
Identification and Dynamics Understanding of Novel Inhibitors of Peptidase Domain of Collagenase G from Clostridium histolyticum

 ,
,  , , , and
, , , and
Abstract
1. Introduction
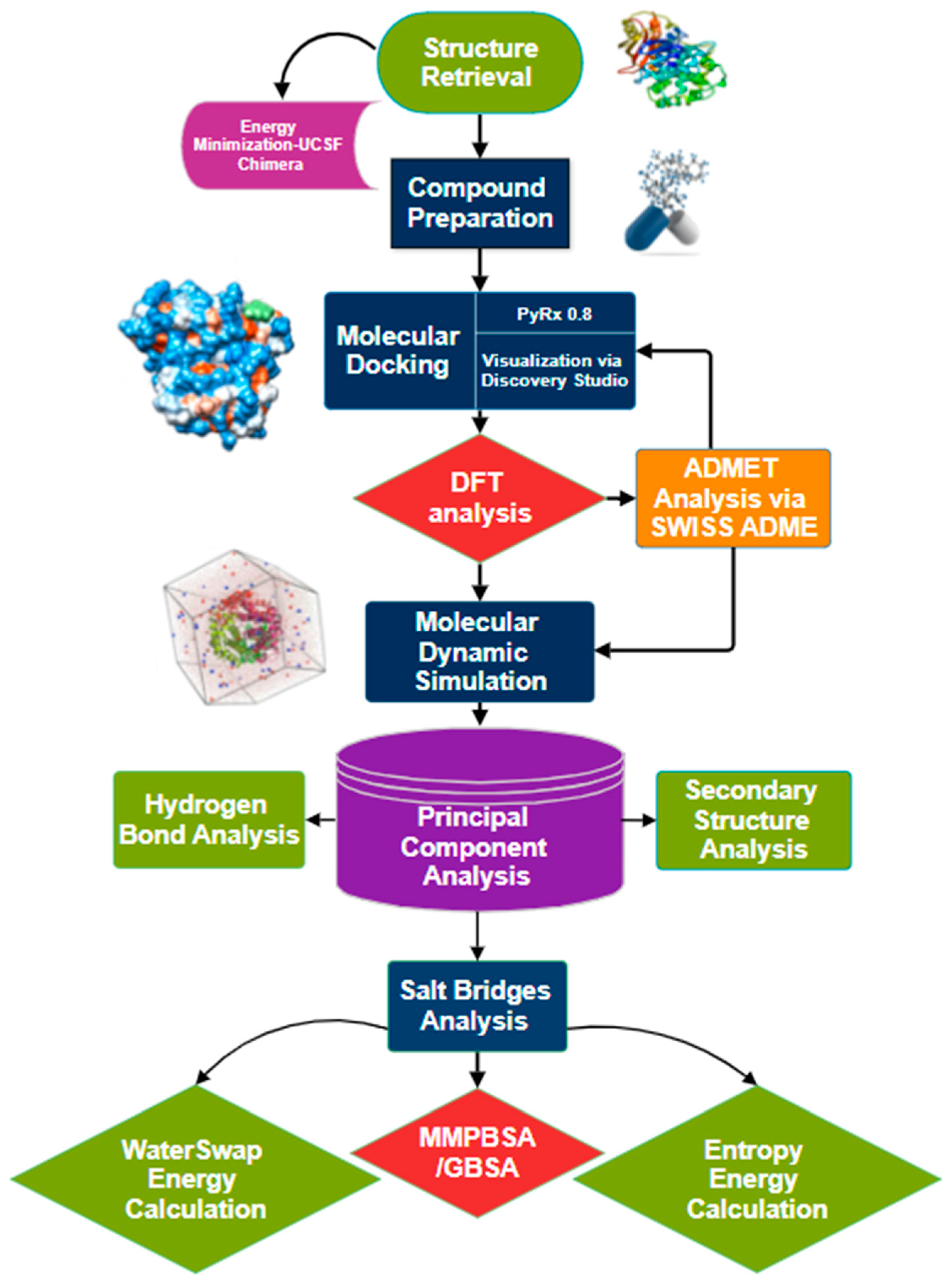
2. Methodology
2.1. Collagenase Enzyme Crystal Structure Retrieval and Preparation
2.2. Ligands Library Selection and Preparation
