Amorphous Mo5O14-Type/Carbon Nanocomposite with Enhanced Electrochemical Capability for Lithium-Ion Batteries


 , , ,
, , , 
Abstract
1. Introduction
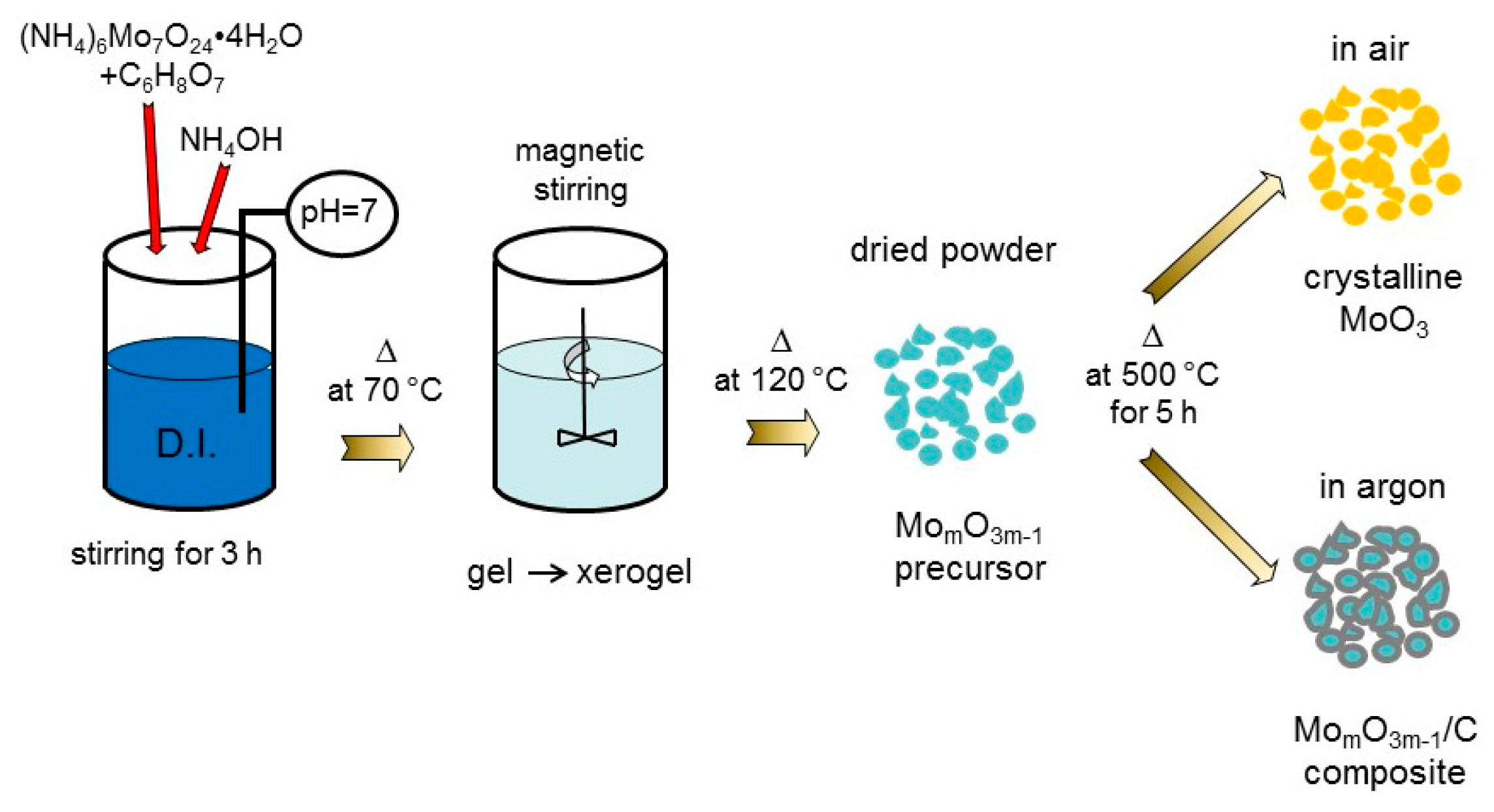
2. Materials and Methods
3. Results
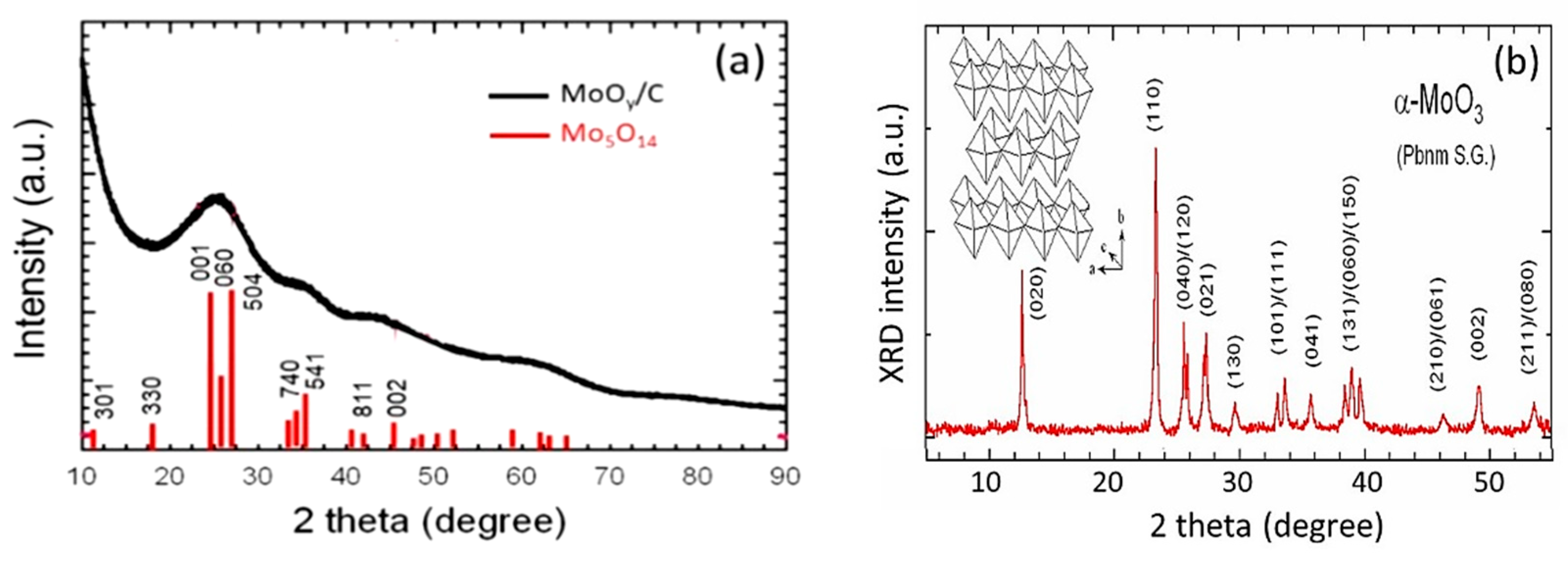
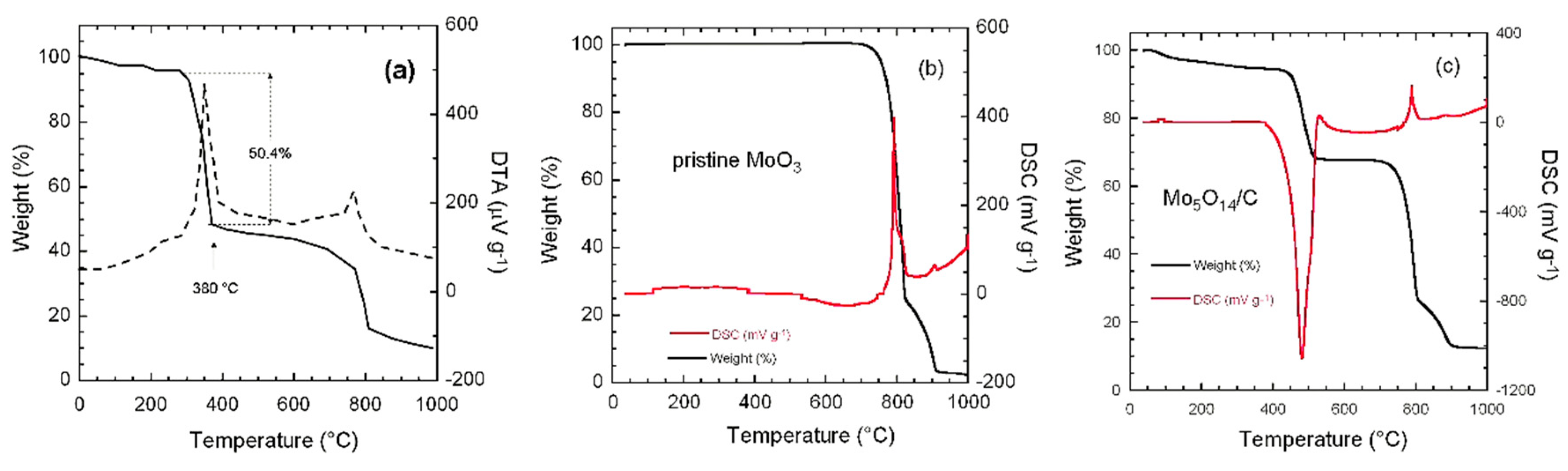
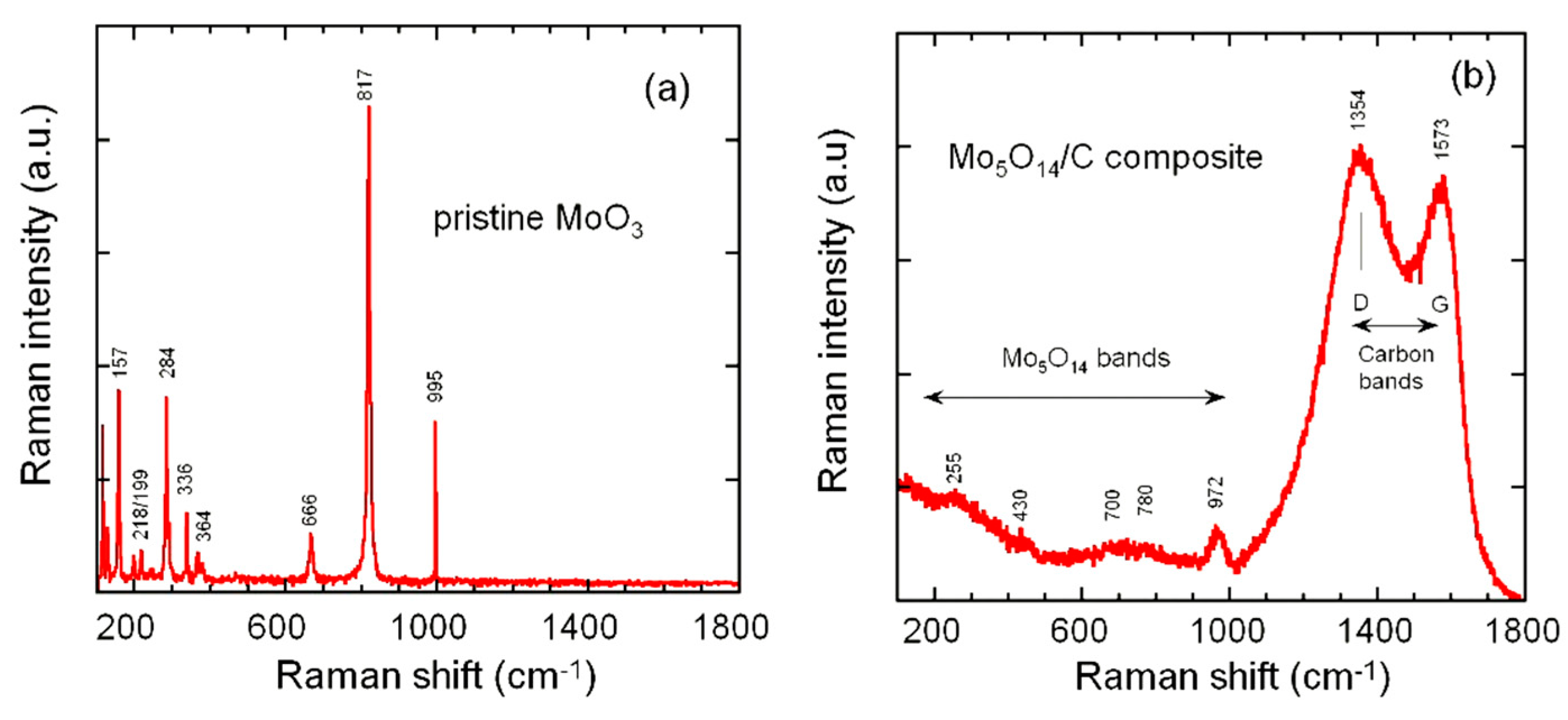
3.1. Structure and Morphology
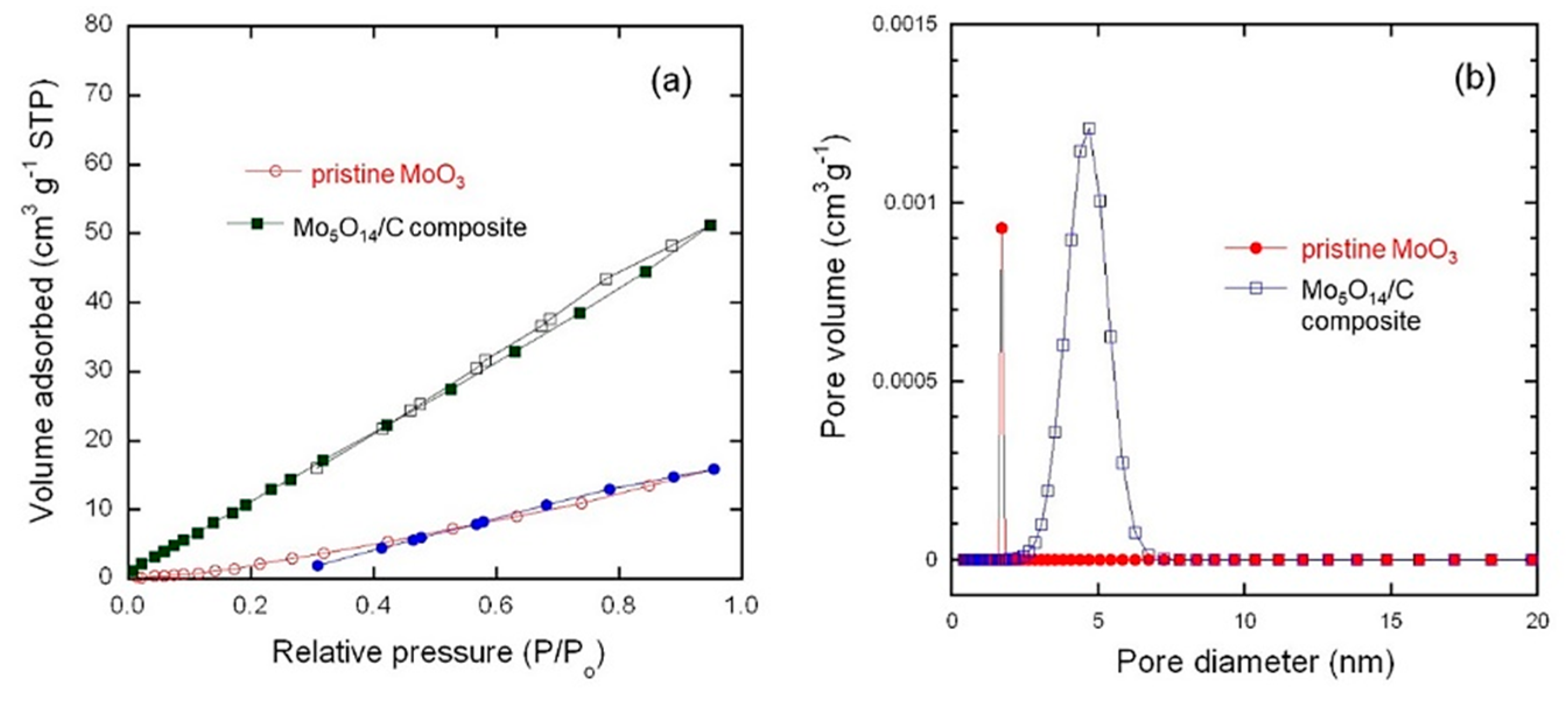
3.2. BET-Surface Area
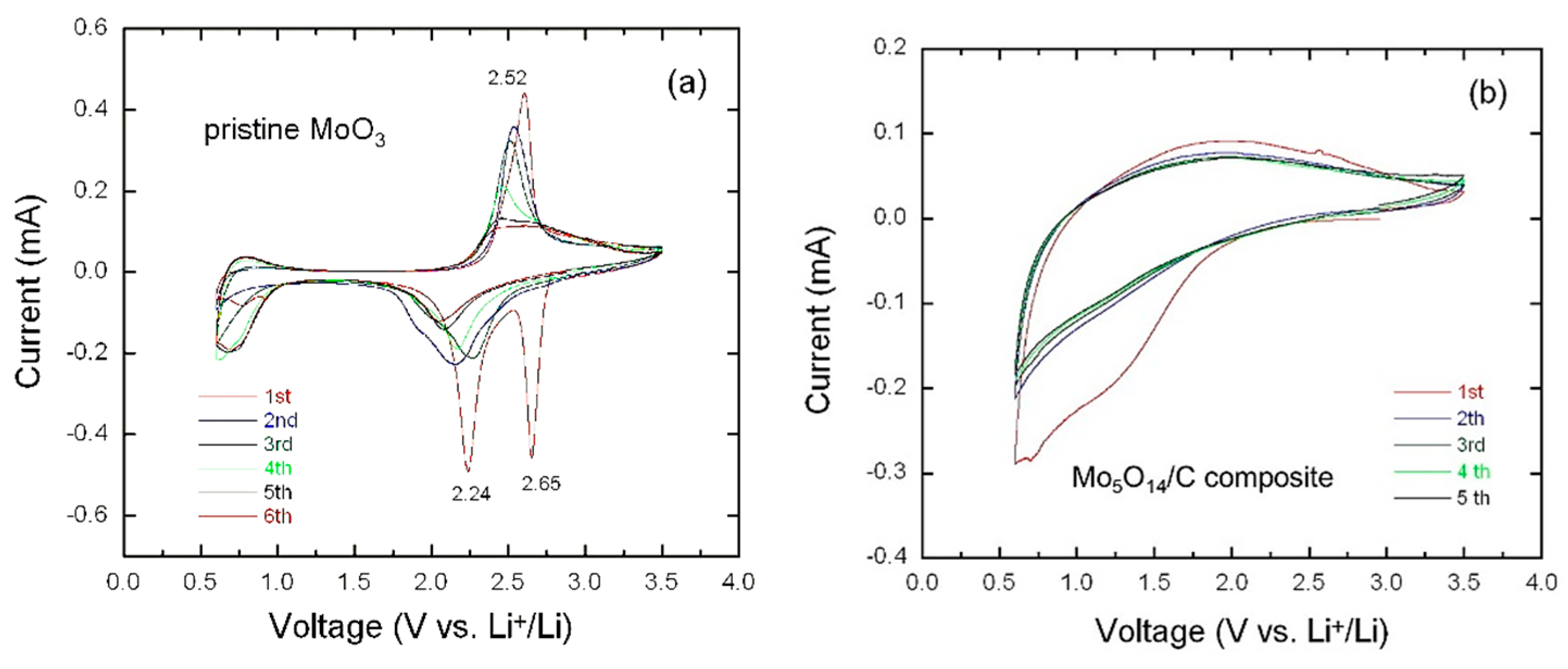
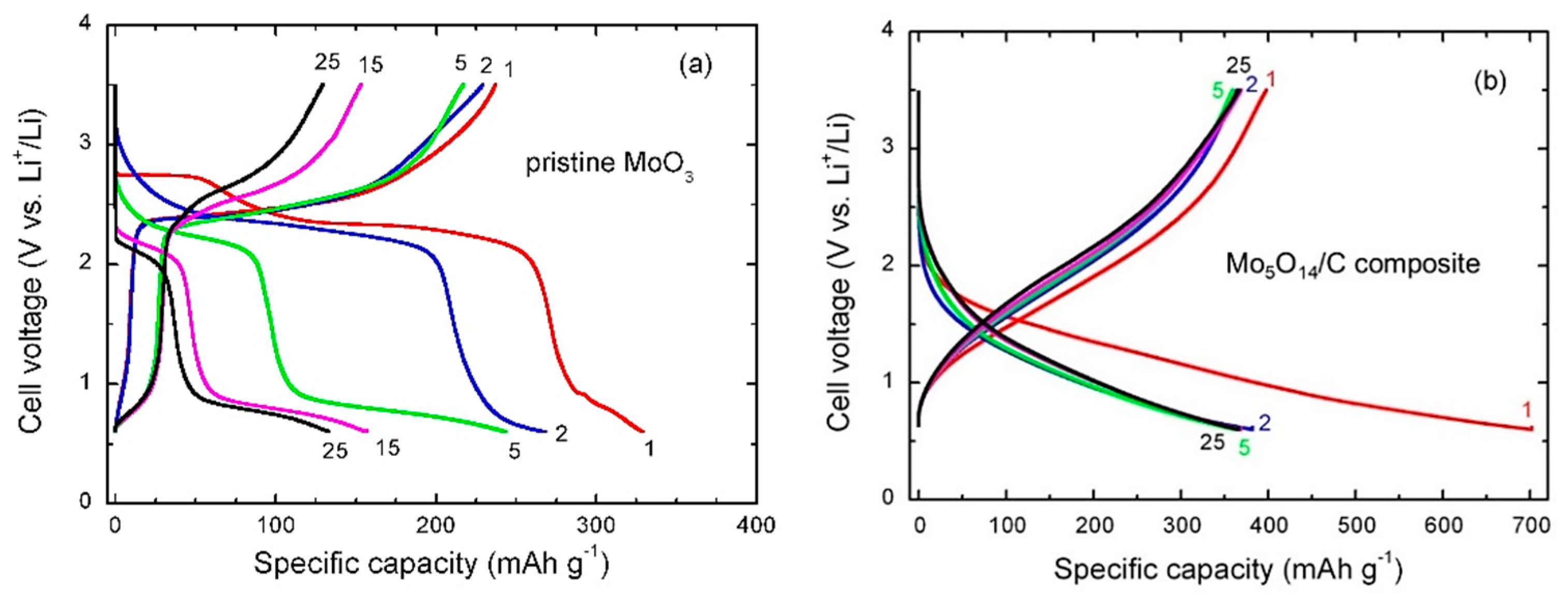
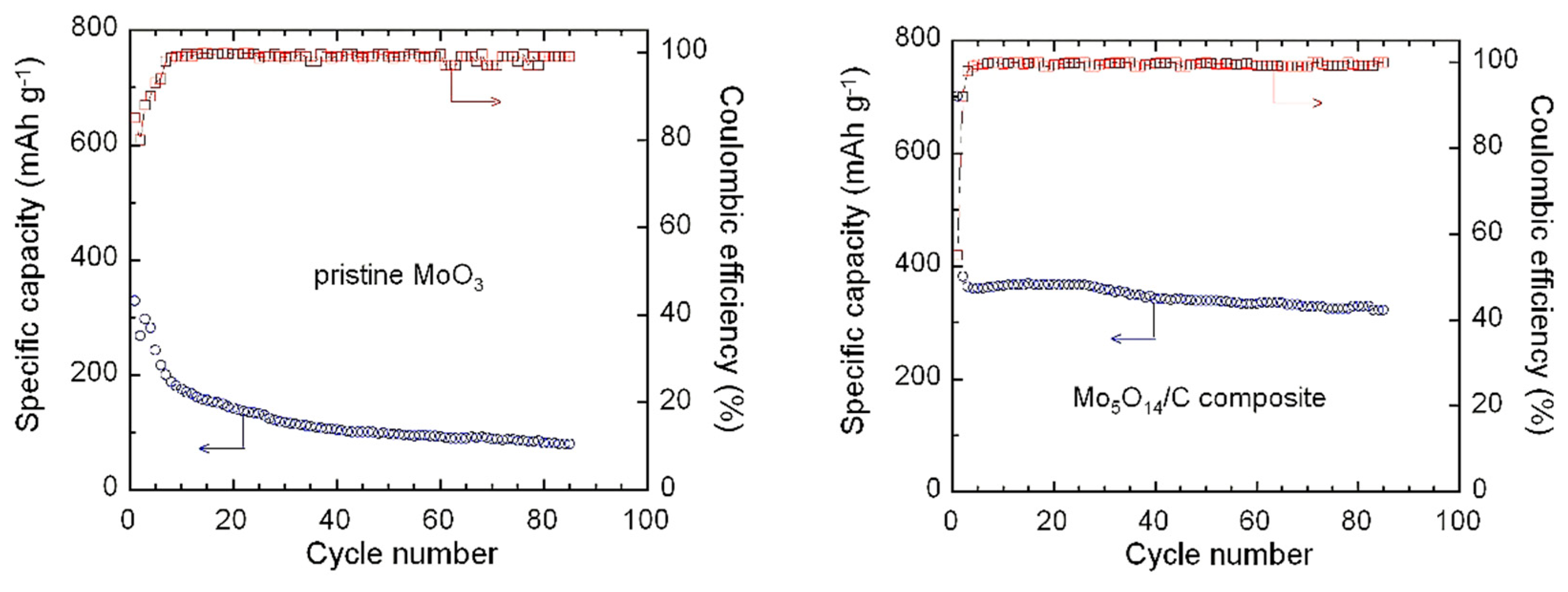
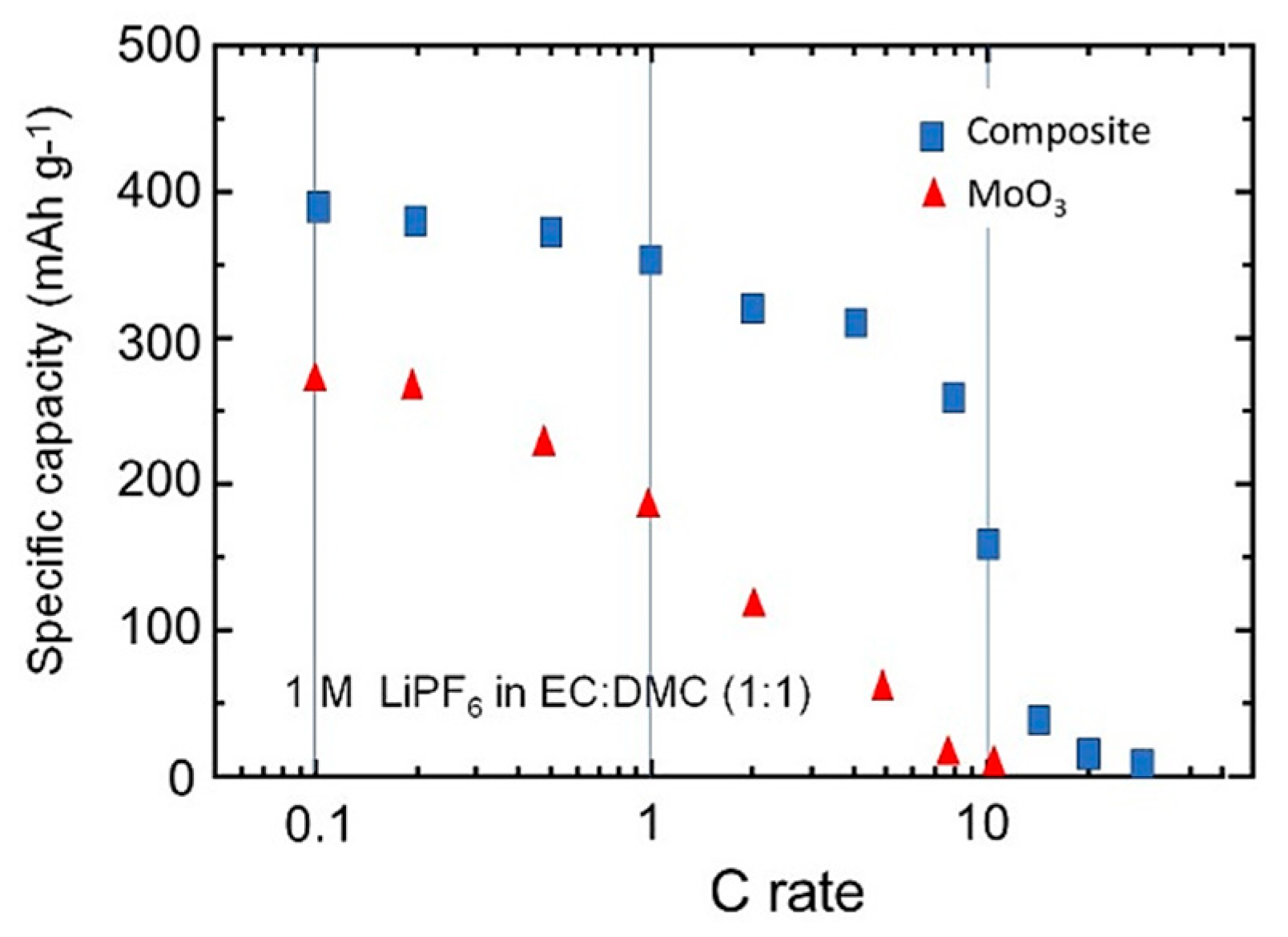
3.3. Electrochemical Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Julien, C.M.; Mauger, A.; Vijh, A.; Zaghib, K. Lithium Batteries: Science and Technology; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Scanlon, D.O.; Watson, G.W.; Payne, D.J.; Atkinson, G.R.; Egdell, R.G.; Law, D.S.L. Theoretical and experimental study of the electronic structures of MoO3 and MoO2. J. Phys. Chem. C 2010, 114, 4636–4645. [Google Scholar] [CrossRef]
- Kihlborg, L. Studies on molybdenum oxides. Acta Chem. Scand. 1959, 13, 954–962. [Google Scholar] [CrossRef]
- Besenhard, J.O.; Schollhorn, R. The discharge reaction mechanism of the MoO3 electrode in organic electrolytes. J. Power Sources 1976, 1, 267–276. [Google Scholar] [CrossRef]
- Cignini, P.; Icovi, M.; Panero, S.; Pistoia, G.; Temperoni, C. Non-stoichiometric molybdenum oxides as cathodes for lithium cells. Part. I. Primary batteries. J. Electroanal. Chem. 1979, 102, 333–342. [Google Scholar] [CrossRef]
