Origin of the Photoluminescence of Metal Nanoclusters: From Metal-Centered Emission to Ligand-Centered Emission



Abstract

{kind=link}
{kind=link}
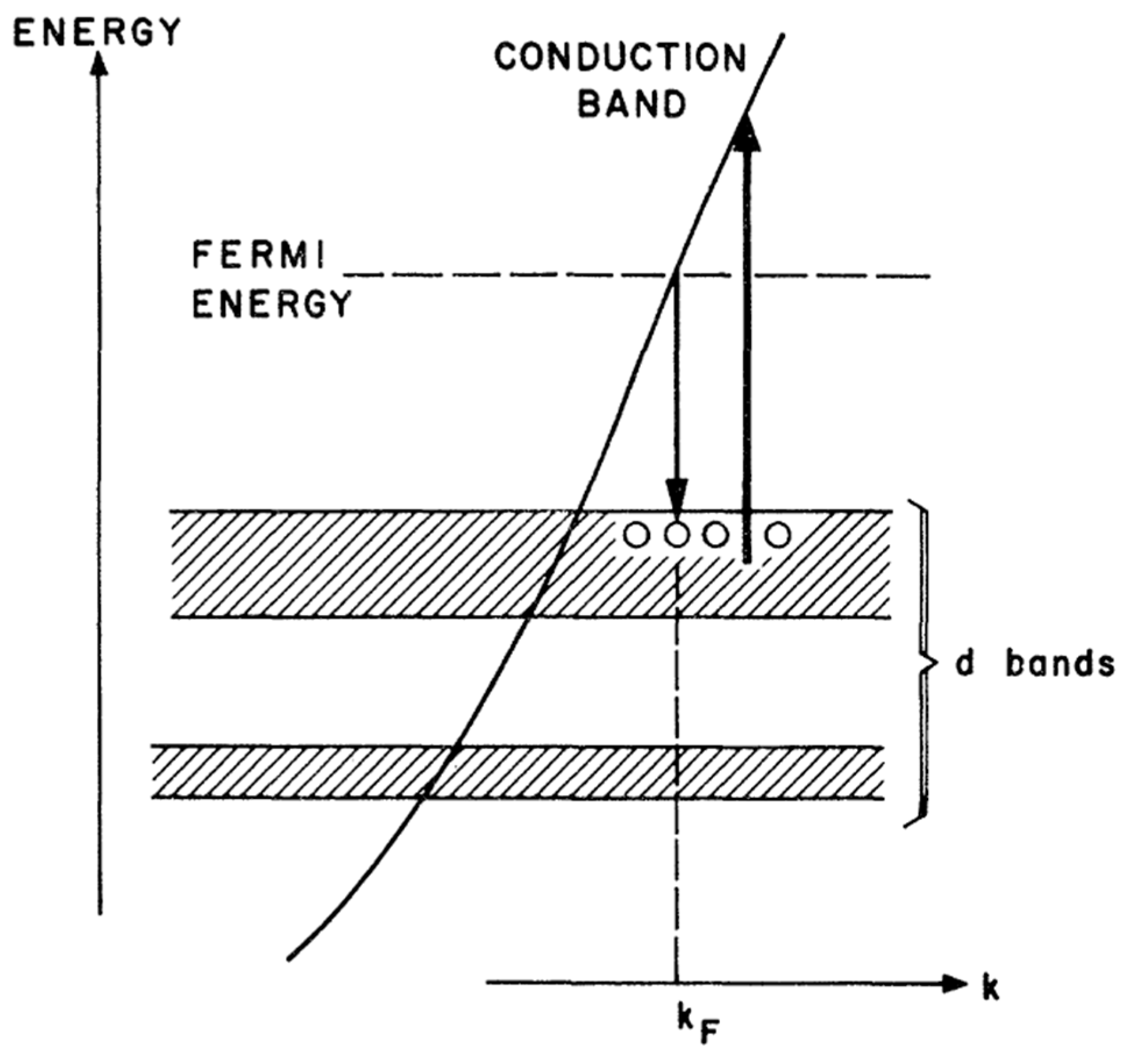{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
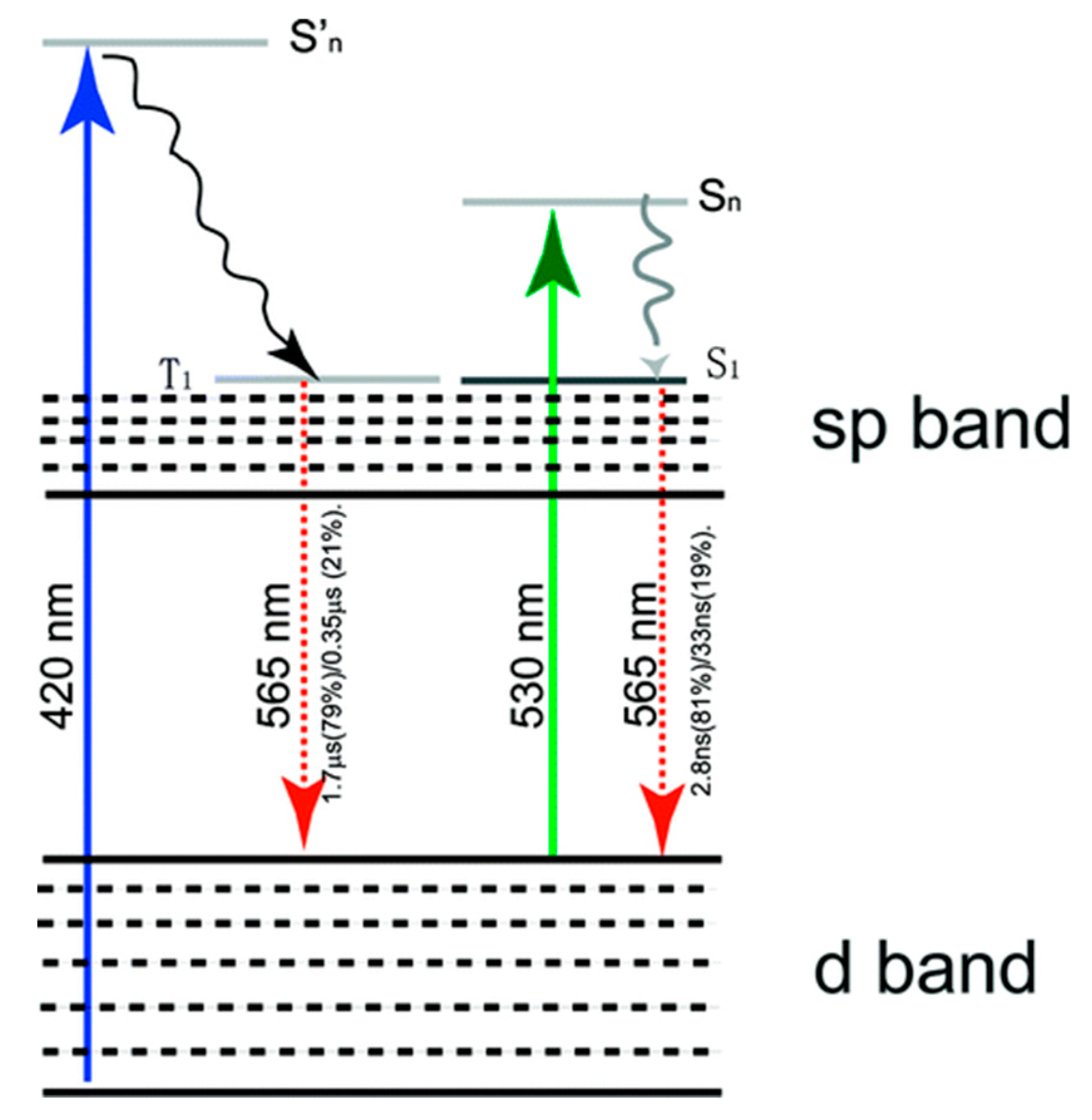{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Photoemission Mechanism of Metal Nanoclusters
2.1. Size-Dependent Photoemission
2.2. Size-Independent Photoemission
2.2.1. Ligand Effect
2.2.2. Metal Valence State Correlated Photoemission
2.2.3. Self-Assembly Governed Photoemission
3. Our P Band Intermediate State (PBIS) Model Dominates the Photoluminescence Emission of MNCs
4. Summary and Outlooks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, J.; Nicovich, P.R.; Dickson, R.M. Highly fluorescent noble-metal quantum dots. Ann. Rev. Phys. Chem. 2007, 58, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Diez, I.; Ras, R.H. Fluorescent silver nanoclusters. Nanoscale 2011, 3, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhou, C.; Yu, M.; Liu, J. Different sized luminescent gold nanoparticles. Nanoscale 2012, 4, 4073–4083. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wen, X.; Toh, Y.-R.; Ma, X.; Tang, J. Fluorescent Metallic Nanoclusters: Electron Dynamics, Structure, and Applications. Part. Part. Syst. Charact. 2015, 32, 142–163. [Google Scholar] [CrossRef]
- Higaki, T.; Li, Q.; Zhou, M.; Zhao, S.; Li, Y.; Li, S.; Jin, R. Toward the Tailoring Chemistry of Metal Nanoclusters for Enhancing Functionalities. Acc. Chem. Res. 2018, 51, 2764–2773. [Google Scholar] [CrossRef]
- Nieto-Ortega, B.; Burgi, T. Vibrational Properties of Thiolate-Protected Gold Nanoclusters. Acc. Chem. Res. 2018, 51, 2811–2819. [Google Scholar] [CrossRef]
- He, X.; Yam, V.W.W. Luminescent gold(I) complexes for chemosensing. Coord. Chem. Rev. 2011, 255, 2111–2123. [Google Scholar] [CrossRef]
- Shang, L.; Dong, S.; Nienhaus, G.U. Ultra-small fluorescent metal nanoclusters: Synthesis and biological applications. Nano Today 2011, 6, 401–418. [Google Scholar] [CrossRef]
- Choi, S.; Dickson, R.M.; Yu, J. Developing luminescent silver nanodots for biological applications. Chem. Soc. Rev. 2012, 41, 1867–1891. [Google Scholar] [CrossRef]
- Guével, X.L. Recent Advances on the Synthesis ofMetal Quantum Nanoclusters and Their Application for Bioimaging. IEEE J. Top. Quant. 2014, 20, 6801312. [Google Scholar]
- Palmal, S.; Jana, N.R. Gold nanoclusters with enhanced tunable fluorescence as bioimaging probes. WIREs Nanomed. Nanobiotechnol. 2014, 6, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, E. Metal nanoclusters: New fluorescent probes for sensors and bioimaging. Nano Today 2014, 9, 132–157. [Google Scholar] [CrossRef]
- Chen, L.Y.; Wang, C.W.; Yuan, Z.; Chang, H.T. Fluorescent gold nanoclusters: Recent advances in sensing and imaging. Anal. Chem. 2015, 87, 216–229. [Google Scholar] [CrossRef]
- Zheng, Y.; Lai, L.; Liu, W.; Jiang, H.; Wang, X. Recent advances in biomedical applications of fluorescent gold nanoclusters. Adv. Colloid Interface Sci. 2017, 242, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, H.; Chen, B.; Zhao, G. Highly fluorescent gold nanoclusters stabilized by food proteins: From preparation to application in detection of food contaminants and bioactive nutrients. Crit. Rev. Food Sci. Nutr. 2018, 58, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Bonacic-Koutecky, V.; Kulesza, A.; Gell, L.; Mitric, R.; Antoine, R.; Bertorelle, F.; Hamouda, R.; Rayane, D.; Broyer, M.; Tabarin, T.; et al. Silver cluster-biomolecule hybrids: From basics towards sensors. Phys. Chem. Chem. Phys. 2012, 14, 9282–9290. [Google Scholar] [CrossRef]
