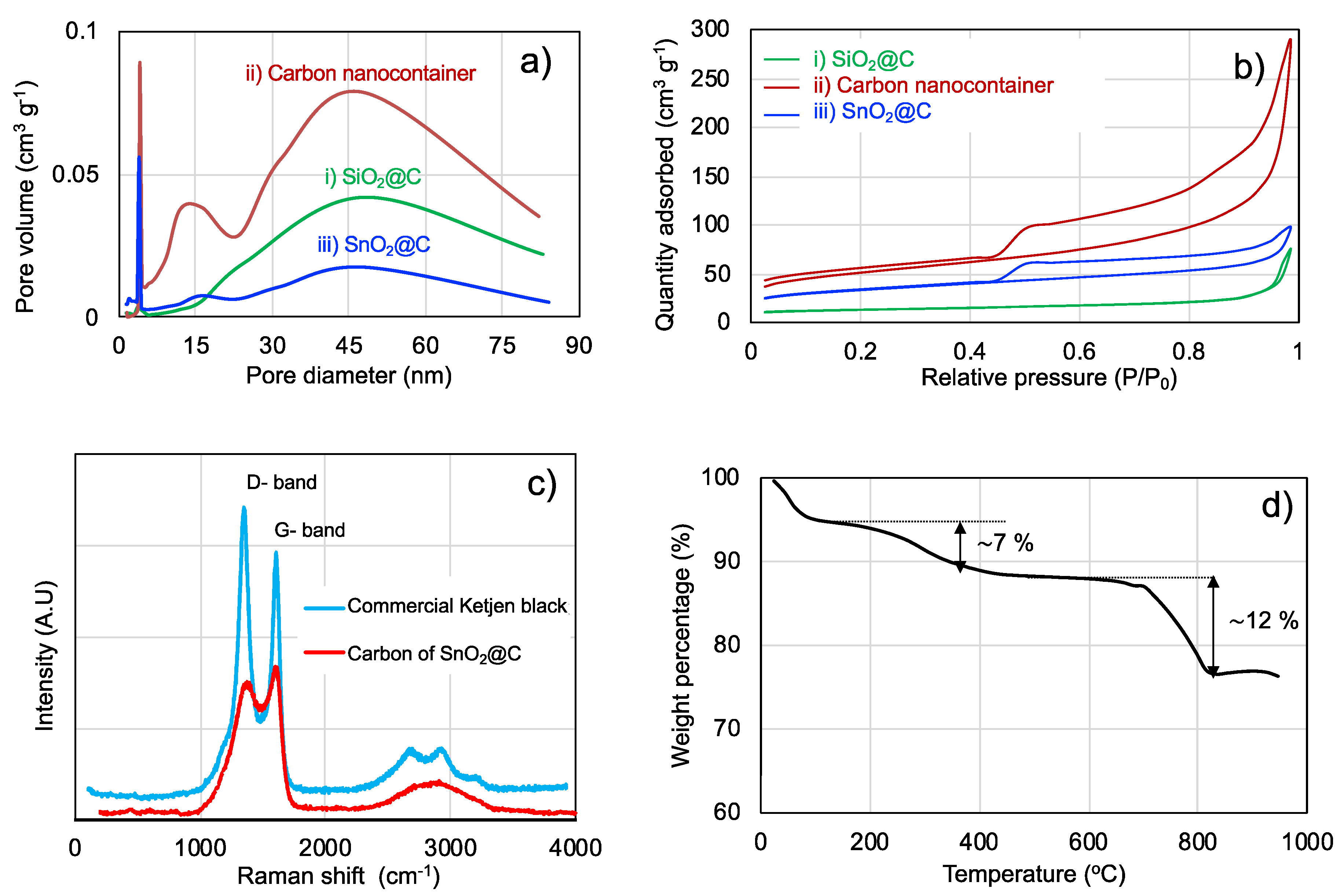
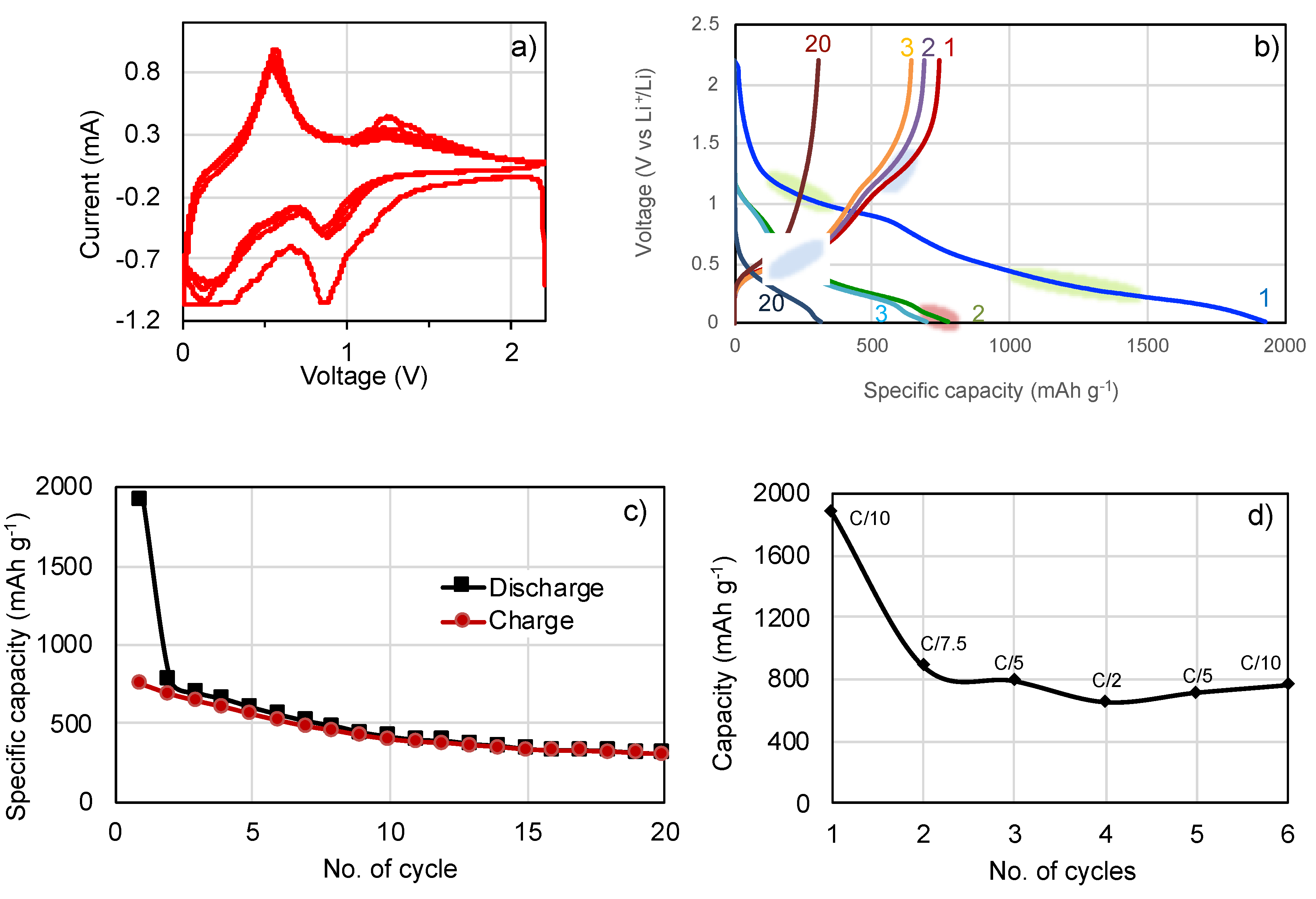
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Burns, J.C.; Stevens, D.A.; Dahn, J.R. In-Situ Detection of Lithium Plating Using High Precision Coulometry. J. Electrochem. Soc. 2015, 162, A959–A964. [Google Scholar] [CrossRef]
- Ghanbari, N.; Waldmann, T.; Kasper, M.; Axmann, P.; Wohlfahrt-Mehrens, M. Detection of Li Deposition by Glow Discharge Optical Emission Spectroscopy in Post-Mortem Analysis. ECS Electrochem. Lett. 2015, 4, A100–A102. [Google Scholar] [CrossRef]
- Zhang, W.-J. A Review of the Electrochemical Performance of Alloy Anodes for Lithium-Ion Batteries. J. Power Sources 2011, 196, 13–24. [Google Scholar] [CrossRef]
- Obrovac, M.N.; Chevrier, V.L. Alloy Negative Electrodes for Li-Ion Batteries. Chem. Rev. 2014, 114, 11444–11502. [Google Scholar] [CrossRef]
- Huggins, R.A. Lithium Alloy Negative Electrodes. J. Power Sources 1999, 81–82, 13–19. [Google Scholar] [CrossRef]
- Goriparti, S.; Miele, E.; De Angelis, F.; Di Fabrizio, E.; Proietti Zaccaria, R.; Capiglia, C. Review on Recent Progress of Nanostructured Anode Materials for Li-Ion Batteries. J. Power Sources 2014, 257, 421–443. [Google Scholar] [CrossRef] [Green Version]
- Winter, M.; Besenhard, J.O. Electrochemical Lithiation of Tin and Tin-Based Intermetallics and Composites. Electrochim. Acta 1999, 45, 31–50. [Google Scholar] [CrossRef]
- Derrien, G.; Hassoun, J.; Panero, S.; Scrosati, B. Nanostructured Sn–C Composite as an Advanced Anode Material in High-Performance Lithium-Ion Batteries. Adv. Mater. 2007, 19, 2336–2340. [Google Scholar] [CrossRef]
- Jung, Y.S.; Lee, K.T.; Ryu, J.H.; Im, D.; Oh, S.M. Sn-Carbon Core-Shell Powder for Anode in Lithium Secondary Batteries. J. Electrochem. Soc. 2005, 152, A1452–A1457. [Google Scholar] [CrossRef]
- Hassoun, J.; Derrien, G.; Panero, S.; Scrosati, B. A Nanostructured Sn–C Composite Lithium Battery Electrode with Unique Stability and High Electrochemical Performance. Adv. Mater. 2008, 20, 3169–3175. [Google Scholar] [CrossRef]