- Icovi, M.; Panero, S.; D’Agate, A.; Pistoia, G.; Temperoni, C. Non-stoichiometric molybdenum oxides as cathodes for lithium cells. Part. II. Secondary batteries. J. Electroanal. Chem. 1979, 102, 343–349. [Google Scholar] [CrossRef]
- Pistoia, G.; Temperoni, C.; Cignini, P.; Icovi, M.; Panero, S. Non-stoichiometric molybdenum oxides as cathodes for lithium cells. Part. III. Cells based on Mo18O52. J. Electroanal. Chem. 1980, 108, 169–180. [Google Scholar] [CrossRef]
- Fiordiponti, P.; Pistoia, G.; Temperoni, C.; Icovi, M.; Panero, S. Non-stoichiometric molybdenum oxides as cathodes for lithium cells. Part. IV. Factors influencing the performance of Li/Mo8O23 batteries. J. Electroanal. Chem. 1980, 108, 181–190. [Google Scholar] [CrossRef]
- Christian, P.A.; Carides, J.N.; DiSalvo, F.J.; Waszczak, J.V. Molybdenum oxide cathodes in secondary lithium cells. J. Electrochem. Soc. 1980, 127, 2315–2319. [Google Scholar] [CrossRef]
- Nazri, G.-A.; Julien, C. Studies of lithium intercalation in heat-treated products obtained from molybdic acid. Ionics 1996, 2, 1–6. [Google Scholar] [CrossRef]
- Julien, C.; Yebka, B. Electrochemical Features of Lithium Batteries Based on Molybdenum-Oxide Compounds. In Materials for Lithium Batteries; Julien, C., Stoynov, Z., Eds.; Springer Science: Dordrecht, The Netherlands, 2000; pp. 263–277. [Google Scholar]
- Mondal, A.K.; Chen, S.; Liu, D.S.H.; Wang, G. Fabrication and enhanced electrochemical performances of MoO3/graphene composite as anode material for lithium-ion batteries. Inter. J. Smart Grid Clean Energy 2014, 3, 142–148. [Google Scholar] [CrossRef]
- Gao, B.; Fan, H.; Zhang, X. Hydrothermal synthesis of single crystal MoO3 nanobelts and their electrochemical properties as cathode electrode materials for rechargeable lithium batteries. J. Phys. Chem. Solids 2012, 73, 423–429. [Google Scholar] [CrossRef]