2.3. Molecular Docking Studies
2.4. Density Functional Theory (DFT)
2.5. ADME and Pharmacokinetics Profile
2.6. Molecular Dynamic Simulation
2.7. Hydrogen Bond Analysis
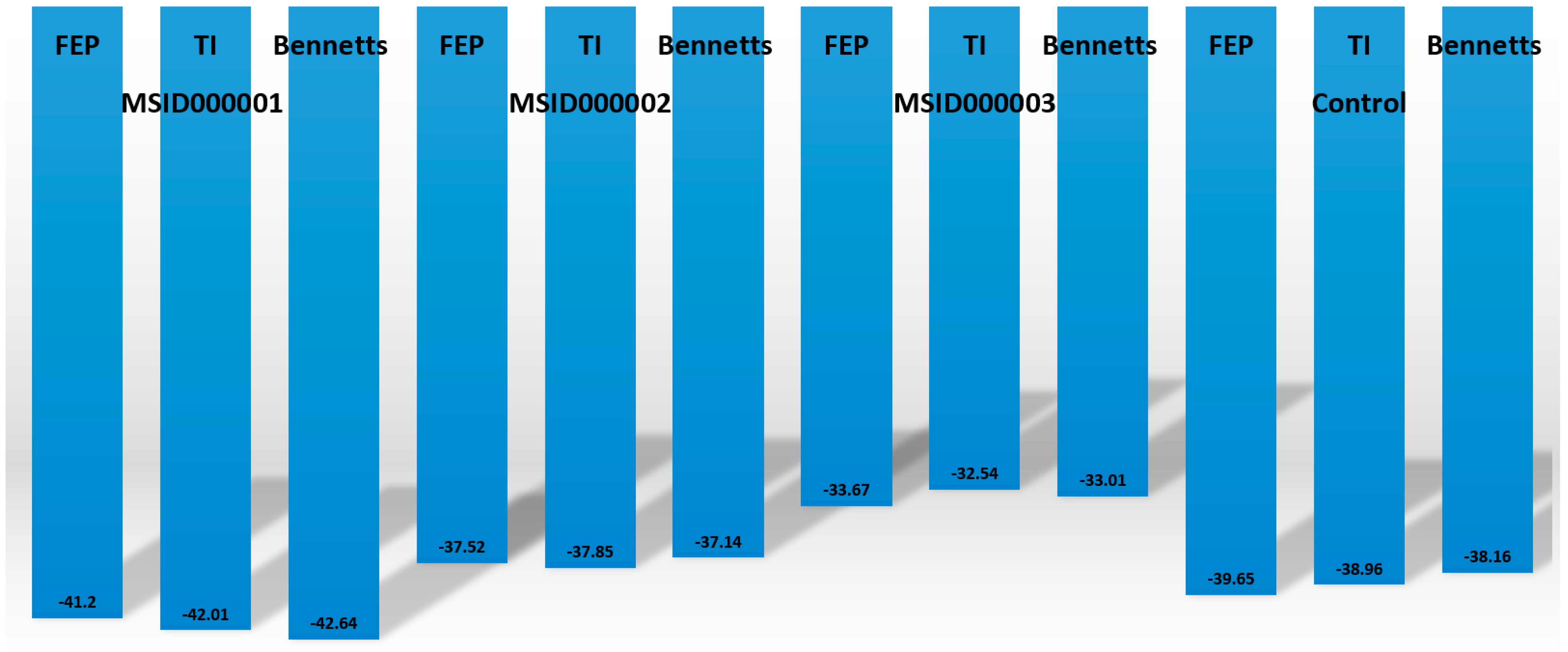
2.8. Calculating Binding Affinities
ΔGasol = ΔGp + ΔGnp
ΔGtotal = ΔEMM + ΔGsol
ΔGbind = ΔEMM + ΔGsol − T
2.9. