Embedding Ordered Mesoporous Carbons into Thermosensitive Hydrogels: A Cutting-Edge Strategy to Vehiculate a Cargo and Control Its Release Profile

 ,
,  ,
,  , ,
, ,  , and
, and 
Abstract
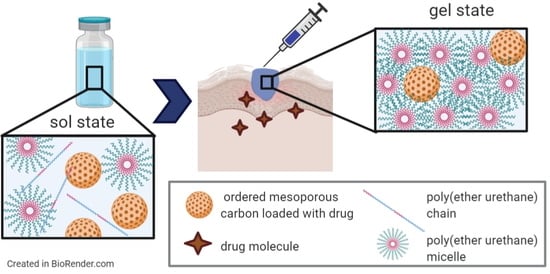
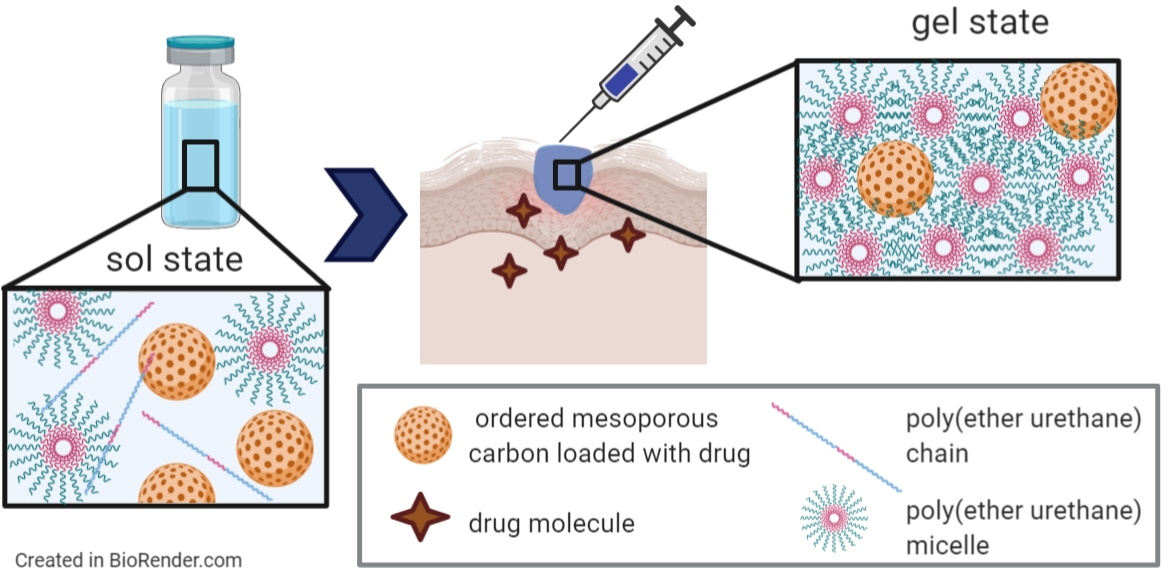
1. Introduction
2. Materials and Methods
2.1. Synthesis of Ibuprofen-Loaded Ordered Mesoporous Carbons
2.1.1. Materials
2.1.2. Rod-Like CMK-3 Type Ordered Mesoporous Carbons
2.1.3. Spherical CMK-1 Ordered Mesoporous Carbon
2.1.4. Drug-Loaded Ordered Mesoporous C3 and C1 Carbons
2.2. Characterization of C3 and C1 Based Materials
2.3. Synthesis of Poly(Ether Urethane)
2.3.1. Materials
2.3.2. Poly(Ether Urethane) Synthesis Protocol
2.4. Chemical Characterization of as-Synthesized Poly(Ether Urethane)
2.5. Design and Characterization of Hybrid Sol-Gel Systems Based on Thermosensitive PEU Hydrogels and OMCs
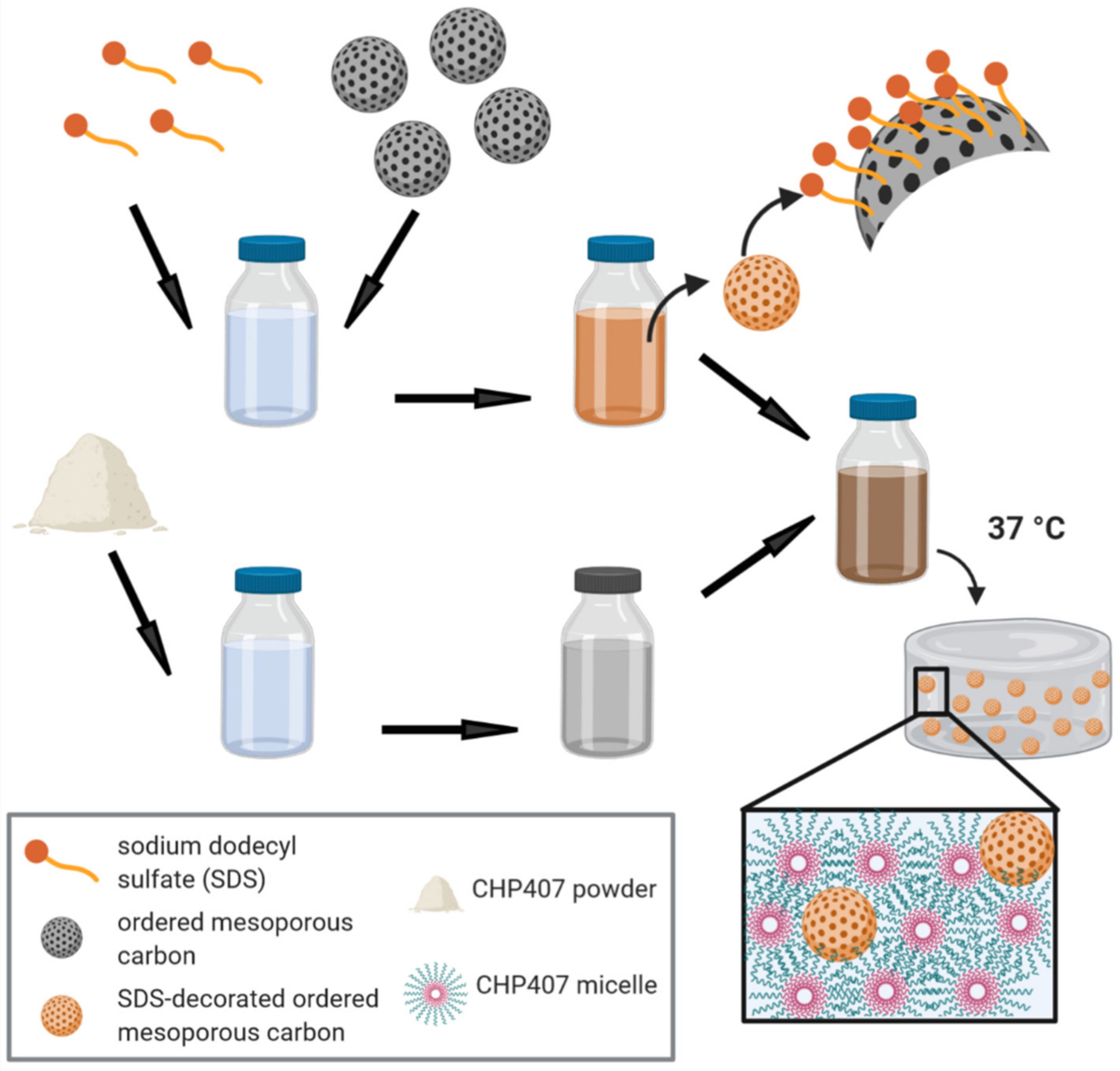
2.5.1. Hydrogel Preparation Protocol
2.5.2. Hybrid Hydrogel Characterization
2.6. Ibuprofen Release from Hybrid Sol-Gel Systems
2.7. Statistical Analysis
3. Results
3.1. Physico-Chemical Characterization of Pristine and IBU-Loaded OMCs
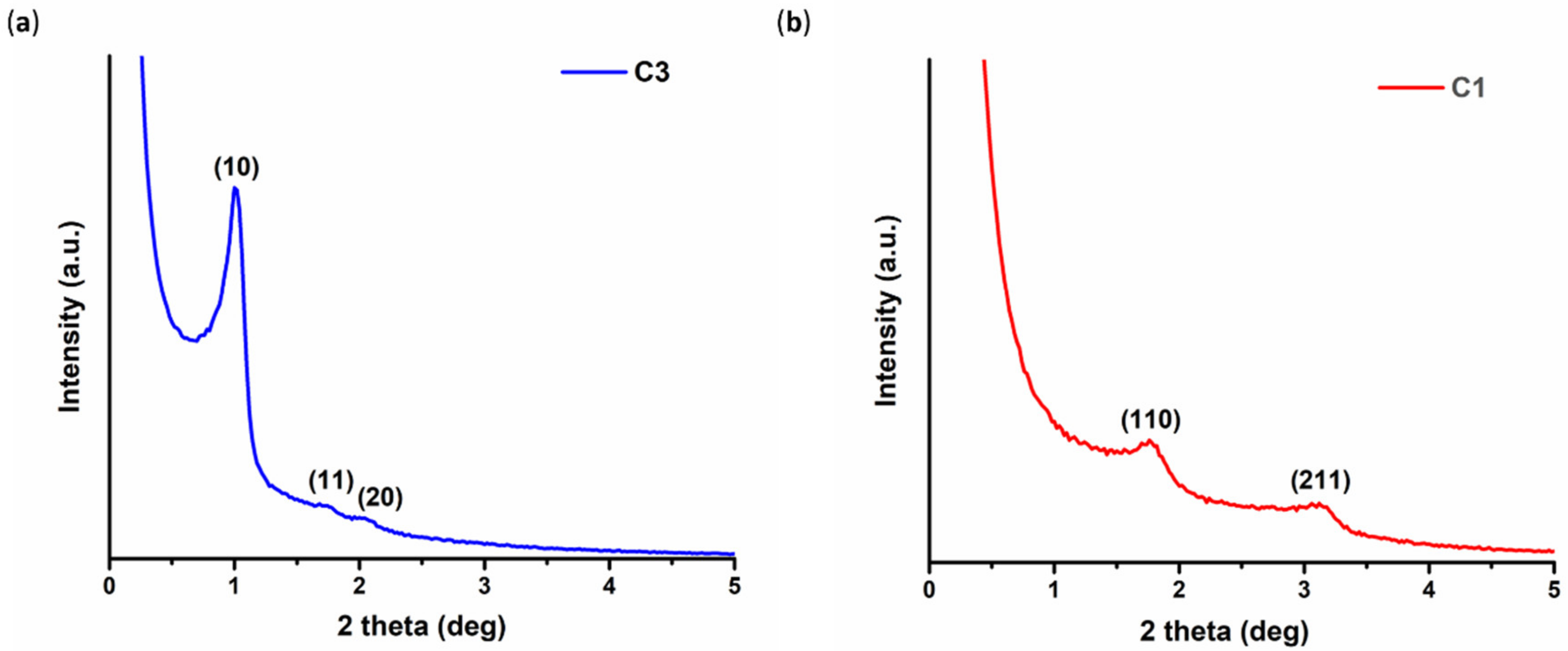
3.1.1. Structural Characterization of Pristine C1 and C3 Carbons
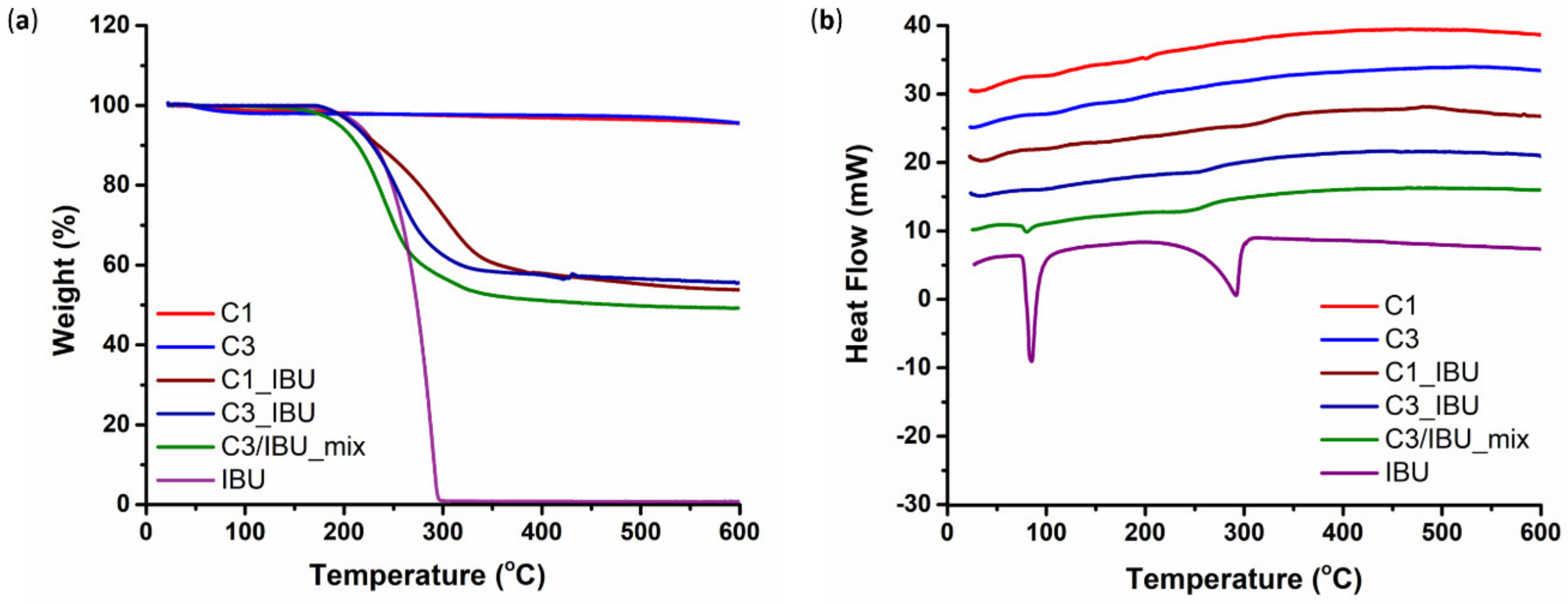
3.1.2. Ibuprofen Loading into C1 and C3 Carbons
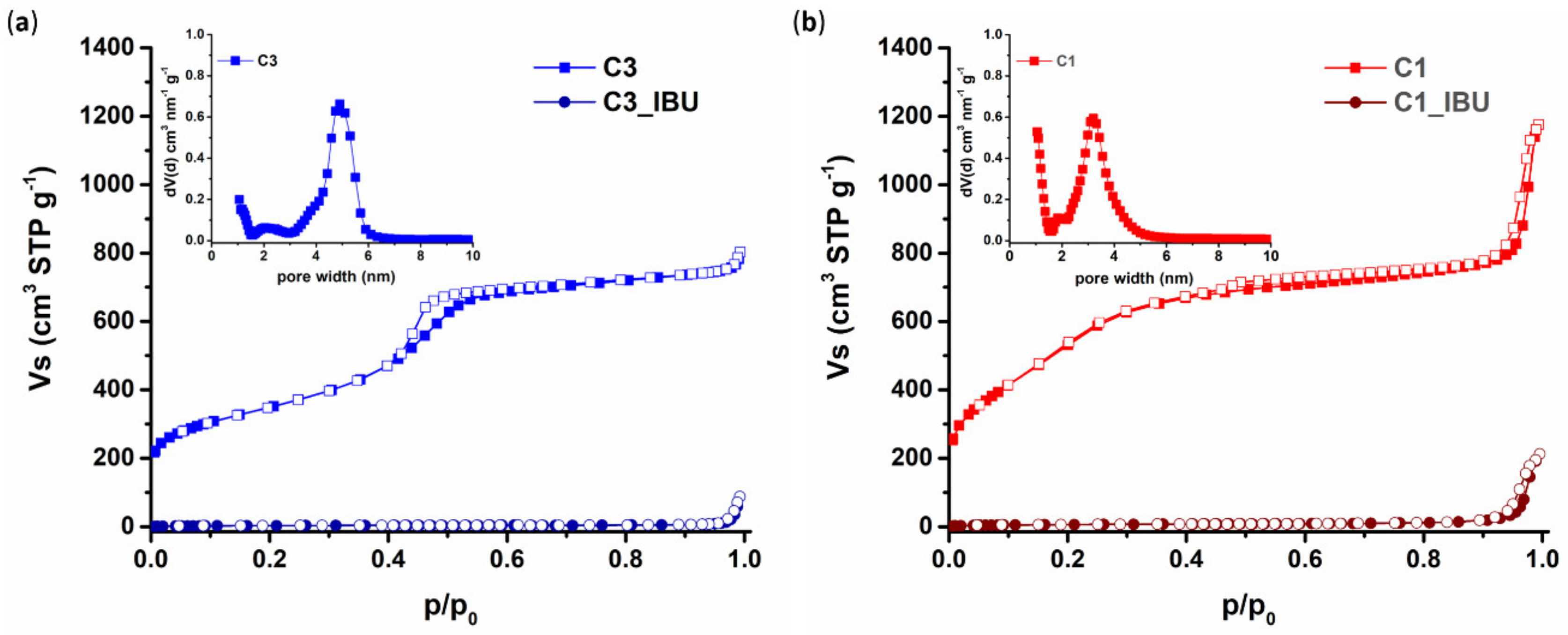
3.1.3. Pore Properties of Plain and IBU-Loaded C1 and C3 Carbons
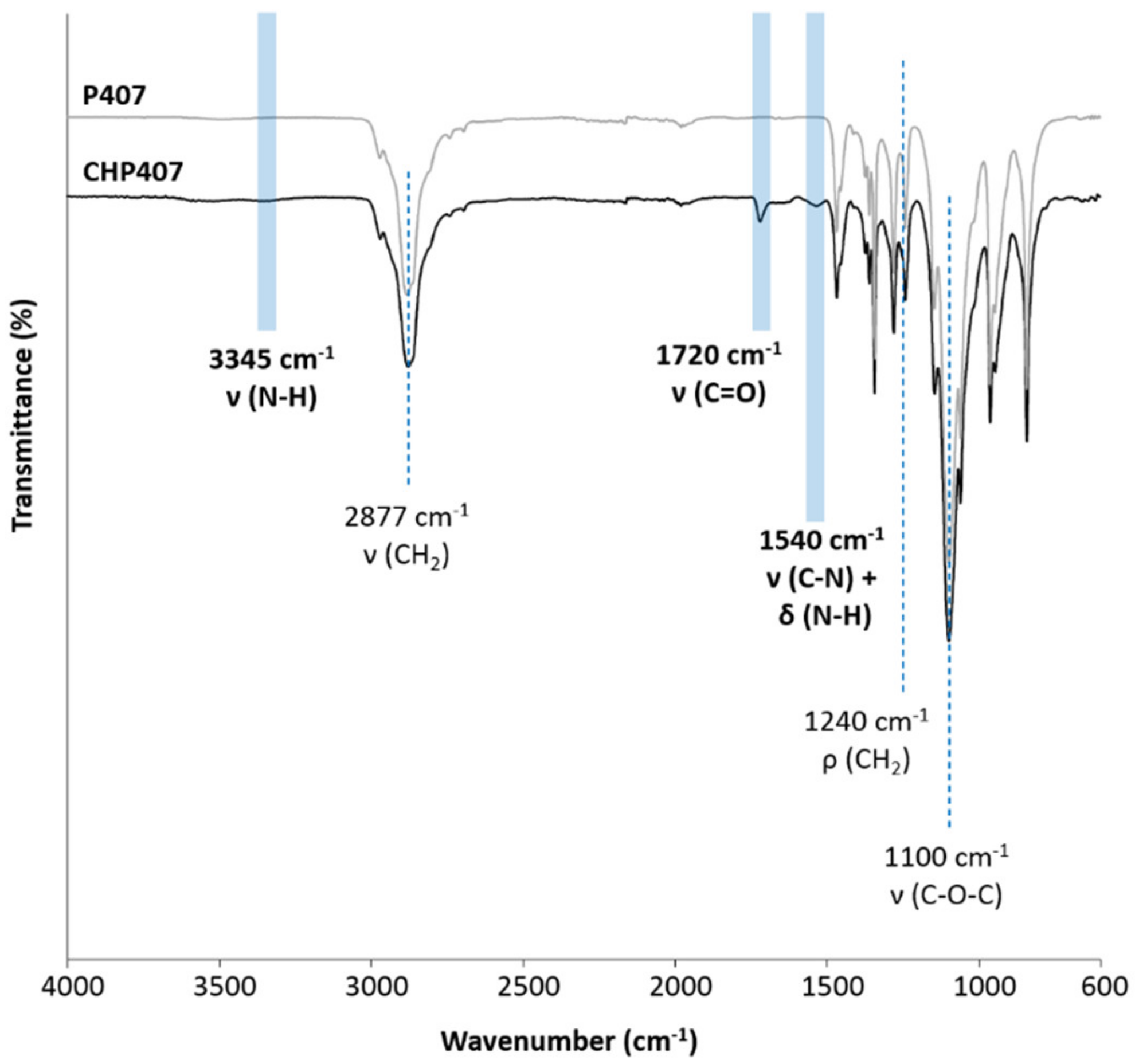
3.2. Chemical Characterization of Synthesized Poly(Ether Urethane)
3.3. Characterization of Hybrid Sol-Gel Systems Based on Thermosensitive PEU Hydrogels and OMCs
3.3.1. Tube Inverting Test
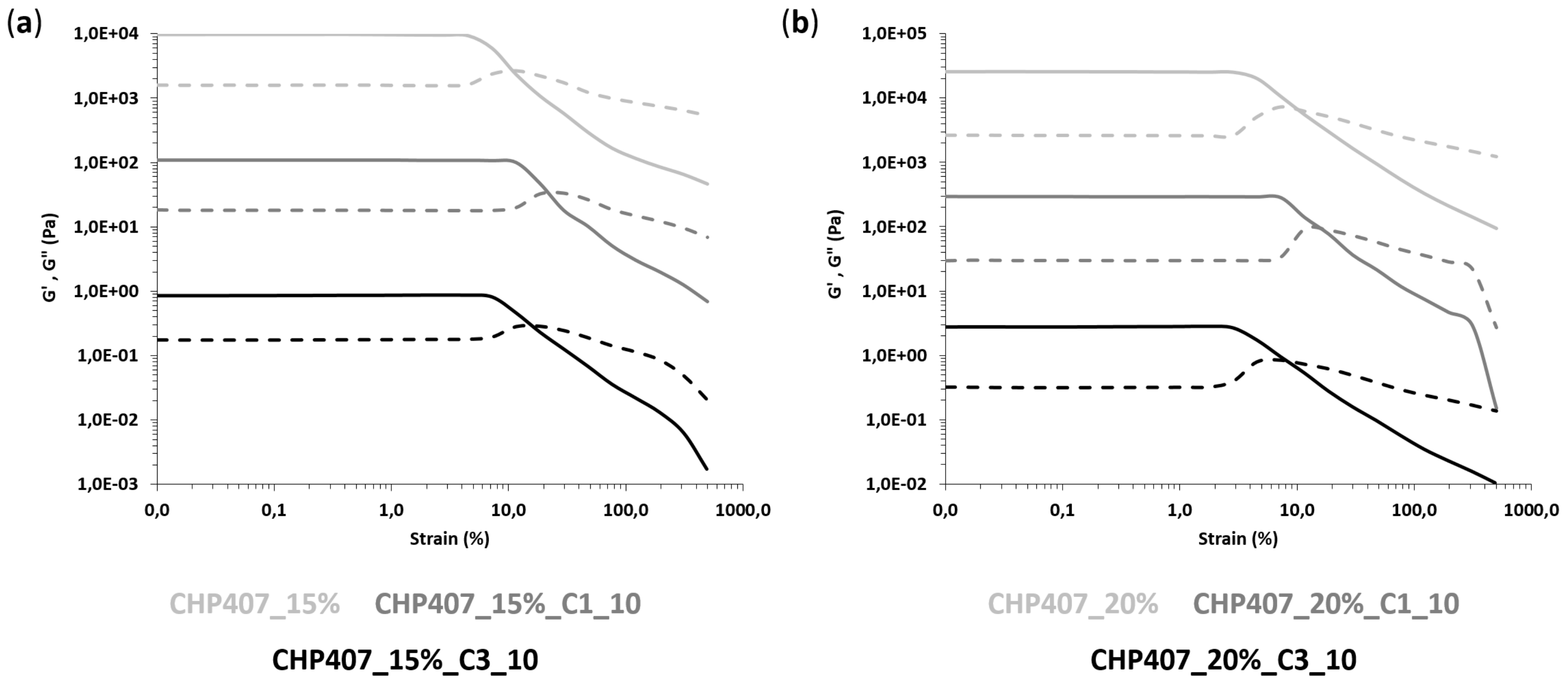
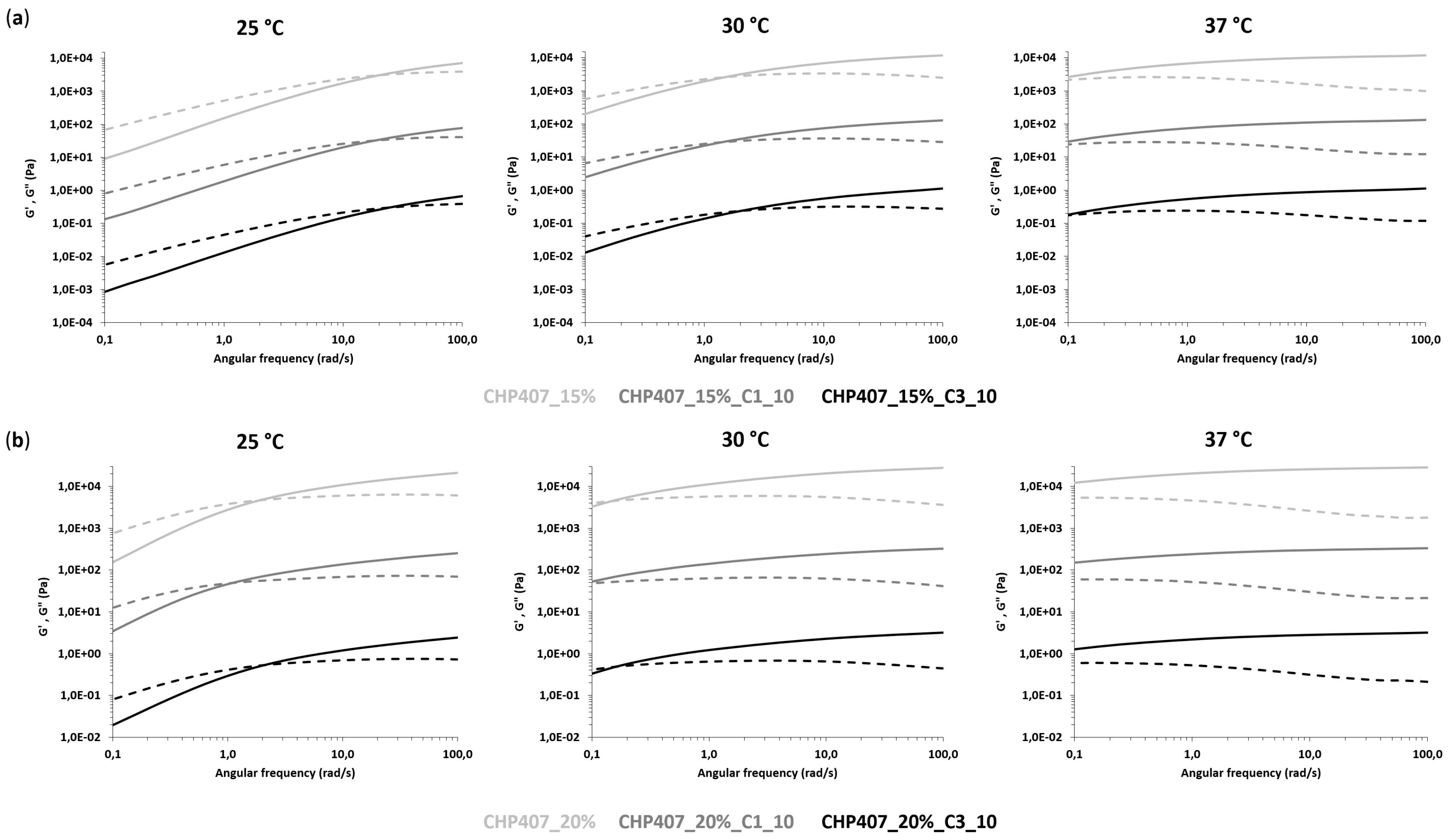
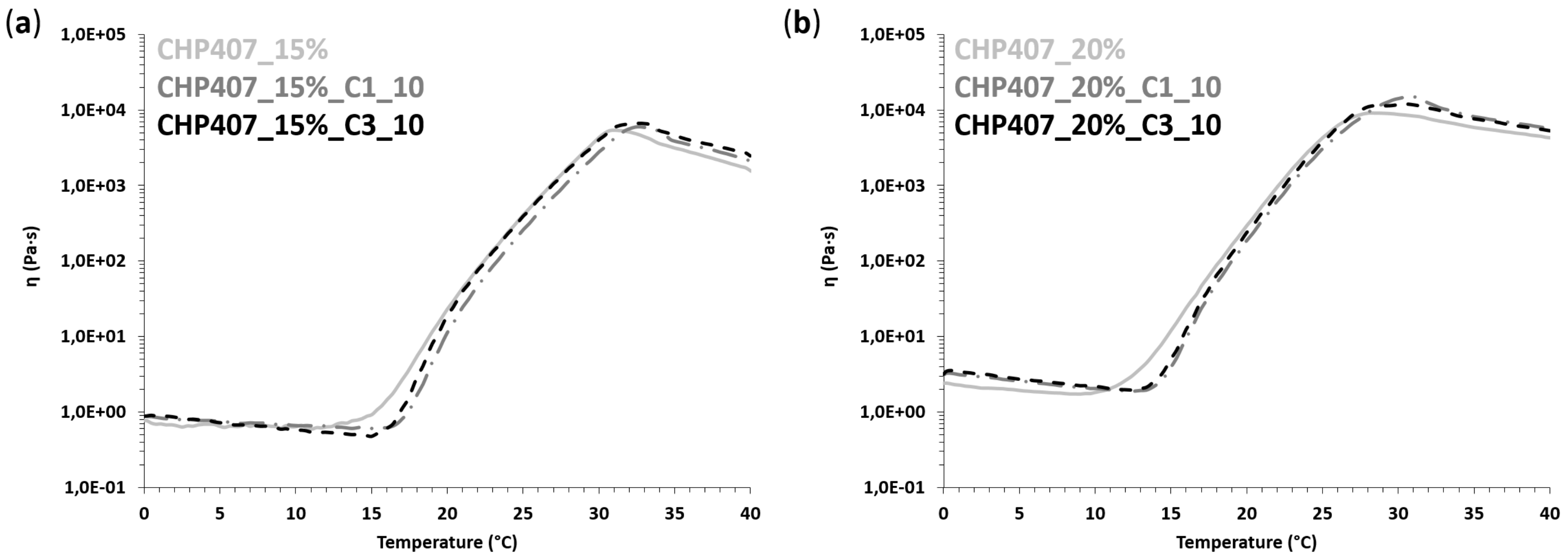
3.3.2. Rheological Characterization
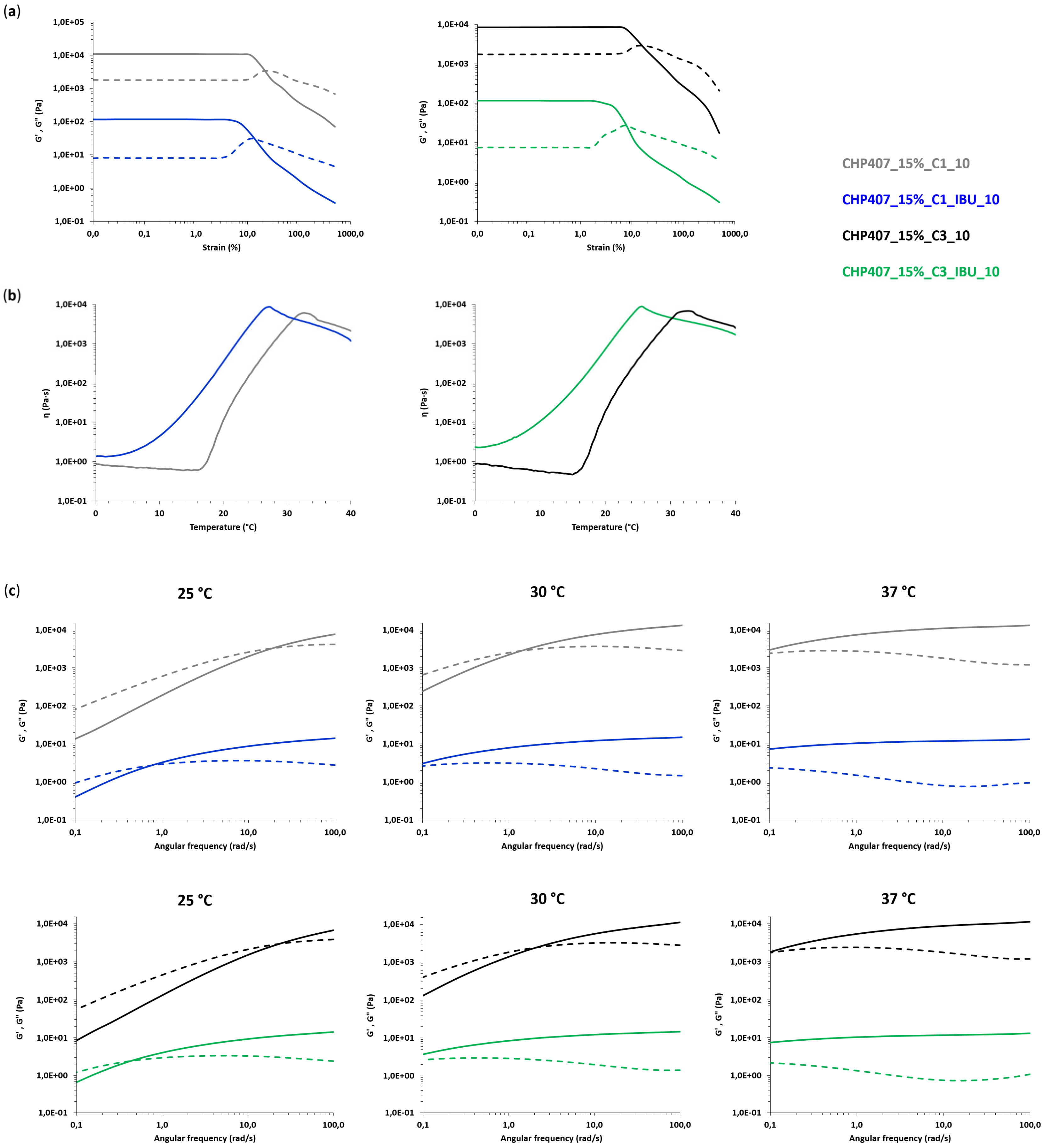
3.4. Characterization of Hybrid Sol-Gel Systems Based on Thermosensitive PEU Hydrogels and OMCs Loaded with Ibuprofen
3.4.1. Rheological Characterization
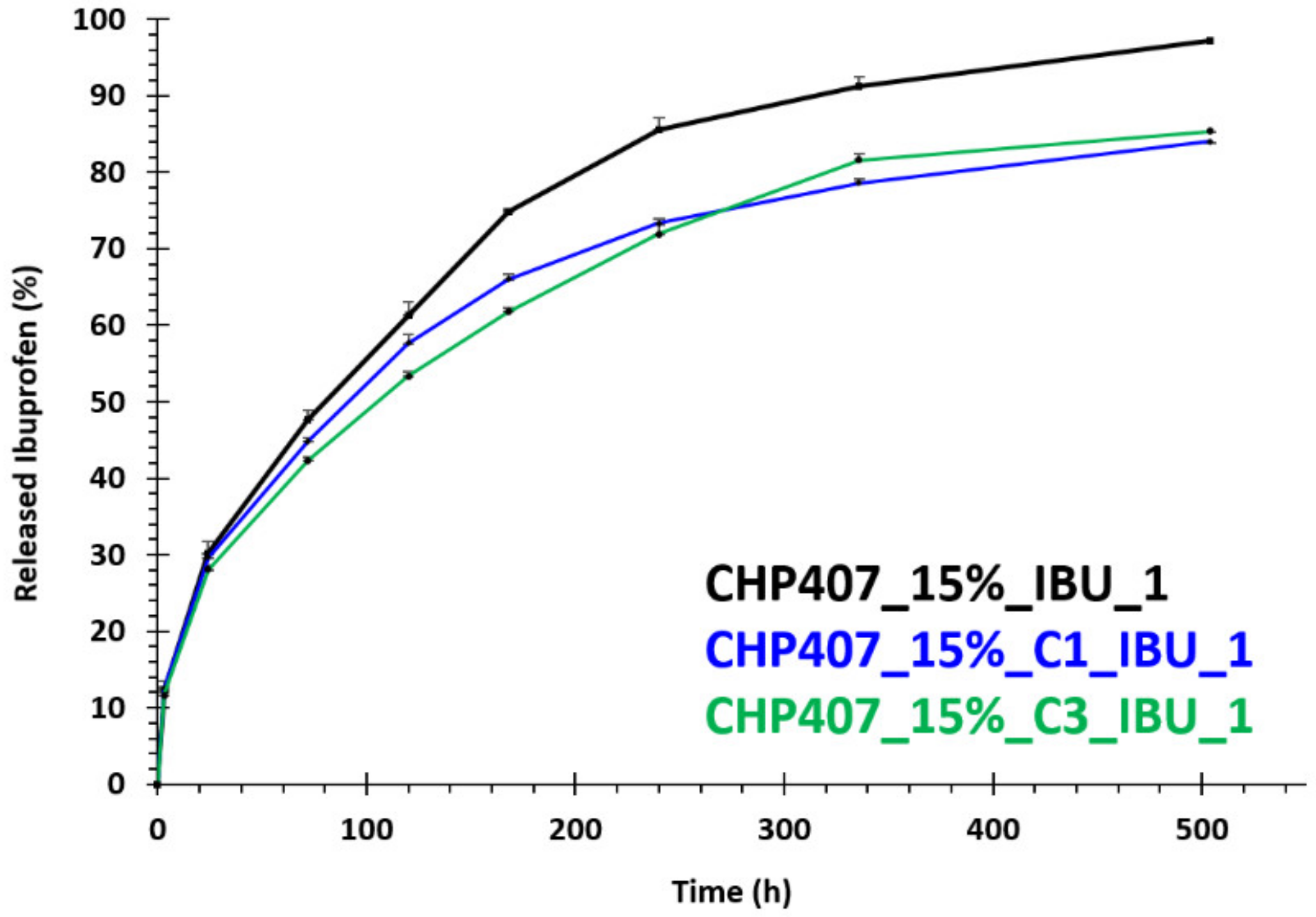
3.4.2. In Vitro Drug Release Tests
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous silica nanoparticles: A comprehensive review on synthesis and recent advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Vallet-Regì, M. Mesoporous silica nanoparticles for drug delivery. Adv. Funct. Mater. 2020, 30, 1902634. [Google Scholar] [CrossRef]