- Jung, Y.S.; Lee, S.; Ahn, D.; Dillon, A.C.; Lee, S.-H. Electrochemical reactivity of ball-milled MoO3-y as anode materials for lithium-ion batteries. J. Power Sources 2009, 188, 286–291. [Google Scholar] [CrossRef]
- Hashem, A.M.; Abbas, S.M.; Abdel-Ghany, A.E.; Eid, A.E.; Abdel-Khalek, A.A.; Indris, S.; Ehrenberg, H.; Mauger, A.; Julien, C.M. Blend formed by oxygen deficient MoO3-δ oxides as lithium-insertion compounds. J. Alloys Compd. 2016, 686, 744–752. [Google Scholar] [CrossRef]
- Xia, Q.; Zhao, H.; Du, Z.; Zeng, Z.; Gao, C.; Zhang, Z.; Du, X.; Kulka, A.; Swierczek, K. Facile synthesis of MoO3/carbon nanobelts as high-performance anode material for lithium ion batteries. Electrochim. Acta 2015, 180, 947–956. [Google Scholar] [CrossRef]
- Liu, C.-L.; Wang, Y.; Zhang, C.; Li, X.-S.; Dong, W.-S. In situ synthesis of α-MoO3/graphene composites as anode materials for lithium ion battery. Mater. Chem. Phys. 2014, 143, 1111–1118. [Google Scholar] [CrossRef]
- Feng, C.; Gao, H.; Zhang, C.; Guo, Z.; Liu, H. Synthesis and electrochemical properties of MoO3/C nanocomposite. Electrochim. Acta 2013, 93, 101–106. [Google Scholar] [CrossRef]
- Xia, Q.; Zhao, H.; Du, Z.; Wang, J.; Zhang, T.; Wang, J.; Lv, P. Synthesis and electrochemical properties of MoO3/C composite as anode material for lithium-ion batteries. J. Power Sources 2013, 226, 107–111. [Google Scholar] [CrossRef]
- Thomas, T.; Jiji, J.; Joy, L.; Mathew, A.; Reshmi, R. Hydrothermal synthesis MoO3-GO nano composites for energy storage application. AIP Conf. Proc. 2019, 2082, 060002. [Google Scholar]