- Goswami, N.; Yao, Q.; Luo, Z.; Li, J.; Chen, T.; Xie, J. Luminescent Metal Nanoclusters with Aggregation-Induced Emission. J. Phys. Chem. Lett. 2016, 7, 962–975. [Google Scholar] [CrossRef]
- New, S.Y.; Lee, S.T.; Su, X.D. DNA-templated silver nanoclusters: Structural correlation and fluorescence modulation. Nanoscale 2016, 8, 17729–17746. [Google Scholar] [CrossRef]
- Li, D.; Chen, Z.; Mei, X. Fluorescence enhancement for noble metal nanoclusters. Adv. Colloid Interface Sci. 2017, 250, 25–39. [Google Scholar] [CrossRef]
- Musnier, B.; Wegner, K.D.; Comby-Zerbino, C.; Trouillet, V.; Jourdan, M.; Hausler, I.; Antoine, R.; Coll, J.L.; Resch-Genger, U.; Le Guevel, X. High photoluminescence of shortwave infrared-emitting anisotropic surface charged gold nanoclusters. Nanoscale 2019, 11, 12092–12096. [Google Scholar] [CrossRef]
- Peric, M.; Sanader Marsic, Z.; Russier-Antoine, I.; Fakhouri, H.; Bertorelle, F.; Brevet, P.F.; le Guevel, X.; Antoine, R.; Bonacic-Koutecky, V. Ligand shell size effects on one- and two-photon excitation fluorescence of zwitterion functionalized gold nanoclusters. Phys. Chem. Chem. Phys. 2019, 21, 23916–23921. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Corpuz, R.D.; Yonezawa, T. Matrix Sputtering Method: A Novel Physical Approach for Photoluminescent Noble Metal Nanoclusters. Acc. Chem. Res. 2017, 50, 2986–2995. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, M.; Ren, J.; Qu, X. Metal nanoclusters: Novel probes for diagnostic and therapeutic applications. Chem. Soc. Rev. 2015, 44, 8636–8663. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Rao, B.; Jiang, W.; Yang, S.; Zhu, M. The photoluminescent metal nanoclusters with atomic precision. Coord. Chem. Rev. 2019, 378, 595–617. [Google Scholar] [CrossRef]
- Kang, X.; Zhu, M. Tailoring the photoluminescence of atomically precise nanoclusters. Chem. Soc. Rev. 2019, 48, 2422–2457. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, C.; Dickson, R.M. Highly Fluorescent, Water-Soluble, Size-Tunable Gold Quantum Dots. Phys. Rev. Lett. 2004, 93, 077402. [Google Scholar] [CrossRef]
- LEE, T.-H.; Gonzalez, J.I.; Zheng, J.; Dickson, R.M. Single-Molecule Optoelectronics. Acc. Chem. Res. 2005, 38, 534–541. [Google Scholar] [CrossRef]
- Jin, R. Quantum sized, thiolate-protected gold nanoclusters. Nanoscale 2010, 2, 343–362. [Google Scholar] [CrossRef]
- Mooradian, A. Photoluminescence of Metals. Phys. Rev. Lett. 1969, 22, 185–187. [Google Scholar] [CrossRef]
- Link, S.; Beeby, A.; FitzGerald, S.; El-Sayed, M.A.; Schaaff, T.G.; Whetten, R.L. Visible to Infrared Luminescence from a 28-Atom Gold Cluster. J. Phys. Chem. B 2002, 106, 3410–3415. [Google Scholar] [CrossRef]
- Forward, J.M.; Bohmann, D.; John, P.; Fackler, J.; Staples, R.J. Luminescence Studies of Gold(I) Thiolate Complexes. Inorg. Chem. 1995, 34, 6330–6336. [Google Scholar] [CrossRef]
- Cha, S.-H.; Kim, J.-U.; Kim, K.-H.; Lee, J.-C. Preparation and Photoluminescent Properties of Gold(I)-Alkanethiolate Complexes Having Highly Ordered Supramolecular Structures. Chem. Mater. 2007, 19, 6297–6303. [Google Scholar] [CrossRef]