- Kim, I.-S.; Blomgren, G.E.; Kumta, P.N. Sn/C Composite Anodes for Li-Ion Batteries. Electrochem. Solid-State Lett. 2004, 7, A44–A48. [Google Scholar] [CrossRef]
- Xu, L.; Kim, C.; Shukla, A.K.; Dong, A.; Mattox, T.M.; Milliron, D.J.; Cabana, J. Monodisperse Sn Nanocrystals as a Platform for the Study of Mechanical Damage during Electrochemical Reactions with Li. Nano Lett. 2013, 13, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Liu, Y.; Lu, Z.; Ma, C.; Ge, T.; Li, W.; Yang, G. The Effect of Passivation Film in Preparation 3D Structural Carbon Paper/Tin Oxide@Carbon as Freestanding Anode for Lithium-Ion Batteries. Appl. Surf. Sci. 2018, 435, 1307–1313. [Google Scholar] [CrossRef]
- Wang, C.-M.; Xu, W.; Liu, J.; Zhang, J.-G.; Saraf, L.V.; Arey, B.W.; Choi, D.; Yang, Z.-G.; Xiao, J.; Thevuthasan, S.; et al. In Situ Transmission Electron Microscopy Observation of Microstructure and Phase Evolution in a SnO2 Nanowire during Lithium Intercalation. Nano Lett. 2011, 11, 1874–1880. [Google Scholar] [CrossRef]
- Larcher, D.; Beattie, S.; Morcrette, M.; Edstrom, K.; Jumas, J.-C.; Tarascon, J.-M. Recent Findings and Prospects in the Field of Pure Metals as Negative Electrodes for Li-Ion Batteries. J. Mater. Chem. 2007, 17, 3759–3772. [Google Scholar] [CrossRef]
- Lou, X.W.; Wang, Y.; Yuan, C.; Lee, J.Y.; Archer, L.A. Template-Free Synthesis of SnO2 Hollow Nanostructures with High Lithium Storage Capacity. Adv. Mater. 2006, 18, 2325–2329. [Google Scholar] [CrossRef]
- Han, S.; Jang, B.; Kim, T.; Oh, S.M.; Hyeon, T. Simple Synthesis of Hollow Tin Dioxide Microspheres and Their Application to Lithium-Ion Battery Anodes. Adv. Func. Mater. 2005, 15, 1845–1850. [Google Scholar] [CrossRef]
- Woo, H.; Wi, S.; Kim, J.; Kim, J.; Lee, S.; Hwang, T.; Kang, J.; Kim, J.; Park, K.; Gil, B.; et al. Complementary Surface Modification by Disordered Carbon and Reduced Graphene Oxide on SnO2 Hollow Spheres as an Anode for Li-Ion Battery. Carbon 2018, 129, 342–348. [Google Scholar] [CrossRef]