- Cao, X.; Zheng, B.; Shi, W.; Yang, J.; Fan, Z.; Luo, Z.; Rui, X.; Chen, B.; Yan, Q.; Zhang, H. Reduced graphene oxide-wrapped MoO3 composites prepared by using metal–organic frameworks as precursor for all-solid-state flexible supercapacitors. Adv. Mater. 2015, 27, 4695–4701. [Google Scholar] [CrossRef]
- Choi, S.H.; Kang, Y.C. Crumpled graphene–molybdenum oxide composite powders: Preparation and application in lithium-ion batteries. ChemSusChem 2014, 7, 523–528. [Google Scholar] [CrossRef]
- Tao, T.; Glushenkov, A.M.; Zhang, C.F.; Zhang, H.Z.; Zhou, D.; Guo, Z.P.; Liu, H.K.; Chen, Q.Y.; Hu, H.P.; Chen, Y. MoO3 nanoparticles dispersed uniformly in carbon matrix: A high capacity composite anode for Li-ion batteries. J. Mater. Chem. 2011, 21, 9350–9355. [Google Scholar] [CrossRef]
- Kihlborg, L. Stabilization of the tunnel structure of Mo5O14 by partial metal atom substitution. Acta Chem. Scand. 1969, 23, 1834–1835. [Google Scholar] [CrossRef]
- Ekström, T. Formation of ternary phases of Mo5O14 and Mo17O47 structure in the molybdenum-wolfram-oxygen system. Mater. Res. Bull. 1972, 7, 19–26. [Google Scholar] [CrossRef]
- Knobl, S.; Zenkovets, G.A.; Kryukova, G.N.; Ovsitser, O.; Niemeyer, D.; Schlögl, R.; Mestl, G. The synthesis and structure of a single-phase, nanocrystalline MoVW mixed oxide catalyst of the Mo5O14 type. J. Catal. 2003, 215, 177–187. [Google Scholar] [CrossRef]
- Phillips, B.; Chang, L.Y. Condensed-phase relations in the system Mo-O. Trans. Met. Soc. AIME 1965, 233, 1433–1436. [Google Scholar]
- Nazri, G.-A.; Julien, C. Far-infrared and Raman studies of orthorhombic MoO3 single crystal. Solid State Ionics 1992, 53–56, 376–382. [Google Scholar] [CrossRef]
- Dieterle, M.; Mestl, G. Raman spectroscopy of molybdenum oxides. Part II. Resonance Raman spectroscopic characterization of the molybdenum oxides Mo4O11 and MoO2. Phys. Chem. Chem. Phys. 2002, 4, 822–826. [Google Scholar] [CrossRef]
- Balendhran, S.; Deng, J.; Ou, J.Z.; Walia, S.; Scott, J.; Tang, J.; Wang, K.L.; Field, M.R.; Russo, S.; Zhuiykov, S.; et al. Enhanced charge carrier mobility in two-dimensional high dielectric molybdenum oxide. Adv. Mater. 2013, 25, 109–114. [Google Scholar] [CrossRef]
- Nazri, G.A.; Julien, C. Heat-treatment studies of molybdenum oxide-monohydrate. Solid State Ionics 1995, 80, 271–275. [Google Scholar] [CrossRef]
- Hardcastle, F.D.; Wachs, I.E. Determination of molybdenum-oxygen bond distances and bond orders by Raman spectroscopy. J. Raman Spectrosc. 1990, 21, 683–691. [Google Scholar] [CrossRef]
- Subrahmanyam, K.S.; Vivekchand, S.R.C.; Govindaraj, A.; Rao, C.N.R. A study of graphenes prepared by different methods: Characterization, properties and solubilization. J. Mater. Chem. 2008, 18, 1517–1523. [Google Scholar] [CrossRef]
- Hu, Z.H.; Srinivasan, M.P.; Ni, Y.M. Preparation of mesoporous high surface area activated carbon. Adv. Mater. 2000, 12, 62–65. [Google Scholar] [CrossRef]
- Rodriguez-Reinoso, F.; Molina-Sabio, M. Activated carbons from lignocellulosic materials by chemical and/or physical activation: An overview. Carbon 1992, 30, 1111–1118. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Sun, Y.X.; Wang, J.; Zhao, B.; Cai, R.; Ran, R.; Shao, Z.P. Binder-free α-MoO3 nanobelt electrode for lithium-ion batteries utilizing van der Waals forces for film formation and connection with current collector. J. Mater. Chem. A 2013, 1, 4736–4746. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Madhavi, S.; Lou, X.W. Ultralong α-MoO3 nanobelts: Synthesis and effect of binder choice on their lithium storage properties. J. Phys. Chem. C 2012, 116, 12508–12513. [Google Scholar] [CrossRef]
- Hashem, A.M.; Askar, M.H.; Winter, M.; Albering, J.H.; Besenhard, J.O. Two-phase reaction mechanism during chemical lithium insertion into α-MoO3. Ionics 2007, 13, 3–8. [Google Scholar] [CrossRef]
- Li, W.Y.; Cheng, F.Y.; Tao, Z.L.; Chen, J. Vapor-transportation preparation and reversible lithium intercalation/deintercalation of alpha-MoO3 microrods. J. Phys. Chem. B 2006, 110, 119–124. [Google Scholar] [CrossRef]
- Leroux, F.; Loene, B.E.; Nazar, L.F. Electrochemical lithium intercalation into a polyaniline/V2O5 nanocomposite. J. Electrochem. Soc. 1996, 143, L181–L183. [Google Scholar] [CrossRef]
- Dong, W.; Mansour, A.N.; Dunn, B. Structural and electrochemical properties of amorphous and crystalline molybdenum oxide aerogels. Solid State Ionics 2001, 144, 31–40. [Google Scholar] [CrossRef]
- Brezesinski, T.; Wang, J.; Tolbert, S.H.; Dunn, B. Ordered mesoporous α-MoO3 with iso-oriented nanocrystalline walls for thin-film pseudo capacitors. Nat. Mater. 2010, 9, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.; Krautz, E. Untersuchungen der elektrischen leifähigkeit und des magnetowiderstandes im system molydän-sauerstoff. Phys. Status Solidi A 1980, 62, 615–624. [Google Scholar] [CrossRef]
- Aso, I.; Nakao, M.; Yamazoe, N.; Seiyama, T. Study of metal oxide catalysts in the olefin oxidation from their reduction behavior. 1. Reduction of various metal oxides with propylene. J. Catal. 1979, 57, 287–295. [Google Scholar] [CrossRef]