- Wu, Z.; Jin, R. On the ligand’s role in the fluorescence of gold nanoclusters. Nano Lett. 2010, 10, 2568–2573. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.A.J.; Li, J.K.; Lee, C.H.; Shen, J.L.; Hsieh, J.T.; Chan, W.H.; Wang, H.-H.; Yeh, H.I.; Chang, W.H. Synthesis of Fluorescent Metallic Nanoclusters towards Biomedical Application: Recent Progress and Present Challenges. J. Med. Biol. Eng. 2009, 29, 276–283. [Google Scholar]
- Jadzinsky, P.D.; Calero, G.; Ackerson, C.J.; Bushnell, D.A.; Kornberg, R.D. Structure of a Thiol Monolayer–Protected Gold Nanoparticle at 1.1 Å Resolution. Science 2007, 318, 430–433. [Google Scholar] [CrossRef]
- Zhu, M.; Aikens, C.M.; Hollander, F.J.; Schatz, G.C.; Jin, R. Correlating the Crystal Structure of A Thiol-Protected Au25 Cluster and Optical Properties. J. Am. Chem. Soc. 2008, 130, 5883–5885. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Y.; Lei, J.; Shi, L.; Wu, X.; Makinen, V.; Lin, S.; Tang, Z.; He, J.; Hakkinen, H.; et al. Ligand-stabilized Au13Cux (x = 2, 4, 8) bimetallic nanoclusters: Ligand engineering to control the exposure of metal sites. J. Am. Chem. Soc. 2013, 135, 9568–9571. [Google Scholar] [CrossRef]
- Aikens, C.M. Electronic and Geometric Structure, Optical Properties, and Excited State Behavior in Atomically Precise Thiolate-Stabilized Noble Metal Nanoclusters. Acc. Chem. Res. 2018, 51, 3065–3073. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, M.; So, W.Y.; Huang, J.; Li, M.; Kauffman, D.R.; Cotlet, M.; Higaki, T.; Peteanu, L.A.; Shao, Z.; et al. A Mono-cuboctahedral Series of Gold Nanoclusters: Photoluminescence Origin, Large Enhancement, Wide Tunability, and Structure-Property Correlation. J. Am. Chem. Soc. 2019, 141, 5314–5325. [Google Scholar] [CrossRef]
- Khatun, E.; Bodiuzzaman, M.; Sugi, K.S.; Chakraborty, P.; Paramasivam, G.; Dar, W.A.; Ahuja, T.; Antharjanam, S.; Pradeep, T. Confining an Ag10 Core in an Ag12 Shell: A Four-Electron Superatom with Enhanced Photoluminescence upon Crystallization. ACS Nano 2019, 13, 5753–5759. [Google Scholar] [CrossRef]
- Jia, X.; Li, J.; Wang, E. Supramolecular self-assembly of morphology-dependent luminescent Ag nanoclusters. Chem. Commun. 2014, 50, 9565–9568. [Google Scholar] [CrossRef]
- Bertorelle, F.; Russier-Antoine, I.; Calin, N.; Comby-Zerbino, C.; Bensalah-Ledoux, A.; Guy, S.; Dugourd, P.; Brevet, P.F.; Sanader, Z.; Krstic, M.; et al. Au10(SG)10: A Chiral Gold Catenane Nanocluster with Zero Confined Electrons. Optical Properties and First-Principles Theoretical Analysis. J. Phys. Chem. Lett. 2017, 8, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Shan, B.; Huang, F.; Yang, S.; Peng, B.; Yuan, E.; Wu, P.; Zhang, K. P band intermediate state (PBIS) tailors photoluminescence emission at confined nanoscale interface. Commun. Chem. 2019, 2, 132. [Google Scholar] [CrossRef]
- Zhou, C.; Sun, C.; Yu, M.; Qin, Y.; Wang, J.; Kim, M.; Zheng, J. Luminescent Gold Nanoparticles with Mixed Valence States Generated from Dissociation of Polymeric Au(I) Thiolates. J. Phys. Chem. C 2010, 114, 7727–7732. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, J.; Gao, Y.; Liu, H.; Li, T.; Zou, H.; Wang, Z.; Zhang, K.; Wang, Y.; Zhang, H.; et al. Assembly-Induced Enhancement of Cu Nanoclusters Luminescence with Mechanochromic Property. J. Am. Chem. Soc. 2015, 137, 12906–12913. [Google Scholar] [CrossRef] [PubMed]