- Liu, D.; Kong, Z.; Liu, X.; Fu, A.; Wang, Y.; Guo, Y.G.; Guo, P.; Li, H.; Zhao, X.S. Spray-Drying-Induced Assembly of Skeleton-Structured SnO2/Graphene Composite Spheres as Superior Anode Materials for High-Performance Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2018, 10, 2515–2525. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, H.C.; Lee, J.Y. Highly Reversible Lithium Storage in Porous SnO2 Nanotubes with Coaxially Grown Carbon Nanotube Overlayers. Adv. Mater. 2006, 18, 645–649. [Google Scholar] [CrossRef]
- Retoux, R.; Brousse, T.; Schleich, D.M. High-Resolution Electron Microscopy Investigation of Capacity Fade in SnO2 Electrodes for Lithium-Ion Batteries. J. Electrochem. Soc. 1999, 146, 2472–2476. [Google Scholar] [CrossRef]
- Li, N.; Martin, C.R.; Scrosati, B. A High-Rate, High-Capacity, Nanostructured Tin Oxide Electrode. Electrochem. Solid-State Lett. 2000, 3, 316–318. [Google Scholar] [CrossRef]
- Chang, S.T.; Leu, I.C.; Liao, C.L.; Yen, J.H.; Hon, M.H. Electrochemical Behavior of Nanocrystalline Tin Oxide Electrodeposited on a Cu Substrate for Li-Ion Batteries. J. Mater. Chem. 2004, 14, 1821–1826. [Google Scholar] [CrossRef]
- Hulteen, J.C.; Martin, C.R. A General Template-Based Method for the Preparation of Nanomaterials. J. Mater. Chem. 1997, 7, 1075–1087. [Google Scholar] [CrossRef]
- Liu, L.; An, M.; Yang, P.; Zhang, J. Superior Cycle Performance and High Reversible Capacity of SnO2/Graphene Composite as an Anode Material for Lithium-Ion Batteries. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Yin, X.M.; Li, C.C.; Zhang, M.; Hao, Q.Y.; Liu, S.; Chen, L.B.; Wang, T.H. One-Step Synthesis of Hierarchical SnO2 Hollow Nanostructures via Self-Assembly for High Power Lithium Ion Batteries. J. Phys. Chem. C 2010, 114, 8084–8088. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, Q.; Wang, C.; Sun, L.; Yi, Z.; Wang, L. Large-Scale Fabrication of Core-Shell Structured C/SnO2 Hollow Spheres as Anode Materials with Improved Lithium Storage Performance. Small 2017, 13, 1701993–1702012. [Google Scholar] [CrossRef]
- Lou, X.W.; Li, C.M.; Archer, L.A. Designed Synthesis of Coaxial SnO2@carbon Hollow Nanospheres for Highly Reversible Lithium Storage. Adv. Mater. 2009, 21, 2536–2539. [Google Scholar] [CrossRef] [Green Version]