- Yi, Y.; An, H.; Zhang, P.; Tian, X.; Yang, P.; Liu, P.; Wang, T.; Qu, L.; Li, M.; Yang, G.; et al. Oxygen deficiency driven conversion of polysulfide by electrocatalysis: MoO3-x nanobelts for an improved lithium-sulfur battery cathode. ChemNanoMat 2019, 5, 926–931. [Google Scholar] [CrossRef]
- Cho, J.S. Large scale process for low crystalline MoO3-carbon composite microspheres prepared by one-step spray pyrolysis for anodes in lithium-ion batteries. Nanomaterials 2019, 9, 539. [Google Scholar] [CrossRef]
- Lu, W.; Chen, X.; Xue, M.; Cui, Y.; Zhuang, Q. Ultrasonic synthesis of α-MoO3 Nanobelt@CNTs composite for lithium battery and its electrochemical performances. Int. J. Electrochem. Sci. 2018, 13, 275–286. [Google Scholar] [CrossRef]
- Ni, J.F.; Wang, G.; Yang, J.; Gao, D.; Chen, J.; Gao, L.; Li, Y. Carbon nanotube-wired and oxygen-deficient MoO3 nanobelts with enhanced lithium-storage capability. J. Power Sources 2014, 247, 90–94. [Google Scholar] [CrossRef]
- Wu, D.; Shen, R.; Yang, R.; Ji, W.; Jiang, M.; Ding, W.; Peng, L. Mixed molybdenum oxides with superior performances as an advanced anode material for lithium-ion batteries. Sci. Rep. 2017, 7, 44697. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET Surface Area (m2 g−1) | Total Pore Volume (cm3 g−1) | Average Pore Size (nm) |
|---|---|---|---|
| P-MoO3 | 4.5 | 0.0024 | 2.19 |
| Mo5O14-type/C | 7.1 | 0.0719 | 4.01 |
| Composite (a) | Cathode Loading (mg cm−2) | Specific Capacity (mAh g−1) | Current Rate | Ref. |
|---|---|---|---|---|
| MoO3/a-C | 1.5 | 630 | 1 A g−1 | [47] |