- Benito, Q.; Le Goff, X.F.; Maron, S.; Fargues, A.; Garcia, A.; Martineau, C.; Taulelle, F.; Kahlal, S.; Gacoin, T.; Boilot, J.P.; et al. Polymorphic copper iodide clusters: Insights into the mechanochromic luminescence properties. J. Am. Chem. Soc. 2014, 136, 11311–11320. [Google Scholar] [CrossRef] [PubMed]
- Wilcoxon, J.P.; Martin, J.E.; Parsapour, F.; Wiedenman, B.; Kelley, D.F. Photoluminescence from nanosize gold clusters. J. Chem. Phys. 1998, 108, 9137–9143. [Google Scholar] [CrossRef]
- Yan, J.; Teo, B.K.; Zheng, N. Surface Chemistry of Atomically Precise Coinage-Metal Nanoclusters: From Structural Control to Surface Reactivity and Catalysis. Acc. Chem. Res. 2018, 51, 3084–3093. [Google Scholar] [CrossRef]
- Harbich, W.; Fedrigo, S.; Buttet, J.; Lindsay, D.M. Deposition of mass selected gold clusters in solid krypton. J. Chem. Phys. 1992, 96, 8104–8108. [Google Scholar] [CrossRef]
- Fedrigo, S.; Harbich, W.; Buttet, J. Optical response of Ag2, Ag3, Au2, and Au3 in argon matrices. J. Chem. Phys. 1993, 99, 5712–5717. [Google Scholar] [CrossRef]
- Zheng, J.; Dickson, R.M. Individual Water-Soluble Dendrimer-Encapsulated Silver Nanodot Fluorescence. J. Am. Chem. Soc. 2002, 124, 13982–13983. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Petty, J.T.; Dickson, R.M. High Quantum Yield Blue Emission from Water-Soluble Au8 Nanodots. J. Am. Chem. Soc. 2003, 125, 7780–7781. [Google Scholar] [CrossRef] [PubMed]
- Angel, L.A.; Majors, L.T.; Dharmaratne, A.C.; Dass, A. Ion Mobility Mass Spectrometry of Au25(SCH2CH2Ph)18 Nanoclusters. ACS Nano 2010, 4, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Qian, H.; Wu, Z.; Zhu, Y.; Zhu, M.; Mohanty, A.; Garg, N. Size Focusing: A Methodology for Synthesizing Atomically Precise Gold Nanoclusters. J. Phys. Chem. Lett. 2010, 1, 2903–2910. [Google Scholar] [CrossRef]
- Udaya Bhaskara Rao, T.; Pradeep, T. Luminescent Ag7 and Ag8 clusters by interfacial synthesis. Angew. Chem. Int. Ed. 2010, 49, 3925–3929. [Google Scholar] [CrossRef]
- Luo, Z.; Yuan, X.; Yu, Y.; Zhang, Q.; Leong, D.T.; Lee, J.Y.; Xie, J. From aggregation-induced emission of Au(I)-thiolate complexes to ultrabright Au(0)@Au(I)-thiolate core-shell nanoclusters. J. Am. Chem. Soc. 2012, 134, 16662–16670. [Google Scholar] [CrossRef]
- Liu, J.; Duchesne, P.N.; Yu, M.; Jiang, X.; Ning, X.; Vinluan, R.D., 3rd; Zhang, P.; Zheng, J. Luminescent Gold Nanoparticles with Size-Independent Emission. Angew. Chem. Int. Ed. 2016, 55, 8894–8898. [Google Scholar] [CrossRef]
- Xie, J.; Zheng, Y.; Ying, J.Y. Protein-Directed Synthesis of Highly Fluorescent Gold Nanoclusters. J. Am. Chem. Soc. 2009, 131, 888–889. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, M.; Yang, J.; Zheng, X.; Cai, W.; Meng, G.; Qian, H.; Wang, H.; Jin, R. Well-defined nanoclusters as fluorescent nanosensors: A case study on Au25(SG)18. Small 2012, 8, 2028–2035. [Google Scholar] [CrossRef]
- Brewer, L.; King, B.A.; Wang, J.L.; Meyer, B.; Moore, G.F. Absorption Spectrum of Silver Atoms in Solid Argon, Krypton, and Xenon. J. Chem. Phys. 1968, 49, 5209–5213. [Google Scholar] [CrossRef]
- Simo, A.; Polte, J.; Pfander, N.; Vainio, U.; Emmerling, F.; Rademann, K. Formation mechanism of silver nanoparticles stabilized in glassy matrices. J. Am. Chem. Soc. 2012, 134, 18824–18833. [Google Scholar] [CrossRef]
- Cremer, G.D.; Coutiño-Gonzalez, E.; Roeffaers, M.B.J.; Moens, B.; Ollevier, J.; Auweraer, M.V.d.; Schoonheydt, R.; Jacobs, P.A.; Schryver, F.C.D.; Hofkens, J.; et al. Characterization of Fluorescence in Heat-Treated Silver-Exchanged Zeolites. J. Am. Chem. Soc. 2009, 131, 3049–3056. [Google Scholar] [CrossRef]
