Double-Emulsion Copolyester Microcapsules for Sustained Intraperitoneal Release of Carboplatin

 ,
,  ,
, 

Abstract
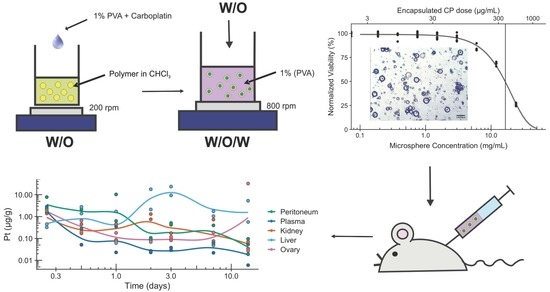
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
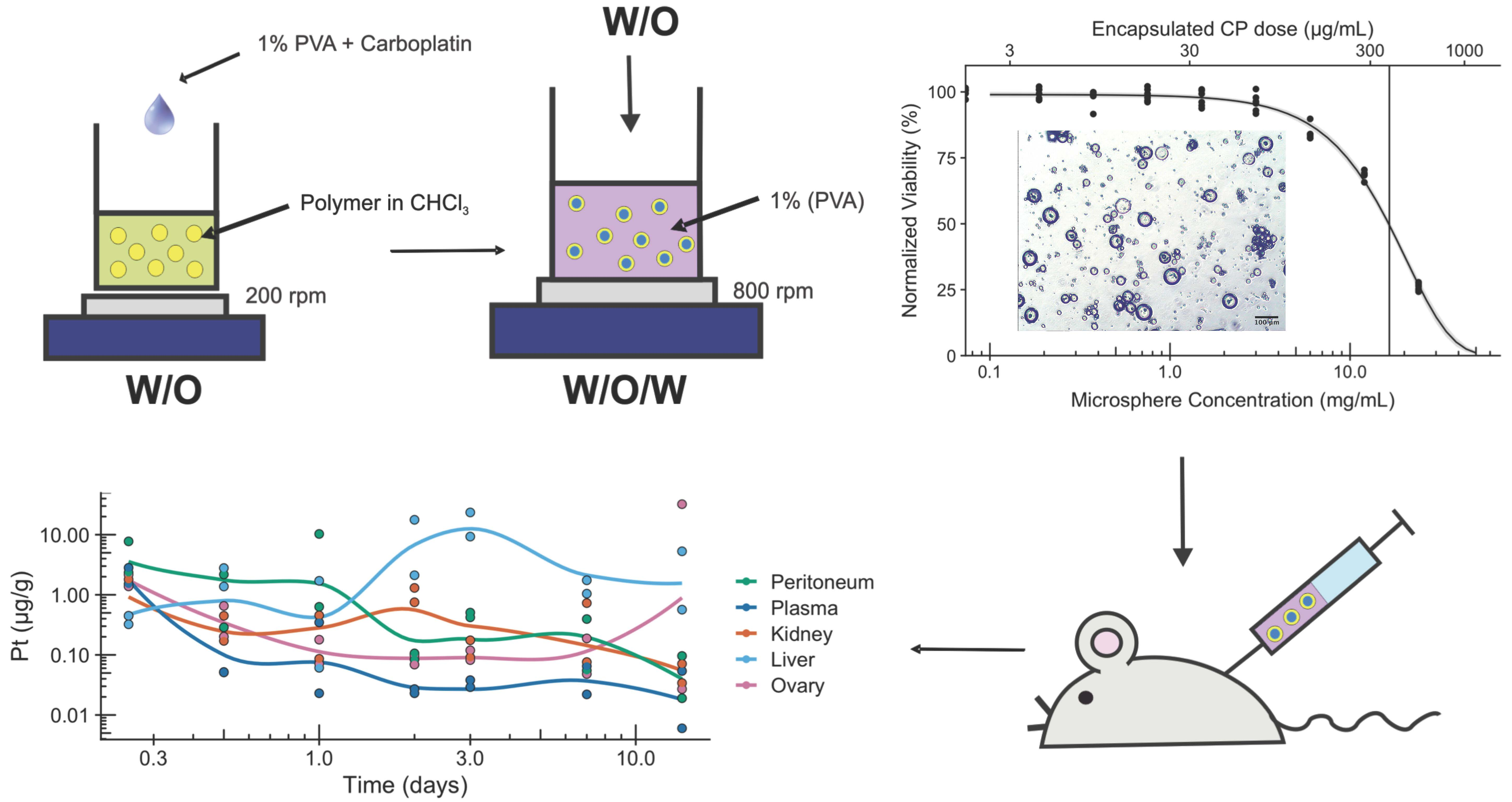
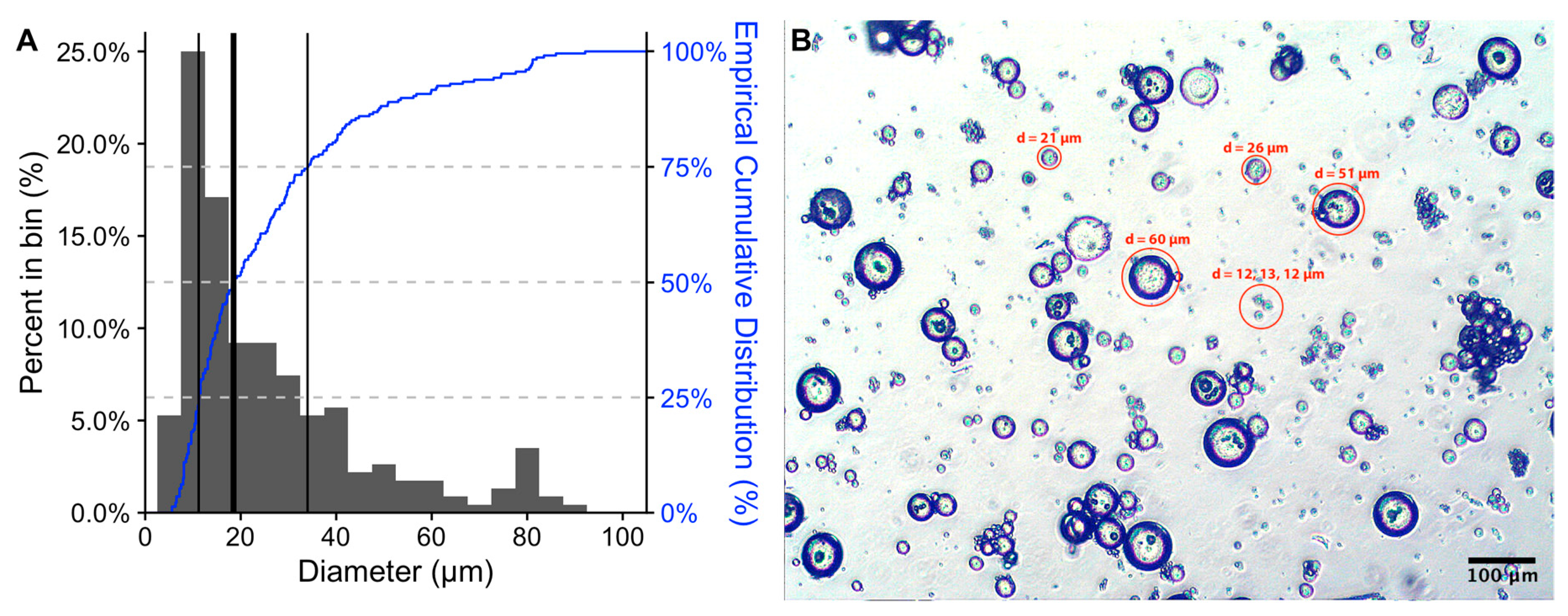
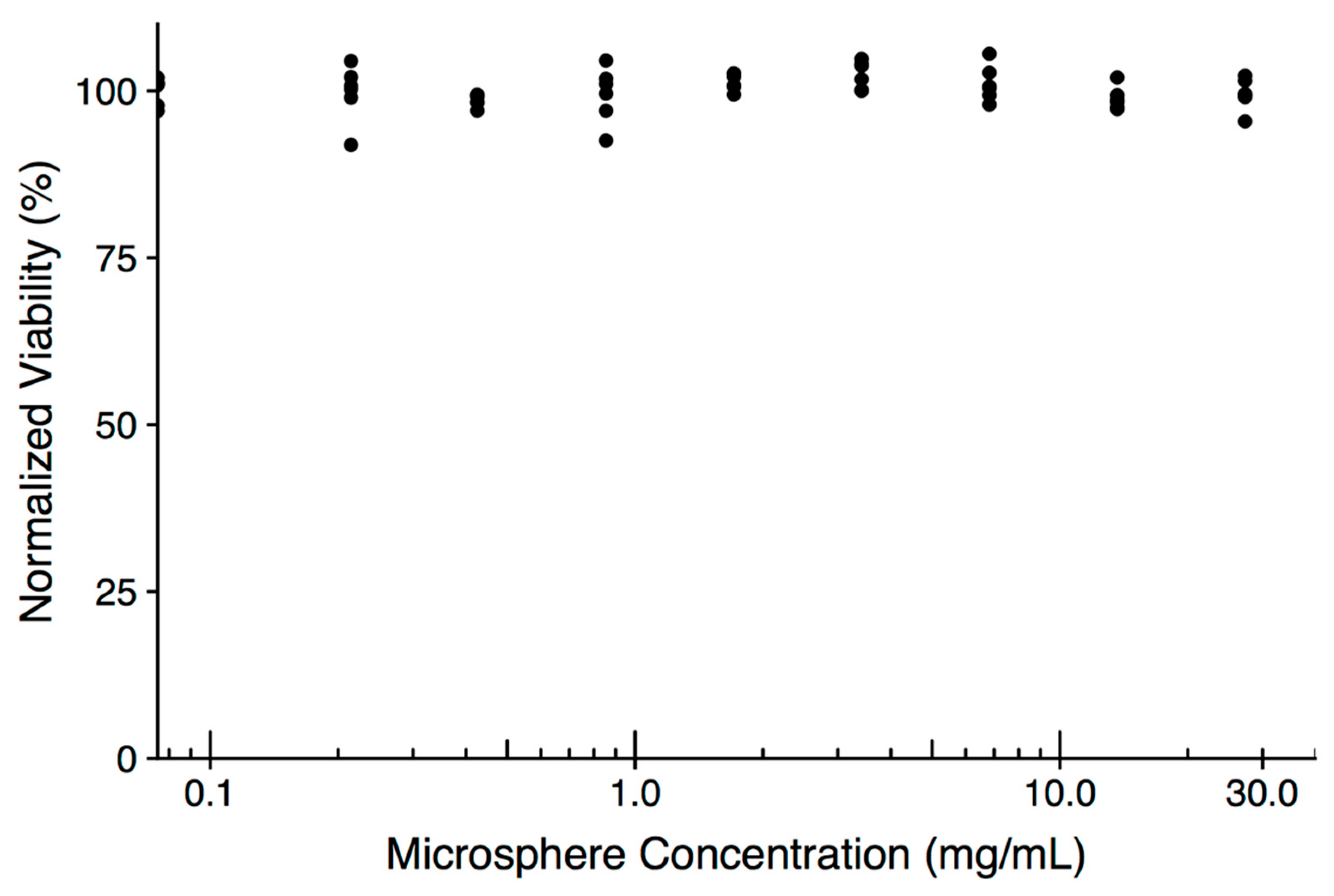
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals and Reagents
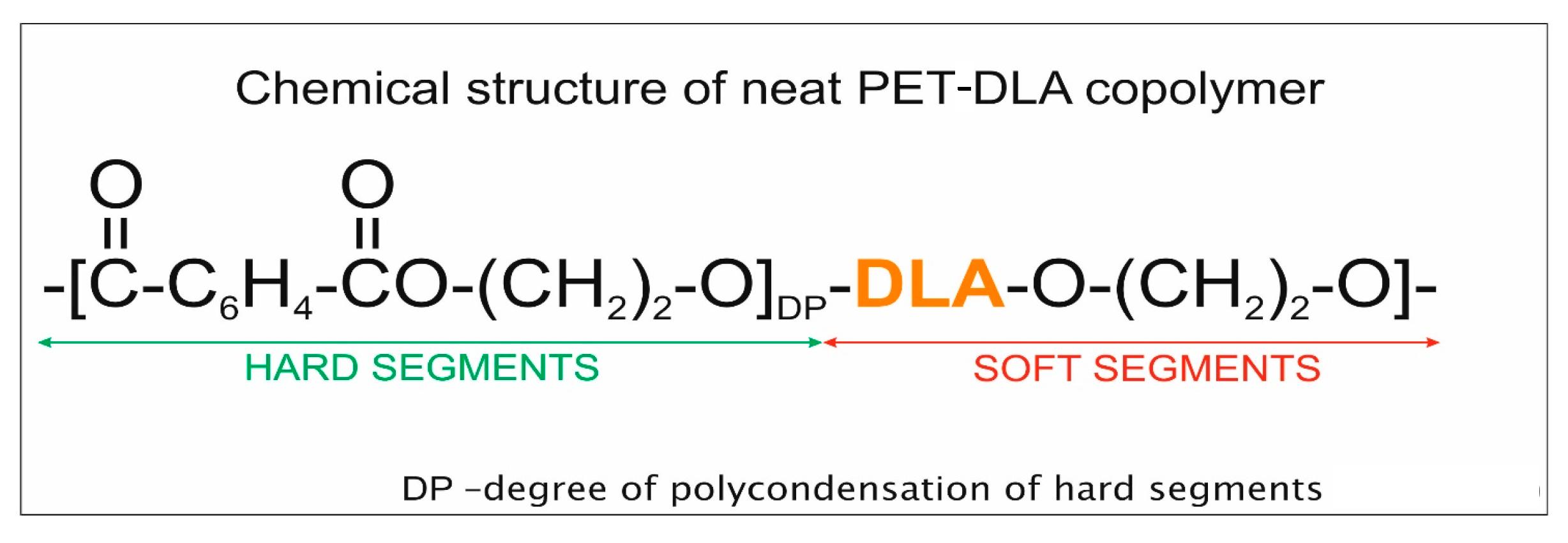