- Saini, K.; Prabhuraj, R.S.; Bandyopadhyaya, R. Development of mesoporous silica nanoparticles of tunable pore diameter for superior gemcitabine drug delivery in pancreatic cancer cells. J. Nanosci. Nanotechnol. 2020, 20, 3084–3096. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.L.; Kuo, H.P.; Johnson, A.; Wu, L.C.; Chang, K.L.B. Curcumin-loaded mesoporous silica nanoparticles markedly enhanced cytotoxicity in hepatocellular carcinoma cells. Int. J. Mol. Sci. 2019, 20, 2918. [Google Scholar] [CrossRef] [PubMed]
- Gulin-Sarfraz, T.; Jonasson, S.; Wigenstam, E.; von Haartman, E.; Bucht, A.; Rosenholm, J.M. Feasibility study of mesoporous silica particles for pulmonary drug delivery: Therapeutic treatment with dexamethasone in a mouse model of airway inflammation. Pharmaceutics 2019, 11, 149. [Google Scholar] [CrossRef]
- Jin, R.; Liu, Z.; Bai, Y.; Zhou, Y.; Chen, X. Multiple-responsive mesoporous silica nanoparticles for highly accurate drugs delivery to tumor cells. ACS Omega 2018, 3, 4306–4315. [Google Scholar] [CrossRef]
- Arriagada, F.; Günther, G.; Morales, J. Nanoantioxidant–based silica particles as flavonoid carrier for drug delivery applications. Pharmaceutics 2020, 12, 302. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, K.; Wang, J.; Zhao, R.; Zhang, Q.; Kong, X. Poly(amidoamine)-modified mesoporous silica nanoparticles as a mucoadhesive drug delivery system for potential bladder cancer therapy. Colloid Surf. B 2020, 189, 110832. [Google Scholar] [CrossRef]
- Chaudhary, Z.; Subramaniam, S.; Khan, G.M.; Abeer, M.M.; Qu, Z.; Janjua, T.; Kumeria, T.; Batra, J.; Popat, A. Encapsulation and controlled release of resveratrol within functionalized mesoporous silica nanoparticles for prostate cancer therapy. Front. Bioeng. Biotechnol. 2019, 7, 225. [Google Scholar] [CrossRef]
- Kundu, M.; Chetterjee, S.; Ghosh, N.; Manna, P.; Das, J.; Sil, P.C. Tumor targeted delivery of umbelliferone via a smart mesoporous silica nanoparticles controlled-release drug delivery system for increased anticancer efficiency. Mater. Sci. Eng. C 2020, 116, 111239. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Song, F.X.; Zhang, L.; Yang, W.; Wang, H.X.; Chen, Q.L. A pH-sensitive drug delivery system based on folic acid-targeted HBP-modified mesoporous silica nanoparticles for cancer therapy. Colloid Surf. B 2020, 590, 124470. [Google Scholar] [CrossRef]