- Lou, X.W.; Yuan, C.; Archer, L.A. Shell-by-Shell Synthesis of Tin Oxide Hollow Colloids with Nanoarchitectured Walls: Cavity Size Tuning and Functionalization. Small 2007, 3, 261–265. [Google Scholar] [CrossRef]
- Guo, H.; Mao, R.; Tian, D.; Wang, W.; Zhao, D.; Yang, X.; Wang, S. Morphology-Controlled Synthesis of SnO2/C Hollow Core-Shell Nanoparticle Aggregates with Improved Lithium Storage. J. Mater. Chem. A 2013, 1, 3652–3658. [Google Scholar] [CrossRef]
- Wu, P.; Du, N.; Zhang, H.; Yu, J.; Qi, Y.; Yang, D. Carbon-coated SnO2 Nanotubes: Template-Engaged Synthesis and Their Application in Lithium-Ion Batteries. Nanoscale 2011, 3, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, P.; Ye, Y.; Wang, H.; Zhou, Y.; Tang, Y.; Lu, T. Designed Synthesis of SnO2@C Yolk–Shell Spheres for High-Performance Lithium Storage. Cryst. Eng. Comm. 2014, 16, 517–521. [Google Scholar] [CrossRef]
- Courtney, I.A.; McKinnon, W.R.; Dahn, J.R. On the Aggregation of Tin in SnO Composite Glasses Caused by the Reversible Reaction with Lithium. J. Electrochem. Soc. 1999, 146, 59–68. [Google Scholar] [CrossRef]
- Kasavajjula, U.; Wang, C.; Appleby, A.J. Nano- and Bulk-Silicon-Based Insertion Anodes for Lithium-Ion Secondary Cells. J. Power Sources 2007, 163, 1003–1039. [Google Scholar] [CrossRef]
- Lee, S.W.; McDowell, M.T.; Choi, J.W.; Cui, Y. Anomalous Shape Changes of Silicon Nanopillars by Electrochemical Lithiation. Nano Lett. 2011, 11, 3034–3039. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wu, Q.; Li, J.; Xiao, X.; Lott, A.; Lu, W.; Sheldon, B.W.; Wu, J. Silicon-Based Nanomaterials for Lithium-Ion Batteries: A Review. Adv. Energy Mater. 2014, 4, 1300882–1300904. [Google Scholar] [CrossRef]
- Priebe, M.; Fromm, K.M. One-Pot Synthesis and Catalytic Properties of Encapsulated Silver Nanoparticles in Silica Nanocontainers. Part. Part. Syst. Charact. 2014, 31, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shah, Z.H.; Zhang, S.; Lu, R. Silica-Based Nanocomposites via Reverse Microemulsions: Classifications, Preparations, and Applications. Nanoscale 2014, 6, 4418–4437. [Google Scholar] [CrossRef]