3.2. Polymer for Drug Encapsulation
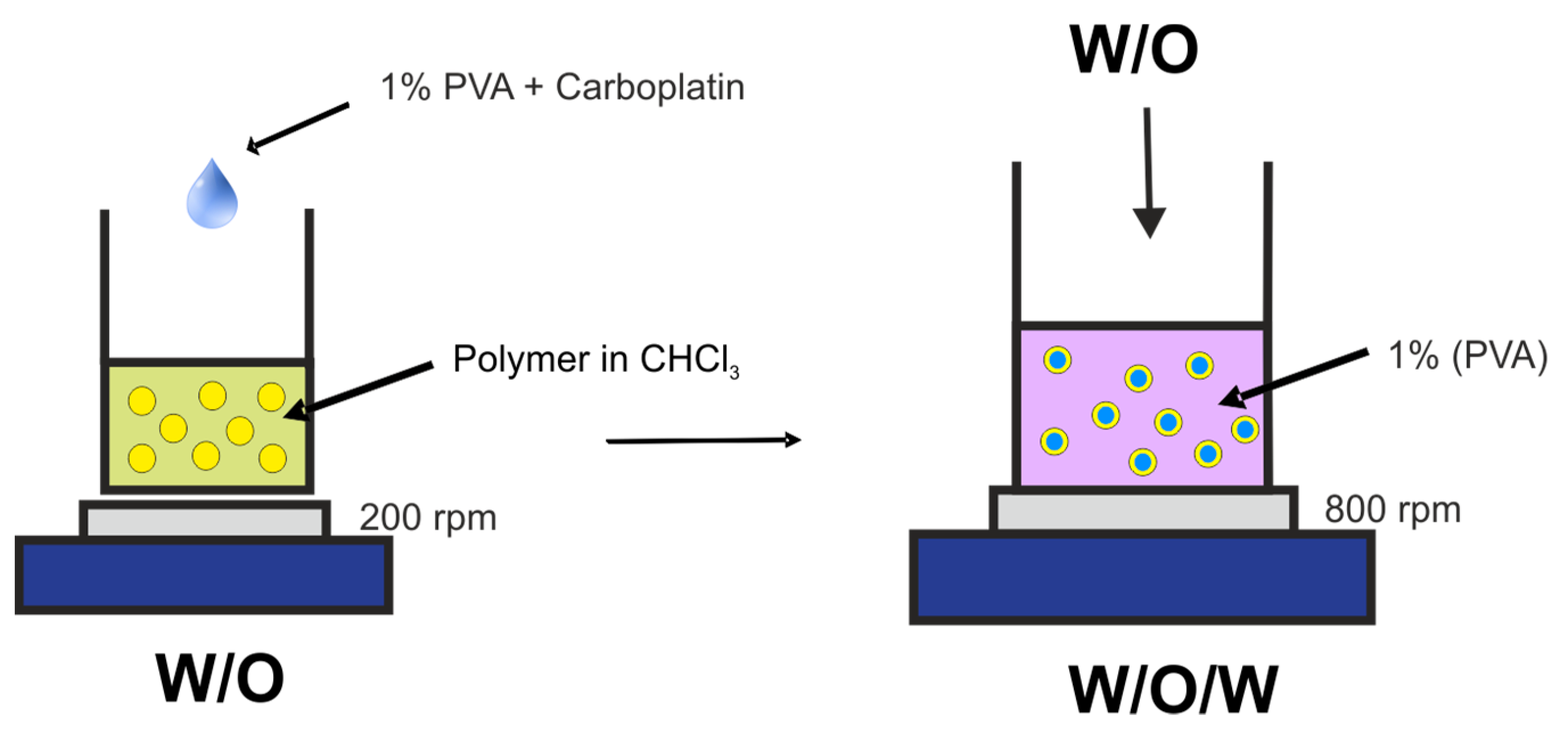
3.3. Preparation of Microspheres
3.4. Morphology of Microspheres
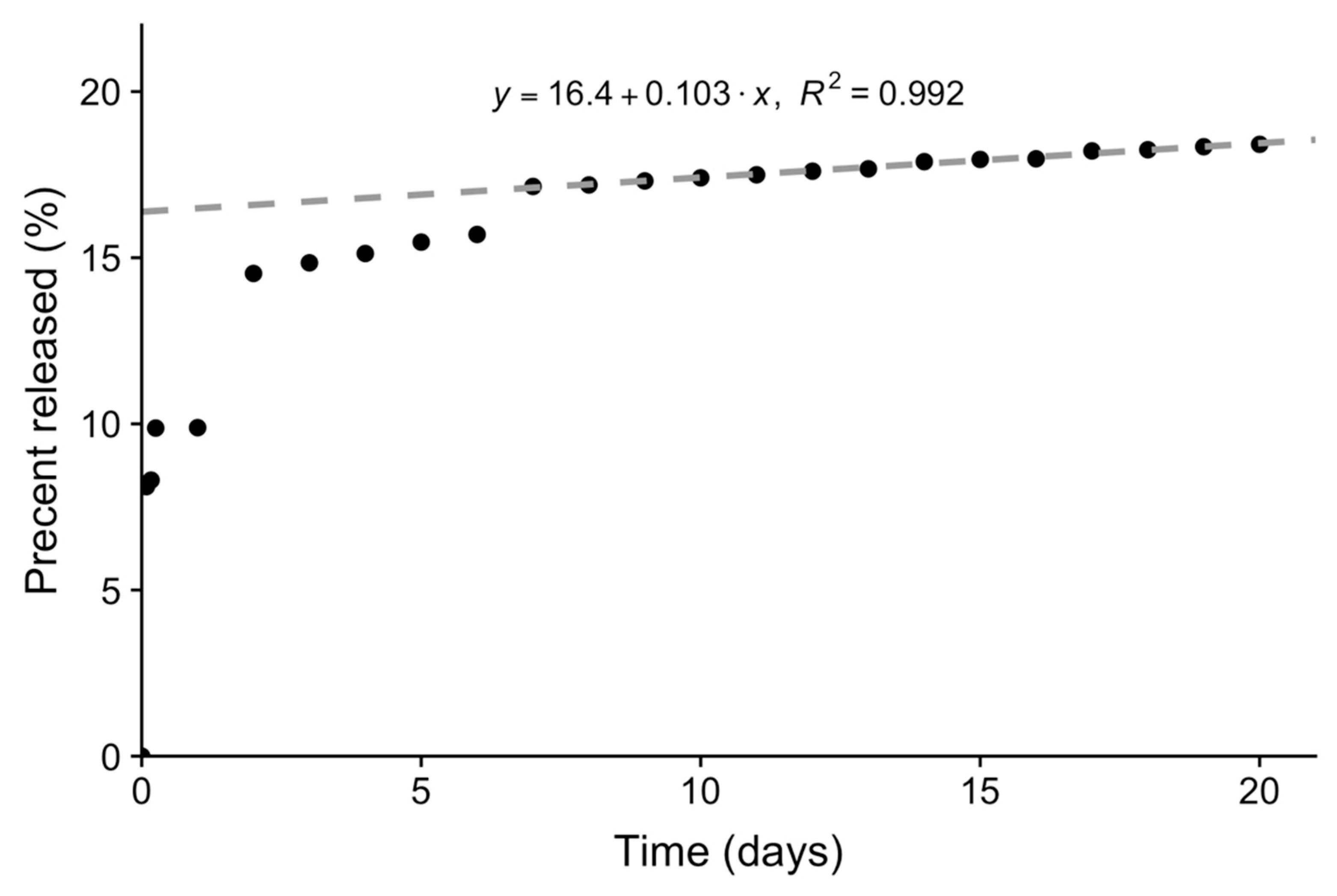
3.5. Loading and Release of Carboplatin In Vitro
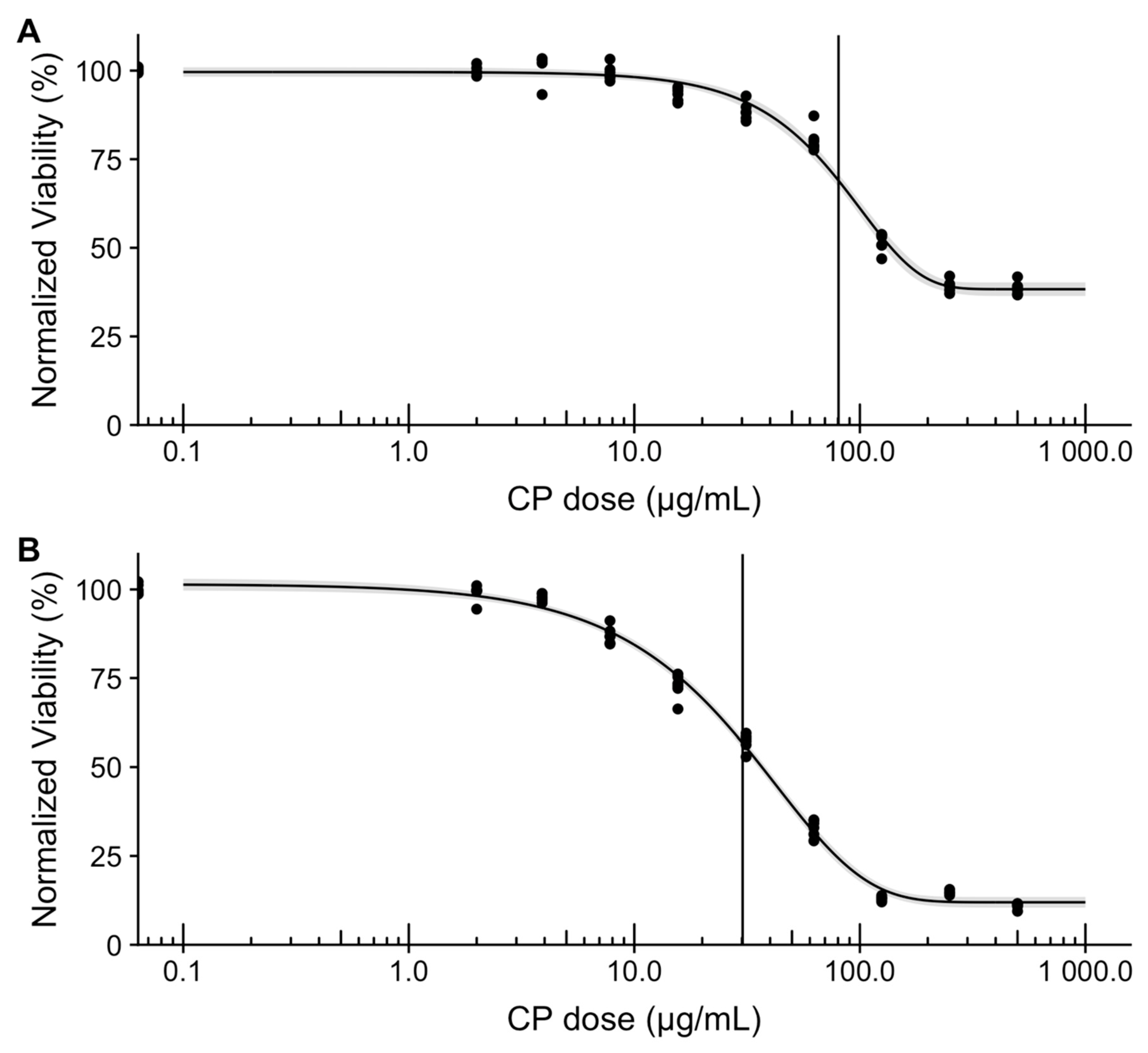
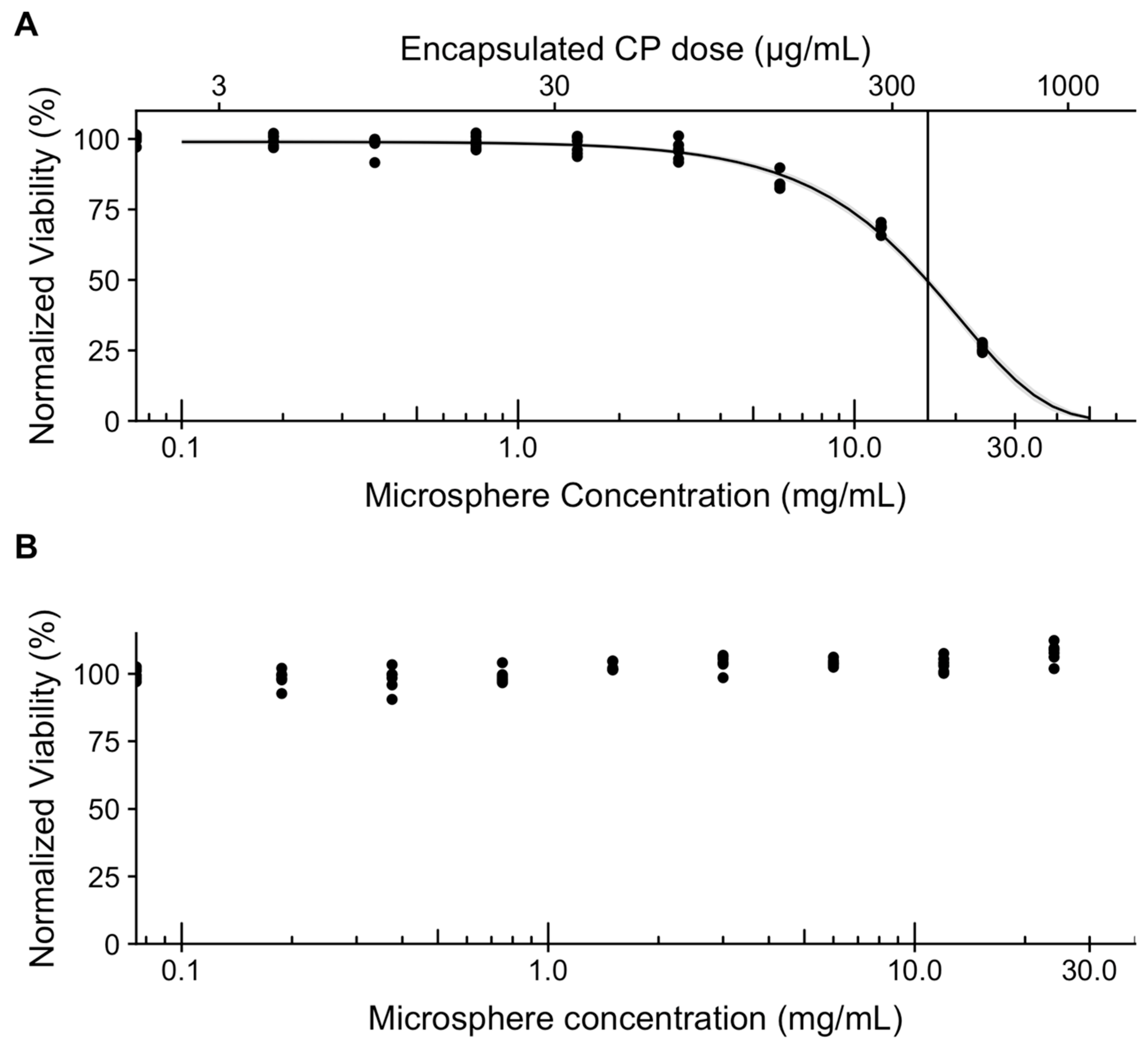
3.6. In Vitro Assessment of Microsphere Cytotoxicity and Drug Delivery
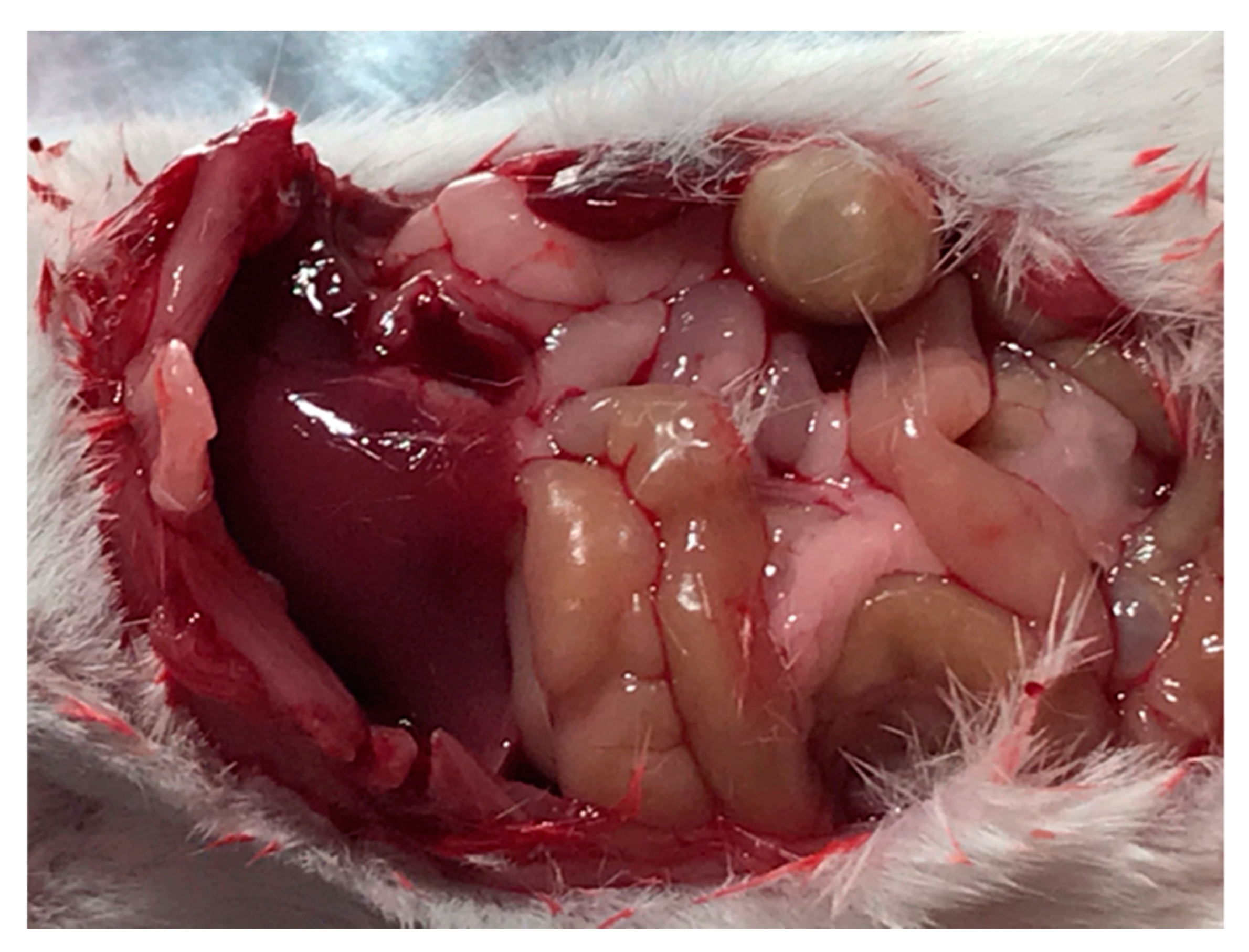
3.7. In Vivo Assessment of Drug Delivery
3.8. Release of Carboplatin In Vivo
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- The World Ovarian Cancer Coalition Atlas: Global Trends in Incidence, Mortality and Survival. Available online: https://www.semanticscholar.org/paper/THE-WORLD-OVARIAN-CANCER-COALITION-ATLAS-GLOBAL-IN/67b106416bcb66f605debf039b56ff37a530a288#citing-papers (accessed on 4 October 2019).
- Luvero, D.; Plotti, F.; Aloisia, A.; Montera, R.; Terranova, C.; De Cicco Nardone, C.; Scaletta, G.; Lopez, S.; Miranda, A.; Capriglione, S.; et al. Ovarian cancer relapse: From the latest scientific evidence to the best practice. Crit. Rev. Oncol. Hematol. 2019, 140, 28–38. [Google Scholar] [CrossRef]
- Marchetti, C.; Pisano, C.; Facchini, G.; Bruni, G.S.; Magazzino, F.P.; Losito, S.; Pignata, S. First-line treatment of advanced ovarian cancer: Current research and perspectives. Expert Rev. Anticancer Ther. 2010, 10, 47–60. [Google Scholar] [CrossRef]
- Jaaback, K.; Johnson, N.; Lawrie, T.A. Intraperitoneal chemotherapy for the initial management of primary epithelial ovarian cancer. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Elferink, F.; van der Vijgh, W.J.; Klein, I.; ten Bokkel Huinink, W.W.; Dubbelman, R.; McVie, J.G. Pharmacokinetics of carboplatin after intraperitoneal administration. Cancer Chemother. Pharmacol. 1988, 21, 57–60. [Google Scholar] [CrossRef]
- Miyagi, Y.; Fujiwara, K.; Kigawa, J.; Itamochi, H.; Nagao, S.; Aotani, E.; Terakawa, N.; Kohno, I. Intraperitoneal carboplatin infusion may be a pharmacologically more reasonable route than intravenous administration as a systemic chemotherapy. A comparative pharmacokinetic analysis of platinum using a new mathematical model after intraperitoneal vs. i. Gynecol. Oncol. 2005, 99, 591–596. [Google Scholar] [CrossRef]
- Raavé, R.; de Vries, R.B.M.; Massuger, L.F.; van Kuppevelt, T.H.; Daamen, W.F. Drug delivery systems for ovarian cancer treatment: A systematic review and meta-analysis of animal studies. PeerJ 2015, 3. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Y.; Zhang, H.; Xu, P.; Liu, J.; Ma, J.; Lv, M.; Li, D.; Katirai, F.; Shen, G.-X.; et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun. 2012, 3, 1282. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Tabata, Y.; Ikada, Y. Phagocytosis of polymer microspheres by macrophages. In New Polymer Materials; Springer: Berlin/Heidelberg, Germany, 2005; pp. 107–141. [Google Scholar]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Li, M.; Rouaud, O.; Poncelet, D. Microencapsulation by solvent evaporation: State of the art for process engineering approaches. Int. J. Pharm. 2008, 363, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Spenlehauer, G.; Vert, M.; Benoît, J.P.; Chabot, F.; Veillard, M. Biodegradable cisplatin microspheres prepared by the solvent evaporation method: Morphology and release characteristics. J. Control. Release 1988, 7, 217–229. [Google Scholar] [CrossRef]
- Verrijk, R.; Smolders, I.J.H.; McVie, J.G.; Begg, A.C. Polymer-coated albumin microspheres as carriers for intravascular tumour targeting of cisplatin. Cancer Chemother. Pharmacol. 1991, 29, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Manunta, M.L.; Gavini, E.; Chessa, G.; Passino, E.S.; Careddu, G.M.; Giua, S.; Mollica, A. Carboplatin Sustained Delivery System Using Injectable Microspheres. J. Vet. Med. Ser. A 2005, 422, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Verrijk, R.; Smolders, I.J.; Bosnie, N.; Begg, A.C. Reduction of systemic exposure and toxicity of cisplatin by encapsulation in poly-lactide-co-glycolide. Cancer Res. 1992, 52, 6653–6656. [Google Scholar]
- Ike, O.; Shimizu, Y.; Wada, R.; Hyon, S.H.; Ikada, Y. Controlled cisplatin delivery system using poly (d, l-lactic acid]. Biomaterials 1992, 13, 230–234. [Google Scholar] [CrossRef]
- Hagiwara, A.; Takahashi, T.; Kojima, O.; Yamaguchi, T.; Sasabe, T.; Lee, M.; Sakakura, C.; Shoubayashi, S.; Ikada, Y.; Hyon, S.-H. Pharmacologic effects of cisplatin microspheres on peritoneal carcinomatosis in rodents. Cancer 1993, 71, 844–850. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kumagai, S.; Nishida, T.; Ushijima, K.; Matsuo, T.; Yakushiji, M.; Hyon, S.H.; Ikada, Y. Experimental and clinical evaluation of cisplatin-containing microspheres as intraperitoneal chemotherapy for ovarian cancer. Anticancer Res. 1998, 18, 2837–2842. [Google Scholar]
- Tokuda, K.; Natsugoe, S.; Shimada, M.; Kumanohoso, T.; Baba, M.; Takao, S.; Nakamura, K.; Yamada, K.; Yoshizawa, H.; Hatate, Y.; et al. Design and testing of a new cisplatin form using a base material by combining poly-D,L-lactic acid and polyethylene glycol acid against peritoneal metastasis. Int. J. Cancer 1998, 76, 709–712. [Google Scholar] [CrossRef]
- Chen, W.; Lu, D.R. Carboplatin -loaded PLGA microspheres for intracerebral injection: Formulation and characterization. J. Microencapsul. 1999, 16, 551–563. [Google Scholar]
- Tamura, T.; Imai, J.; Matsumoto, A.; Tanimoto, M.; Suzuki, A.; Horikiri, Y.; Suzuki, T.; Yoshino, H.; Ike, O. Organ distribution of cisplatin after intraperitoneal administration of cisplatin-loaded microspheres. Eur. J. Pharm. Biopharm. 2002, 54, 1–7. [Google Scholar] [CrossRef]
- Fujiyama, J.; Nakase, Y.; Osaki, K.; Sakakura, C.; Yamagishi, H.; Hagiwara, A. Cisplatin incorporated in microspheres: Development and fundamental studies for its clinical application. J. Control. Release 2003, 89, 397–408. [Google Scholar] [CrossRef]
- Lu, B.; Zhang, J.; Yang, H. Lung-targeting microspheres of carboplatin. Int. J. Pharm. 2003, 265, 1–11. [Google Scholar] [CrossRef]
- Gunji, S.; Obama, K.; Matsui, M.; Tabata, Y.; Sakai, Y. A novel drug delivery system of intraperitoneal chemotherapy for peritoneal carcinomatosis using gelatin microspheres incorporating cisplatin. Surgery 2013, 154, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Singh, S.K. Diffusion-Controlled Release. In Controlled Release A Quantitative Treatment; Springer: Berlin/Heidelberg, Germany, 1989; pp. 9–88. [Google Scholar]
- Chudecka-Głaz, A.; Szczeblińska, J.; Cymbaluk-Płoska, A.; Kohn, J.; El Fray, M. New poly (ester-amide) copolymers modified with polyether (PEAE) for anticancer drug encapsulation. J. Microencapsul. 2016, 33. [Google Scholar] [CrossRef] [PubMed]
- Piegat, A.; El Fray, M. Poly (ethylene terephthalate) modification with the monomer from renewable resources. Polimery 2007, 52, 885–888. [Google Scholar] [CrossRef]
- El Fray, M.; Piegat, A.; Prowans, P. Influence of TiO 2 Nanoparticles Incorporated into Elastomeric Polyesters on their Biocompatibility In Vitro and In Vivo. Adv. Eng. Mater. 2009, 11, 200–203. [Google Scholar] [CrossRef]
- Jawad, H.; El Fray, M.; Boccaccini, A.R.; Harding, S.E.; Wright, J.S.; Chen, Q.; Piegat, A.; Ali, N.N. Nanocomposite Elastomeric Biomaterials for Myocardial Tissue Engineering Using Embryonic Stem Cell-derived Cardiomyocytes. Adv. Eng. Mater. 2010, 12, 664–674. [Google Scholar] [CrossRef]
- Kohane, D.S.; Tse, J.Y.; Yeo, Y.; Padera, R.; Shubina, M.; Langer, R. Biodegradable polymeric microspheres and nanospheres for drug delivery in the peritoneum. J. Biomed. Mater. Res. Part A 2006, 77, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Shimizu, M.; Kukizaki, M. Particle control of emulsion by membrane emulsification and its applications. Adv. Drug Deliv. Rev. 2000, 45, 47–56. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, M.-J.; Chu, L.-Y. Microfluidic approach for encapsulation via double emulsions. Curr. Opin. Pharmacol. 2014, 18, 35–41. [Google Scholar] [CrossRef] [PubMed]
- ISO. Biological Evaluation of Medical Devices—Part 5: Tests for in Vitro Cytotoxicity; ISO: Geneva, Switzerland, 2009. [Google Scholar]
- DrugBank Carboplatin. Available online: http://www.drugbank.ca/drugs/DB00958 (accessed on 4 October 2019).