- Li, S.; Dai, W.; Yin, Z.Z.; Gao, J.; Wu, D.; Kong, Y. Synthesis of oxidized pullulan coated mesoporous silica for pH-sensitive drug delivery. Eur. Polym. J. 2020, 122, 109399. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, C.; Li, R.; Ruan, L. Microwave thermal-triggered drug delivery using thermosensitive peptide-coated core–shell mesoporous silica nanoparticles. J. Mater. Sci. 2020, 55, 6118–6129. [Google Scholar] [CrossRef]
- Saini, K.; Bandyopadhyaya, R. Transferrin-conjugated polymer-coated mesoporous silica nanoparticles loaded with gemcitabine for killing pancreatic cancer cells. ACS Appl. Nano Mater. 2020, 3, 229–240. [Google Scholar] [CrossRef]
- Bari, A.; Bloise, N.; Fiorilli, S.; Novajra, G.; Vallet-Regì, M.; Bruni, G.; Torres-Pardo, A.; Gonzàlez-Calbet, J.M.; Visai, L.; Vitale-Brovarone, C. Copper-containing mesoporous bioactive glass nanoparticles as multifunctional agent for bone regeneration. Acta Biomater. 2020, 55, 493–504. [Google Scholar] [CrossRef]
- Gu, J.; Huang, M.; Liu, J.; Li, Y.; Zhao, W.; Shi, J. Calcium doped mesoporous silica nanoparticles as efficient alendronate delivery vehicles. New J. Chem. 2012, 36, 1717–1720. [Google Scholar] [CrossRef]
- Philippart, A.; Gómez-Cerezo, N.; Arcos, D.; Salinas, A.J.; Boccardi, E.; Vallet-Regì, M.; Boccaccini, A.R. Novel ion-doped mesoporous glasses for bone tissue engineering: Study of their structural characteristics influenced by the presence of phosphorous oxide. J. Non-Cryst. Solids 2017, 455, 90–97. [Google Scholar] [CrossRef]
- Shi, M.; Chen, Z.; Farnagh, S.; Friis, T.; Mao, X.; Xiao, Y.; Wu, C. Copper-doped mesoporous silica nanospheres, a promising immunomodulatory agent for inducing osteogenesis. Acta Biomater. 2016, 30, 334–344. [Google Scholar] [CrossRef]
- Kaya, S.; Cresswell, M.; Boccaccini, A.R. Mesoporous silica-based bioactive glasses for antibiotic-free antibacterial applications. Mater. Sci. Eng. C 2018, 83, 99–107. [Google Scholar] [CrossRef]
- Zhao, Q.; Lin, Y.; Han, N.; Li, X.; Geng, H.; Wang, X.; Cui, Y.; Wang, S. Mesoporous carbon nanomaterials in drug delivery and biomedical application. Drug Deliv. 2017, 24, 94–107. [Google Scholar] [CrossRef]
- Mehmood, A.; Ghafar, H.; Yaqoob, S.; Gohar, U.F.; Ahmad, B. Mesoporous silica nanoparticles: A review. J. Develop. Drugs 2017, 6, 2. [Google Scholar] [CrossRef]
- Kargozar, S.; Kermani, F.; Beidokhti, S.M.; Hamzehlou, S.; Verné, E.; Ferraris, S.; Baino, F. Functionalization and surface modifications of bioactive glasses (BGs): Tailoring of the biological response working on the outermost surface layer. Materials 2019, 12, 3696. [Google Scholar] [CrossRef]
- Gisbert-Garzarán, M.; Berkmann, J.C.; Giasafaki, D.; Lozano, D.; Spyrou, K.; Manzano, M.; Steriotis, T.; Duda, G.N.; Schmidt-Bleek, K.; Charalambopoulou, G.; et al. Engineered pH-responsive mesoporous carbon nanoparticles for drug delivery. ACS Appl. Mater. Interfaces 2020, 12, 14946–14957. [Google Scholar] [CrossRef]
- Torre, E.; Giasafaki, D.; Steriotis, T.; Cassinelli, C.; Morra, M.; Fiorilli, S.; Vitale-Brovarone, C.; Charalambopoulou, G.; Iviglia, G. Silver decorated mesoporous carbons for the treatment of acute and chronic wounds, in a tissue regeneration context. Int J. Nanomed. 2019, 14, 10147–10164. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yang, G.; Ran, F.; Gao, T.; Sun, C.; Zhao, Q.; Wang, S. Polymer-functionalized mesoporous carbon nanoparticles on overcoming multiple barriers and improving oral bioavailability of Probucol. Carbohydr. Polym. 2020, 229, 115508. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Chen, Y.; Zhang, Z.; Lei, L.; Zhu, F.; Yang, W.; Guo, Y.; Chu, M. Fluorescent hollow mesoporous carbon spheres for drug loading and tumor treatment through 980-nm laser and microwave co-irradiation. Biomaterials 2020, 248, 120009. [Google Scholar] [CrossRef]
- Baumann, M.D.; Kang, C.E.; Stanwick, J.C.; Wang, Y.; Kim, H.; Lapitsky, Y.; Shoichet, M.S. An injectable drug delivery platform for sustained combination therapy. J. Control Release 2009, 138, 205–213. [Google Scholar] [CrossRef]
- Ding, Y.; Zhao, A.; Liu, T.; Wang, Y.N.; Gao, Y.; Li, J.A.; Yang, P. An injectable nanocomposite hydrogel for potential application of vascularization and tissue repair. Ann. Biomed. Eng. 2020, 48, 1511–1523. [Google Scholar] [CrossRef]
- Zhao, F.; Wu, D.; Yao, D.; Guo, R.; Wang, W.; Dong, D.; Zhang, J. An injectable particle-hydrogel hybrid system for glucose-regulatory insulin delivery. Acta Biomater. 2017, 64, 334–345. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Phan, V.H.G.; Lee, D.S.; Thambi, T.; Huynh, D.P. Bioresorbable pH-and temperature-responsive injectable hydrogels-incorporating electrosprayed particles for the sustained release of insulin. Polym. Degrad. Stab. 2019, 162, 36–46. [Google Scholar] [CrossRef]
- Thakur, S.; Singh, H.; Singh, A.; Kaur, S.; Sharma, A.; Singh, S.K.; Kaur, S.J.; Kaur, G.; Jain, S.K. Thermosensitive injectable hydrogel containing carboplatin loaded nanoparticles: A dual approach for sustained and localized delivery with improved safety and therapeutic efficacy. J. Drug Deliv. Sci. Tec. 2020, 58, 101817. [Google Scholar] [CrossRef]
- Zhu, M.; Zhu, Y.; Zhang, L.; Shi, J. Preparation of chitosan / mesoporous silica nanoparticle composite hydrogels for sustained co-delivery of biomacromolecules and small chemical drugs. Sci. Technol. Adv. Mater. 2013, 14, 045005. [Google Scholar] [CrossRef]
- Lunter, D.J. Evaluation of mesoporous silica particles as drug carriers in hydrogels. Pharm. Dev. Technol. 2018, 23, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liu, H.; Deng, H.; Xiao, L.; Qin, C.; Du, Y.; Shi, X. A study of chitosan hydrogel with embedded mesoporous silica nanoparticles loaded by ibuprofen as a dual stimuli-responsive drug release system for surface coating of titanium implants. Colloid Surf. B 2014, 123, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Boffito, M.; Pontremoli, C.; Fiorilli, S.; Laurano, R.; Ciardelli, G.; Vitale-Brovarone, C. Injectable thermosensitive formulation based on polyurethane hydrogel/mesoporous glasses for sustained co-delivery of functional ions and drugs. Pharmaceutics 2019, 11, 501. [Google Scholar] [CrossRef] [PubMed]
- Pontremoli, C.; Boffito, M.; Fiorilli, S.; Laurano, R.; Torchio, A.; Bari, A.; Tonda-Turo, C.; Ciardelli, G.; Vitale-Brovarone, C. Hybrid injectable platforms for the in situ delivery of therapeutic ions from mesoporous glasses. Chem. Eng. J. 2018, 340, 103–113. [Google Scholar] [CrossRef]
- Boffito, M.; Torchio, A.; Tonda-Turo, C.; Laurano, R.; Gisbert-Garzaràn, M.; Berkmann, J.C.; Cassino, C.; Manzano, M.; Duda, G.N.; Vallet-Regì, M.; et al. Hybrid injectable sol-gel systems based on thermo-sensitive polyurethane hydrogels carrying pH-sensitive mesoporous silica nanoparticles for the controlled and triggered release of therapeutic agents. Front. Bioeng. Biotechnol. 2020, 8, 384. [Google Scholar] [CrossRef]
- Malgras, V.; Tang, J.; Wang, J.; Kim, J.; Torad, N.L.; Dutta, S.; Ariga, K.; Hossain, M.S.A.; Yamauchi, Y.; Wu, K.C.W. Fabrication of nanoporous carbon materials with hard- and soft-templating approaches: A review. J. Nanosci. Nanotechnol. 2019, 19, 3673–3685. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G. Triblock copolymer syntheses of mesoporous silica with periodic 50–300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef]
- Jun, S.; Joo, S.H.; Ryoo, R.; Kruk, M.; Jaroniec, M.; Liu, Z.; Ohsuna, T.; Terasaki, O. Synthesis of new, nanoporous carbon with hexagonally ordered mesostructure. J. Am. Chem. Soc. 2000, 122, 10712–10713. [Google Scholar] [CrossRef]
- Stöber, A.; Fink, E.; Bohn, J. Controlled growth of monodisperse silica spheres in the micron size range. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Kim, T.W.; Chung, P.W.; Slowing, I.I.; Tsunoda, M.; Yeung, E.S.; Lin, V.S.Y. Structurally ordered mesoporous carbon nanoparticles as transmembrane delivery vehicle in human cancer cells. Nano Lett. 2008, 8, 3724–3727. [Google Scholar] [CrossRef] [PubMed]
- Laurano, R.; Boffito, M.; Torchio, A.; Cassino, C.; Chiono, V.; Ciardelli, G. Plasma treatment of polymer powder as an effective tool to functionalize polymers: Case study application on an amphiphilic polyurethane. Polymers 2019, 11, 2109. [Google Scholar] [CrossRef] [PubMed]
- Boffito, M.; Gioffredi, E.; Chiono, V.; Calzone, S.; Ranzato, E.; Martinotti, S.; Ciardelli, G. Novel polyurethane-based thermosensitive hydrogels as drug release and tissue engineering platforms: Design and in vitro charaterization. Polym. Int. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Boffito, M.; Grivet Brancot, A.; Lima, O.; Bronco, S.; Sartori, S.; Ciardelli, G. Injectable thermosensitive gels for the localized and controlled delivery of biomolecules in tissue engineering/regenerative medicine. Biomed. Sci. Eng. 2019, 3, 9–19. [Google Scholar] [CrossRef]
- Kaneda, M.; Tsubakiyama, T.; Carlsson, A.; Sakamoto, Y.; Ohsuna, T.; Terasaki, O.; Joo, S.H.; Ryoo, R. Structural study of mesoporous MCM-48 and carbon networks synthesized in the spaces of MCM-48 by electron crystallography. J. Phys. Chem. B 2002, 106, 1256–1266. [Google Scholar] [CrossRef]
- Shen, S.C.; Ng, W.K.; Chia, L.; Dong, Y.C.; Tan, R.B.H. Stabilized amorphous state of ibuprofen by co-spray drying with mesoporous sba-15 to enhance dissolution properties. J. Pharm. Sci. 2010, 99, 1997–2007. [Google Scholar] [CrossRef]
- Karavasili, C.; Amanatiadou, E.P.; Sygellou, L.; Giasafaki, D.K.; Steriotis, T.A.; Charalambopoulou, G.C.; Vizirianakis, I.S.; Fatouros, D.G. Development of new drug delivery system based on ordered mesoporous carbons: Characterisation and cytocompatibility studies. J. Mater. Chem. B 2013, 1, 3167–3174. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Laurano, R.; Cassino, C.; Ciardelli, G.; Chiono, V.; Boffito, M. Polyurethane-based thiomers: A new multifunctional copolymer platform for biomedical applications. React. Funct. Polym. 2020, 146, 104413. [Google Scholar] [CrossRef]
- Aoki, T.; Kawashima, M.; Katono, H.; Sanui, K.; Ogata, N.; Okano, T.; Sakurai, Y. Temperature-responsive interprenetrating polymer networks constructed with poly (acrylic acid) and poly (N, N-dimethylacrylamide). Macromolecules 1994, 27, 947–952. [Google Scholar] [CrossRef]
- Laurano, R.; Boffito, M. Thermosensitive micellar hydrogels as vehicles to deliver drugs with different wettability. Front. Bioeng. Biotechnol. 2020, 8, 708. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHP407 (% w/v) | OMC | Acronym | |
|---|---|---|---|
| Type | Concentration (mg/mL) | ||
| 15 | - | - | CHP407_15% |
| 15 | C1 | 10 | CHP407_15%_C1_10 |
| 15 | C3 | CHP407_15%_C3_10 | |
| 15 | C1_IBU | CHP407_15%_C1_IBU_10 | |
| 15 | C3_IBU | CHP407_15%_C3_IBU_10 | |
| 20 | - | - | CHP407_20% |
| 20 | C1 | 10 | CHP407_20%_C1_10 |
| 20 | C3 | CHP407_20%_C3_10 | |
| Sample Acronym | SBET (m2/g) | TPV (cm3/g) 1 | Vmicro (cm3/g) | Vmeso (cm3/g) | Pore Width (nm) |
|---|---|---|---|---|---|
| C3 | 1244 | 1.14 | 0.18 | 0.96 | 4.9 |
| C1 | 1956 | 1.19 | 0.24 | 0.95 | 3.2 |
| C3_IBU | 11 | 0.02 | 0.00 | 0.01 | - |
| C1_IBU | 22 | 0.07 | 0.00 | 0.06 | - |
| SAMPLE | γL (%) | ΔG’-G’’ (Pa) | YS (Pa) |
|---|---|---|---|
| CHP407_15% | 4.5 | 8109 | 407 |
| CHP407_15%_C1_10 | 11.6 | 9008 | 1110 |
| CHP407_15%_C3_10 | 6.2 | 6770 | 675 |
| CHP407_20% | 2.8 | 22,860 | 921 |
| CHP407_20%_C1_10 | 7.3 | 26,649 | 1940 |
| CHP407_20%_C3_10 | 3.0 | 24,809 | 920 |
| SAMPLE | 25 °C | 30 °C | 37 °C | |||
|---|---|---|---|---|---|---|
| ωG’/G’’_cross (rad/s) | ΔG’-G’’_100rad/s (Pa) | ωG’/G’’_cross (rad/s) | ΔG’-G’’_100rad/s (Pa) | ωG’/G’’_cross (rad/s) | ΔG’-G’’_100rad/s (Pa) | |
| CHP407_15% | 20.8 | 3140 | 1.5 | 9190 | - * | 10,812 |
| CHP407_15%_C1_10 | 18.6 | 3610 | 1.4 | 10,130 | - * | 11,990 |
| CHP407_15%_C3_10 | 23.4 | 2870 | 2.1 | 8420 | - * | 10,130 |
| CHP407_20% | 1.9 | 14,860 | 0.1 | 24,060 | - * | 26,710 |