- Lin, C.H.; Chang, J.H.; Yeh, Y.Q.; Wu, S.H.; Liu, Y.H.; Mou, C.Y. Formation of Hollow Silica Nanospheres by Reverse Microemulsion. Nanoscale 2015, 7, 9614–9626. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Wu, S.-H.; Tseng, C.-T.; Hung, Y.; Chang, C.; Mou, C.-Y. Synthesis of Hollow Silica Nanospheres with a Microemulsion as the Template. Chem. Commun. 2009, 3542–3544. [Google Scholar] [CrossRef]
- Wu, S.-H.; Tseng, C.-T.; Lin, Y.-S.; Lin, C.-H.; Hung, Y.; Mou, C.-Y. Catalytic Nano-Rattle of Au@hollow silica: Towards a Poison-Resistant Nanocatalyst. J. Mater. Chem. 2011, 21, 789–794. [Google Scholar] [CrossRef]
- Yuan, D.; Yuan, X.; Zou, W.; Zeng, F.; Huang, X.; Zhou, S. Synthesis of Graphitic Mesoporous Carbon from Sucrose as a Catalyst Support for Ethanol Electro-oxidation. J. Mater. Chem. 2012, 22, 17820–17826. [Google Scholar] [CrossRef]
- Yu, L.; Falco, C.; Weber, J.; White, R.J.; Howe, J.Y.; Titirici, M.-M. Carbohydrate-Derived Hydrothermal Carbons: A Thorough Characterization Study. Langmuir 2012, 28, 12373–12383. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Chen, J.S.; Qi, G.; Duan, X.; Wang, Z.; Giannelis, E.P.; Archer, L.A.; Lou, X.W. Formation of SnO2 Hollow Nanospheres inside Mesoporous Silica Nanoreactors. J. Am. Chem. Soc. 2011, 133, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Tuinstra, F.; Koenig, J.L. Raman Spectrum of Graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman Spectra of Disordered and Amorphous Carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef] [Green Version]
- Courtney, I.A.; Dahn, J.R. Electrochemical and In Situ X-Ray Diffraction Studies of the Reaction of Lithium with Tin Oxide Composites. J. Electrochem. Soc. 1997, 144, 2045–2052. [Google Scholar] [CrossRef]
- Cheng, B.; Russell, J.M.; Shi, W.; Zhang, L.; Samulski, E.T. Large-Scale, Solution-Phase Growth of Single-Crystalline SnO2 Nanorods. J. Am. Chem. Soc. 2004, 126, 5972–5973. [Google Scholar] [CrossRef]