- Grandjean, D.; Coutiño-Gonzalez, E.; Cuong, N.T.; Fron, E.; Baekelant, W.; Aghakhani, S.; Schlexer, P.; D’Acapito, F.; Banerjee, D.; Roeffaers, M.B.J.; et al. Origin of the bright photoluminescence of few-atom silver clusters confined in LTA zeolites. Science 2018, 361, 686–690. [Google Scholar] [CrossRef]
- Fenwick, O.; Coutiño-Gonzalez, E.; Grandjean, D.; Baekelant, W.; Richard, F.; Bonacchi, S.; De Vos, D.; Lievens, P.; Roeffaers, M.; Hofkens, J.; et al. Tuning the energetics and tailoring the optical properties of silver clusters confined in zeolites. Nature Mater. 2016, 15, 1017–1022. [Google Scholar] [CrossRef]
- Richards, C.I.; Choi, S.; Hsiang, J.-C.; Antoku, Y.; Vosch, T.; Bongiorno, A.; Tzeng, Y.-L.; Dickson, R.M. Oligonucleotide-Stabilized Ag Nanocluster Fluorophores. J. Am. Chem. Soc. 2008, 130, 5038–5039. [Google Scholar] [CrossRef]
- Yeh, H.C.; Sharma, J.; Han, J.J.; Martinez, J.S.; Werner, J.H. A DNA-silver nanocluster probe that fluoresces upon hybridization. Nano Lett. 2010, 10, 3106–3110. [Google Scholar] [CrossRef]
- Pyo, K.; Thanthirige, V.D.; Kwak, K.; Pandurangan, P.; Ramakrishna, G.; Lee, D. Ultrabright Luminescence from Gold Nanoclusters: Rigidifying the Au(I)-Thiolate Shell. J. Am. Chem. Soc. 2015, 137, 8244–8250. [Google Scholar] [CrossRef]
- Londoño-Larrea, P.; Vanegas, J.P.; Cuaran-Acosta, D.; Zaballos-García, E.; Pérez-Prieto, J. Water-Soluble Naked Gold Nanoclusters Are Not Luminescent. Chem. Eur. J. 2017, 23, 8137–8141. [Google Scholar] [CrossRef]
- Zhu, M.; Eckenhoff, W.T.; Pintauer, T.; Jin, R. Conversion of Anionic [Au25(SCH2CH2Ph)18]− Cluster to Charge Neutral Cluster via Air Oxidation. J. Phys. Chem. C 2008, 112, 14221–14224. [Google Scholar] [CrossRef]
- Negishi, Y.; Chaki, N.K.; Shichibu, Y.; Whetten, R.L.; Tsukuda, T. Origin of Magic Stability of Thiolated Gold Clusters: A Case Study on Au25(SC6H13)18. J. Am. Chem. Soc. 2007, 129, 11322–11323. [Google Scholar] [CrossRef]
- Shang, L.; Dong, S. Facile preparation of water-soluble fluorescent silver nanoclusters using a polyelectrolyte template. Chem. Commun. 2008, 1088–1090. [Google Scholar] [CrossRef]
- Xie, Z.; Sun, P.; Wang, Z.; Li, H.; Yu, L.; Sun, D.; Chen, M.; Bi, Y.; Xin, X.; Hao, J. Metal-Organic Gels from Silver Nanoclusters with Aggregation-Induced Emission and Fluorescence-to-Phosphorescence Switching. Angew. Chem. Int. Ed. 2019, 58, 1–6. [Google Scholar]
- Luo, J.; Xie, Z.; Lam, J.W.Y.; Cheng, L.; Tang, B.Z.; Chen, H.; Qiu, C.; Kwok, H.S.; Zhan, X.; Liu, Y.; et al. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 2001, 1740–1741. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lam, J.W.Y.; Kwok, R.T.K.; Liu, B.; Tang, B.Z. Aggregation-induced emission: Fundamental understanding and future developments. Mater. Horiz. 2019, 6, 428–433. [Google Scholar] [CrossRef]
- Saigusa, H.; Lim, E.C. Excited-State Dynamics of Aromatic Clusters: Correlation between Exciton Interactions and Excimer Formation Dynamics. J. Phys. Chem. 1995, 99, 15738–15747. [Google Scholar] [CrossRef]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef]
- Jia, X.; Yang, X.; Li, J.; Li, D.; Wang, E. Stable Cu nanoclusters: From an aggregation-induced emission mechanism to biosensing and catalytic applications. Chem. Commun. 2014, 50, 237–239. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, D.; Zhang, H. Self-Assembly Driven Aggregation-Induced Emission of Copper Nanoclusters: A Novel Technology for Lighting. ACS Appl. Mater. Interfaces 2017, 10, 12071–12080. [Google Scholar] [CrossRef]