| CHP407_20%_C1_10 | 1.1 | 18,190 | - * | 28,230 | - * | 31,250 |
| CHP407_20%_C3_10 | 2.1 | 16,670 | 0.2 | 26,940 | - * | 29,900 |
| SAMPLE | Tonset (°C) | η0 °C (Pa∙s) |
|---|---|---|
| CHP407_15% | 12.4 | 0.8 |
| CHP407_15%_C1_10 | 15.7 | 0.9 |
| CHP407_15%_C3_10 | 15.4 | 0.9 |
| CHP407_20% | 9.7 | 2.4 |
| CHP407_20%_C1_10 | 13.7 | 3.1 |
| CHP407_20%_C3_10 | 13.4 | 3.2 |
| Rheological Parameter | CHP407_15%_C1_IBU_10 | CHP407_15%_C3_IBU_10 |
|---|---|---|
| Strain sweep test results | ||
| γL (%) | 4.5 | 6.2 |
| ΔG’-G’’ (Pa) | 10,874 | 10,717 |
| YS (Pa) | 607 | 308 |
| Frequency sweep test results | ||
| ωG’/G’’_cross (rad/s) 25 °C | 0.8 | 0.4 |
| ωG’/G’’_cross (rad/s) 30 °C | - * | - * |
| ωG’/G’’_cross (rad/s) 37 °C | - * | - * |
| Temperature ramp test results | ||
| Tonset (°C) | - ** | - ** |
| η0 °C (Pa∙s) | 1.3 | 2.3 |
| η25 °C (Pa∙s) | 4394 | 7914 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boffito, M.; Laurano, R.; Giasafaki, D.; Steriotis, T.; Papadopoulos, A.; Tonda-Turo, C.; Cassino, C.; Charalambopoulou, G.; Ciardelli, G. Embedding Ordered Mesoporous Carbons into Thermosensitive Hydrogels: A Cutting-Edge Strategy to Vehiculate a Cargo and Control Its Release Profile. Nanomaterials 2020, 10, 2165. https://doi.org/10.3390/nano10112165
Boffito M, Laurano R, Giasafaki D, Steriotis T, Papadopoulos A, Tonda-Turo C, Cassino C, Charalambopoulou G, Ciardelli G. Embedding Ordered Mesoporous Carbons into Thermosensitive Hydrogels: A Cutting-Edge Strategy to Vehiculate a Cargo and Control Its Release Profile. Nanomaterials. 2020; 10(11):2165. https://doi.org/10.3390/nano10112165
Chicago/Turabian StyleBoffito, Monica, Rossella Laurano, Dimitra Giasafaki, Theodore Steriotis, Athanasios Papadopoulos, Chiara Tonda-Turo, Claudio Cassino, Georgia Charalambopoulou, and Gianluca Ciardelli. 2020. "Embedding Ordered Mesoporous Carbons into Thermosensitive Hydrogels: A Cutting-Edge Strategy to Vehiculate a Cargo and Control Its Release Profile" Nanomaterials 10, no. 11: 2165. https://doi.org/10.3390/nano10112165
APA StyleBoffito, M., Laurano, R., Giasafaki, D., Steriotis, T., Papadopoulos, A., Tonda-Turo, C., Cassino, C., Charalambopoulou, G., & Ciardelli, G. (2020). Embedding Ordered Mesoporous Carbons into Thermosensitive Hydrogels: A Cutting-Edge Strategy to Vehiculate a Cargo and Control Its Release Profile. Nanomaterials, 10(11), 2165. https://doi.org/10.3390/nano10112165