- Hu, R.; Chen, D.; Waller, G.; Ouyang, Y.; Chen, Y.; Zhao, B.; Rainwater, B.; Yang, C.; Zhu, M.; Liu, M. Dramatically Enhanced Reversibility of Li2O in SnO2-Based Electrodes: The Effect of Nanostructure on High Initial Reversible Capacity. Energy Environ. Sci. 2016, 9, 595–603. [Google Scholar] [CrossRef]
- Fattakhova-Rohlfing, D.; Zoller, F.; Bohm, D.; Bein, T. Tin Oxide-Based Nanomaterials and Their Application as Anodes in Lithium-Ion Batteries and Beyond. ChemSusChem 2019, 12, 4140–4159. [Google Scholar]
- Xie, J.; Imanishi, N.; Hirano, A.; Takeda, Y.; Yamamoto, O.; Zhao, X.B.; Cao, G.S. Li-Ion Diffusion Behavior in Sn, SnO and SnO2 Thin Films Studied by Galvanostatic Intermittent Titration Technique. Solid State Ion. 2010, 181, 1611–1615. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Li, Q.; Wang, C.; Chen, A.; Zhang, Z.; Fan, R.; Yin, L. Ordered Mesoporous SnO2 with a Highly Crystalline State as an Anode Material for Lithium Ion Batteries with Enhanced Electrochemical Performance. CrystEngComm 2013, 15, 3696–3704. [Google Scholar] [CrossRef]
- Lou, X.W.; Deng, D.; Lee, J.Y.; Archer, L.A. Preparation of SnO2/Carbon Composite Hollow Spheres and Their Lithium Storage Properties. Chem. Mater. 2008, 20, 6562–6566. [Google Scholar] [CrossRef]
- Deng, D.; Lee, J.Y. Hollow Core–Shell Mesospheres of Crystalline SnO2 Nanoparticle Aggregates for High Capacity Li+ Ion Storage. Chem. Mater. 2008, 20, 1841–1846. [Google Scholar] [CrossRef]
- Sun, X.; Liu, J.; Li, Y. Oxides@C Core−Shell Nanostructures: One-Pot Synthesis, Rational Conversion, and Li Storage Property. Chem. Mater. 2006, 18, 3486–3494. [Google Scholar] [CrossRef]
- Yan, J.; Song, H.; Zhang, H.; Yan, J.; Chen, X.; Wang, F.; Yang, H.; Gomi, M. TixSn1−xO3 Solid Solution as an Anode Material in Lithium-Ion Batteries. Electrochim. Acta 2012, 72, 186–191. [Google Scholar] [CrossRef]
- Richardson, T.J.; Chen, G. Solid Solution Lithium Alloy Cermet Anodes. J. Power Sources 2007, 174, 810–812. [Google Scholar] [CrossRef] [Green Version]
- Dahn, J.R.; Mar, R.E.; Abouzeid, A. Combinatorial Study of Sn1−xCox (0 < x < 0.6) and [Sn0.55Co0.45]1-yCy (0 < y < 0.5) Alloy Negative Electrode Materials for Li-Ion Batteries. J. Electrochem. Soc. 2006, 153, A361–A365. [Google Scholar] [CrossRef]