| MoO3/CNTs (b) | - | 300 | C/5 | [48] |
| MoO3/C | - | ~600 | 40 mA g−1 | [18] |
| Mo3O8/CNTs | - | ~450 | 80 mA g−1 | [49] |
| Carbon-free MMO (c) | - | 300 | 0.1 A g−1 | [50] |
| MoO3/C | - | ~420 | 0.2C | [23] |
| MoO3/27 wt.% Gr | - | ~450 | 50 mA g−1 | [17] |
| MoO3/47 wt.% Gr | - | ~480 | 50 mA g−1 | [17] |
| MoO3/C | 1.5 | ~350 | 0.1 A g−1 | [19] |
| MoO3/Gr | 1.2 | ~550 | 2 A g−1 | [22] |
| Mo5O14/C | 2.1 | 703 | C/10 | this work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashem, A.M.; Abdel-Ghany, A.E.; El-Tawil, R.S.; Indris, S.; Ehrenberg, H.; Mauger, A.; Julien, C.M. Amorphous Mo5O14-Type/Carbon Nanocomposite with Enhanced Electrochemical Capability for Lithium-Ion Batteries. Nanomaterials 2020, 10, 8. https://doi.org/10.3390/nano10010008
Hashem AM, Abdel-Ghany AE, El-Tawil RS, Indris S, Ehrenberg H, Mauger A, Julien CM. Amorphous Mo5O14-Type/Carbon Nanocomposite with Enhanced Electrochemical Capability for Lithium-Ion Batteries. Nanomaterials. 2020; 10(1):8. https://doi.org/10.3390/nano10010008
Chicago/Turabian StyleHashem, Ahmed M., Ashraf E. Abdel-Ghany, Rasha S. El-Tawil, Sylvio Indris, Helmut Ehrenberg, Alain Mauger, and Christian M. Julien. 2020. "Amorphous Mo5O14-Type/Carbon Nanocomposite with Enhanced Electrochemical Capability for Lithium-Ion Batteries" Nanomaterials 10, no. 1: 8. https://doi.org/10.3390/nano10010008
APA StyleHashem, A. M., Abdel-Ghany, A. E., El-Tawil, R. S., Indris, S., Ehrenberg, H., Mauger, A., & Julien, C. M. (2020). Amorphous Mo5O14-Type/Carbon Nanocomposite with Enhanced Electrochemical Capability for Lithium-Ion Batteries. Nanomaterials, 10(1), 8. https://doi.org/10.3390/nano10010008