- Pénzváltó, Z.; Lánczky, A.; Lénárt, J.; Meggyesházi, N.; Krenács, T.; Szoboszlai, N.; Denkert, C.; Pete, I.; Győrffy, B. MEK1 is associated with carboplatin resistance and is a prognostic biomarker in epithelial ovarian cancer. BMC Cancer 2014, 14, 837. [Google Scholar] [CrossRef] [PubMed]
- Helland, Ø.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjørge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [PubMed]
- Arung, W.; Meurisse, M.; Detry, O. Pathophysiology and prevention of postoperative peritoneal adhesions. World J. Gastroenterol. 2011, 17, 4545–4553. [Google Scholar] [CrossRef]
- Staniszewski, Z.; El Fray, M. Influence of thermally exfoliated graphite on physicochemical, thermal and mechanical properties of copolyester nanocomposites. Polimery 2016, 61, 482–489. [Google Scholar] [CrossRef]
- Staniszewski, Z.; Piegat, A.; Okroj, W.; Walkowiak-Przybylo, M.; Jakubowski, W.; Walkowiak, B.; Budner, B.; Mroz, W.; Sobolewski, P.; El Fray, M. The effect of carbon nanoparticles on biological properties of polyester nanocomposites. J. Biomater. Appl. 2017, 31, 1328–1336. [Google Scholar] [CrossRef]
- Staniszewski, Z.; Sobolewski, P.; Piegat, A.; El Fray, M. The effects of nano-sized carbon fillers on the physico-chemical, mechanical, and biological properties of polyester nanocomposites. Eur. Polym. J. 2018, 107, 189–201. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Lu, B.; Zhang, J.Q.; Yang, H. Nonphospholipid Vesicles of Carboplatin for Lung Targeting. Drug Deliv. 2003, 10, 87–94. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Sittampalam, G., Coussens, N., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Holbeck, S.L.; Collins, J.M.; Doroshow, J.H. Analysis of FDA-Approved Anti-Cancer Agents in the NCI60 Panel of Human Tumor Cell Lines. Mol. Cancer Ther. 2010, 9, 1451. [Google Scholar] [CrossRef]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [PubMed]
- Jandial, D.D.; Messer, K.; Farshchi-heydari, S.; Pu, M.; Howell, S.B. Gynecologic Oncology Tumor platinum concentration following intraperitoneal administration of cisplatin versus carboplatin in an ovarian cancer model. Gynecol. Oncol. 2009, 115, 362–366. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Caffrey, P.B.; Frenkel, G.D. Prevention of Carboplatin-induced Resistance in Human Ovarian Tumor Xenografts by Selenite. Anticancer Res. 2013, 33, 4249–4254. [Google Scholar] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cymbaluk-Płoska, A.; Sobolewski, P.; Chudecka-Głaz, A.; Wiśniewska, E.; Łapczuk, J.; Frankowski, M.; Droździk, M.; El Fray, M. Double-Emulsion Copolyester Microcapsules for Sustained Intraperitoneal Release of Carboplatin. J. Funct. Biomater. 2019, 10, 55. https://doi.org/10.3390/jfb10040055
Cymbaluk-Płoska A, Sobolewski P, Chudecka-Głaz A, Wiśniewska E, Łapczuk J, Frankowski M, Droździk M, El Fray M. Double-Emulsion Copolyester Microcapsules for Sustained Intraperitoneal Release of Carboplatin. Journal of Functional Biomaterials. 2019; 10(4):55. https://doi.org/10.3390/jfb10040055
Chicago/Turabian StyleCymbaluk-Płoska, Aneta, Peter Sobolewski, Anita Chudecka-Głaz, Ewa Wiśniewska, Joanna Łapczuk, Marcin Frankowski, Marek Droździk, and Miroslawa El Fray. 2019. "Double-Emulsion Copolyester Microcapsules for Sustained Intraperitoneal Release of Carboplatin" Journal of Functional Biomaterials 10, no. 4: 55. https://doi.org/10.3390/jfb10040055
APA StyleCymbaluk-Płoska, A., Sobolewski, P., Chudecka-Głaz, A., Wiśniewska, E., Łapczuk, J., Frankowski, M., Droździk, M., & El Fray, M. (2019). Double-Emulsion Copolyester Microcapsules for Sustained Intraperitoneal Release of Carboplatin. Journal of Functional Biomaterials, 10(4), 55. https://doi.org/10.3390/jfb10040055