- Kucinskis, G.; Bajars, G.; Kleperis, J. Graphene in Lithium Ion Battery Cathode Materials: A review. J. Power Sources 2013, 240, 66–79. [Google Scholar] [CrossRef]
- Winter, M.; Besenhard, J.O.; Spahr, M.E.; Novák, P. Insertion Electrode Materials for Rechargeable Lithium Batteries. Adv. Mater. 1998, 10, 725–763. [Google Scholar] [CrossRef]
- Vargas C, O.A.; Caballero, A.; Morales, J. Can the Performance of Graphene Nanosheets for Lithium Storage in Li-Ion Batteries be Predicted? Nanoscale 2012, 4, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Han, W.Q. Metallic Sn-Based Anode Materials: Application in High-Performance Lithium-Ion and Sodium-Ion Batteries. Adv. Sci. 2017, 4, 1700298–1700318. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET Surface Area (m2 g−1) | Pore Volume (mL g−1) | Mean Pore Size (nm) | Presence of Micro-Pores (<4 nm) |
|---|---|---|---|---|
| (i) SiO2@C | 43 | 0.11 | 11.4 | x |
| (ii) Carbon nanocontainers | 167 | 0.45 | 8.7 | o |
| (iii) SnO2@C nano-rattles | 110 | 0.16 | 4.7 | o |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharajan, S.; Kwon, N.H.; Brodard, P.; Fromm, K.M. A Nano-Rattle SnO2@carbon Composite Anode Material for High-Energy Li-ion Batteries by Melt Diffusion Impregnation. Nanomaterials 2020, 10, 804. https://doi.org/10.3390/nano10040804
Maharajan S, Kwon NH, Brodard P, Fromm KM. A Nano-Rattle SnO2@carbon Composite Anode Material for High-Energy Li-ion Batteries by Melt Diffusion Impregnation. Nanomaterials. 2020; 10(4):804. https://doi.org/10.3390/nano10040804
Chicago/Turabian StyleMaharajan, Sivarajakumar, Nam Hee Kwon, Pierre Brodard, and Katharina M. Fromm. 2020. "A Nano-Rattle SnO2@carbon Composite Anode Material for High-Energy Li-ion Batteries by Melt Diffusion Impregnation" Nanomaterials 10, no. 4: 804. https://doi.org/10.3390/nano10040804
APA StyleMaharajan, S., Kwon, N. H., Brodard, P., & Fromm, K. M. (2020). A Nano-Rattle SnO2@carbon Composite Anode Material for High-Energy Li-ion Batteries by Melt Diffusion Impregnation. Nanomaterials, 10(4), 804. https://doi.org/10.3390/nano10040804