- Jia, X.; Li, J.; Wang, E. Cu nanoclusters with aggregation induced emission enhancement. Small 2013, 9, 3873–3879. [Google Scholar] [CrossRef]
- Wu, Z.; Dong, C.; Li, Y.; Hao, H.; Zhang, H.; Lu, Z.; Yang, B. Self-assembly of Au15 into single-cluster-thick sheets at the interface of two miscible high-boiling solvents. Angew. Chem. Int. Ed. 2013, 52, 9952–9955. [Google Scholar] [CrossRef]
- Ai, L.; Liu, Z.; Zhou, D.; Liu, J.; Zou, H.; Wu, Z.; Liu, Y.; Zhang, H.; Yang, B. Copper inter-nanoclusters distance-modulated chromism of self-assembly induced emission. Nanoscale 2017, 9, 18845–18854. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, Z.; Tian, Y.; Li, Y.; Ai, L.; Li, T.; Zou, H.; Liu, Y.; Zhang, X.; Zhang, H.; et al. Engineering the Self-Assembly Induced Emission of Cu Nanoclusters by Au(I) Doping. ACS Appl. Mater. Interfaces 2017, 9, 24899–24907. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, H.; Li, T.; Liu, J.; Yin, J.; Mohammed, O.F.; Bakr, O.M.; Liu, Y.; Yang, B.; Zhang, H. Contribution of Metal Defects in the Assembly Induced Emission of Cu Nanoclusters. J. Am. Chem. Soc. 2017, 139, 4318–4321. [Google Scholar] [CrossRef] [PubMed]
- White-Morris, R.L.; Olmstead, M.M.; Balch, A.L. Orange Luminescence and Structural Properties of Three Isostructural Halocyclohexylisonitrilegold(I) Complexes. Inorg. Chem. 2003, 42, 6741–6748. [Google Scholar] [CrossRef]
- Schmidbaur, H. The fascinating implications of new results in gold chemistry. Gold Bull. 1990, 23, 11–21. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, T.; Pan, H.; Yuan, Y.; Chen, L.; Liu, M.; Zhang, K.; Zhang, S.; Wu, P.; Xu, J. Photoemission mechanism of water-soluble silver nanoclusters: Ligand-to-metal-metal charge transfer vs. strong coupling between surface plasmon and emitters. J. Am. Chem. Soc. 2014, 136, 1686–1689. [Google Scholar] [CrossRef]
- Yang, T.; Dai, S.; Tan, H.; Zong, Y.; Liu, Y.; Chen, J.; Zhang, K.; Wu, P.; Zhang, S.; Xu, J.; et al. Mechanism of Photoluminescence in Ag Nanoclusters: Metal-Centered Emission versus Synergistic Effect in Ligand-Centered Emission. J. Phys. Chem. C 2019, 123, 18638–18645. [Google Scholar] [CrossRef]
- Yang, T.; Dai, S.; Yang, S.; Chen, L.; Liu, P.; Dong, K.; Zhou, J.; Chen, Y.; Pan, H.; Zhang, S.; et al. Interfacial Clustering-Triggered Fluorescence-Phosphorescence Dual Solvoluminescence of Metal Nanoclusters. J. Phys. Chem. Lett. 2017, 8, 3980–3985. [Google Scholar] [CrossRef]
- Newberry, R.W.; Orke, S.J.; Raines, R.T. n→pi* Interactions Are Competitive with Hydrogen Bonds. Org. Lett. 2016, 18, 3614–3617. [Google Scholar] [CrossRef]
- Kilgore, H.R.; Raines, R.T. n→π* Interactions Modulate the Properties of Cysteine Residues and Disulfide Bonds in Proteins. J. Am. Chem. Soc. 2018, 140, 17606–17611. [Google Scholar] [CrossRef]
- León, I.; Alonso, E.R.; Cabezas, C.; Mata, S.; Alonso, J.L. Unveiling the n→π* interactions in dipeptides. Commun. Chem. 2019, 2, 3. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, L.L.; Jiang, J.G.; Calin, N.; Lam, K.F.; Zhang, S.J.; Wu, H.H.; Wu, G.D.; Albela, B.; Bonneviot, L.; et al. Facile large-scale synthesis of monodisperse mesoporous silica nanospheres with tunable pore structure. J. Am. Chem. Soc. 2013, 135, 2427–2430. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Xing, J.L.; Pang, J.L.; Jiang, S.H.; Lam, K.F.; Yang, T.Q.; Xue, Q.S.; Zhang, K.; Wu, P. Facile synthesis of size controllable dendritic mesoporous silica nanoparticles. ACS Appl. Mater. Interfaces 2014, 6, 22655–22665. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-C.; Yu, Y.-J.; Peng, B.; Ma, S.-Y.; Ning, T.-Y.; Shan, B.-Q.; Yang, T.-Q.; Xue, Q.-S.; Zhang, K.; Wu, P. A dual-templating strategy for the scale-up synthesis of dendritic mesoporous silica nanospheres. Green Chem. 2017, 19, 5575–5581. [Google Scholar] [CrossRef]
- Zhang, K.; Yang, T.Q.; Shan, B.Q.; Liu, P.C.; Peng, B.; Xue, Q.S.; Yuan, E.H.; Wu, P.; Albela, B.; Bonneviot, L. Dendritic and core-shell-corona mesoporous sister nanospheres from polymer-surfactant-silica self-entanglement. Chem. Eur. J. 2018, 24, 478–486. [Google Scholar] [CrossRef]
- Shan, B.-Q.; Xing, J.-L.; Yang, T.-Q.; Peng, B.; Hao, P.; Zong, Y.-X.; Chen, X.-Q.; Xue, Q.-S.; Zhang, K.; Wu, P. One-pot co-condensation strategy for dendritic mesoporous organosilica nanospheres with fine size and morphology control. CrystEngComm 2019, 21, 4030–4035. [Google Scholar] [CrossRef]
- Peng, B.; Zong, Y.-X.; Nie, M.-Z.; Shan, B.-Q.; Yang, T.-Q.; Hao, P.; Ma, S.-Y.; Lam, K.-F.; Zhang, K. Interfacial charge shielding directs the synthesis of dendritic mesoporous silica nanospheres by a dual-templating approach. New J. Chem. 2019, 43, 15777–15784. [Google Scholar] [CrossRef]
- Yang, T.-Q.; Ning, T.-Y.; Peng, B.; Shan, B.-Q.; Zong, Y.-X.; Hao, P.; Yuan, E.-H.; Chen, Q.-M.; Zhang, K. Interfacial electron transfer promotes photo-catalytic reduction of 4-nitrophenol by Au/Ag2O nanoparticles confined in dendritic mesoporous silica nanospheres. Catal. Sci. Technol. 2019, 9, 5786–5792. [Google Scholar] [CrossRef]
- Zhang, K.; Yuan, N.-H.; Xu, L.-L.; Xue, Q.-S.; Luo, C.; Albela, B.; Bonneviot, L. A New Method for Synthesis of High-Quality MCM-48 Mesoporous Silicas and the Mode of Action of the Template. Eur. J. Inorg. Chem. 2012, 4183–4189. [Google Scholar] [CrossRef]
- Zhang, K.; Albela, B.; He, M.-Y.; Wang, Y.; Bonneviot, L. Tetramethyl ammonium as masking agent for molecular stencil patterning in the confined space of the nano-channels of 2D hexagonal-templated porous silicas. Phys. Chem. Chem. Phys. 2009, 11, 2912–2921. [Google Scholar] [CrossRef]
- Zhang, K.; Lam, K.F.; Albela, B.; Xue, T.; Khrouz, L.; Hou, Q.W.; Yuan, E.H.; He, M.Y.; Bonneviot, L. Mononuclear-dinuclear equilibrium of grafted copper complexes confined in the nanochannels of MCM-41 silica. Chem. Eur. J. 2011, 17, 14258–14266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, Y.; Hou, Q.-W.; Yuan, E.-H.; Jiang, J.-G.; Albela, B.; He, M.-Y.; Bonneviot, L. Novel synthesis and molecularly scaled surface hydrophobicity control of colloidal mesoporous silica. Microporous Mesoporous Mater. 2011, 143, 401–405. [Google Scholar] [CrossRef]
- Campbell, E.K.; Holz, M.; Gerlich, D.; Maier, J.P. Laboratory confirmation of C60+ as the carrier of two diffuse interstellar bands. Nature 2015, 523, 322–323. [Google Scholar] [CrossRef] [PubMed]
- Holmlid, L. The diffuse interstellar band carriers in interstellar space: All intense bands calculated from He doubly excited states embedded in Rydberg Matter. Mon. Not. R. Astron. Soc. 2008, 384, 764–774. [Google Scholar] [CrossRef][Green Version]
- Röhr, J.A.; Sá, J.; Konezny, S.J. The role of adsorbates in the green emission and conductivity of zinc oxide. Commun. Chem. 2019, 2, 52. [Google Scholar] [CrossRef]
- Liu, L.; Wu, X.; Wang, L.; Xu, X.; Gan, L.; Si, Z.; Li, J.; Zhang, Q.; Liu, Y.; Zhao, Y.; et al. Atomic palladium on graphitic carbon nitride as a hydrogen evolution catalyst under visible light irradiation. Commun. Chem. 2019, 2, 18. [Google Scholar] [CrossRef]
- Fisher, A.A.E.; Osborne, M.A.; Day, I.J.; Lucena Alcalde, G. Measurement of ligand coverage on cadmium selenide nanocrystals and its influence on dielectric dependent photoluminescence intermittency. Commun. Chem. 2019, 2, 63. [Google Scholar] [CrossRef]
- Bubeck, C.; Widenmeyer, M.; Richter, G.; Coduri, M.; Goering, E.; Yoon, S.; Weidenkaff, A. Tailoring of an unusual oxidation state in a lanthanum tantalum(IV) oxynitride via precursor microstructure design. Commun. Chem. 2019, 2, 134. [Google Scholar] [CrossRef]
- Wakasa, M.; Yago, T.; Sonoda, Y.; Katoh, R. Structure and dynamics of triplet-exciton pairs generated from singlet fission studied via magnetic field effects. Commun. Chem. 2018, 1, 9. [Google Scholar] [CrossRef]
- Noblet, T.; Dreesen, L.; Boujday, S.; Méthivier, C.; Busson, B.; Tadjeddine, A.; Humbert, C. Semiconductor quantum dots reveal dipolar coupling from exciton to ligand vibration. Commun. Chem. 2018, 1, 76. [Google Scholar] [CrossRef]
- Paris, C.; Floris, A.; Aeschlimann, S.; Neff, J.; Kling, F.; Kühnle, A.; Kantorovich, L. Kinetic control of molecular assembly on surfaces. Commun. Chem. 2018, 1, 66. [Google Scholar] [CrossRef]
- Jin, C.; Ma, L.; Sun, W.; Han, P.; Tan, X.; Wu, H.; Liu, M.; Jin, H.; Wu, Z.; Wei, H.; et al. Single-atom nickel confined nanotube superstructure as support for catalytic wet air oxidation of acetic acid. Commun. Chem. 2019, 2, 135. [Google Scholar] [CrossRef]
- Zhao, Q.; Fu, L.; Jiang, D.; Ouyang, J.; Hu, Y.; Yang, H.; Xi, Y. Nanoclay-modulated oxygen vacancies of metal oxide. Commun. Chem. 2019, 2, 11. [Google Scholar] [CrossRef]
- Tabor, E.; Lemishka, M.; Sobalik, Z.; Mlekodaj, K.; Andrikopoulos, P.C.; Dedecek, J.; Sklenak, S. Low-temperature selective oxidation of methane over distant binuclear cationic centers in zeolites. Commun. Chem. 2019, 2, 71. [Google Scholar] [CrossRef]


















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.-Q.; Peng, B.; Shan, B.-Q.; Zong, Y.-X.; Jiang, J.-G.; Wu, P.; Zhang, K. Origin of the Photoluminescence of Metal Nanoclusters: From Metal-Centered Emission to Ligand-Centered Emission. Nanomaterials 2020, 10, 261. https://doi.org/10.3390/nano10020261
Yang T-Q, Peng B, Shan B-Q, Zong Y-X, Jiang J-G, Wu P, Zhang K. Origin of the Photoluminescence of Metal Nanoclusters: From Metal-Centered Emission to Ligand-Centered Emission. Nanomaterials. 2020; 10(2):261. https://doi.org/10.3390/nano10020261
Chicago/Turabian StyleYang, Tai-Qun, Bo Peng, Bing-Qian Shan, Yu-Xin Zong, Jin-Gang Jiang, Peng Wu, and Kun Zhang. 2020. "Origin of the Photoluminescence of Metal Nanoclusters: From Metal-Centered Emission to Ligand-Centered Emission" Nanomaterials 10, no. 2: 261. https://doi.org/10.3390/nano10020261
APA StyleYang, T.-Q., Peng, B., Shan, B.-Q., Zong, Y.-X., Jiang, J.-G., Wu, P., & Zhang, K. (2020). Origin of the Photoluminescence of Metal Nanoclusters: From Metal-Centered Emission to Ligand-Centered Emission. Nanomaterials, 10(2), 261. https://doi.org/10.3390/nano10020261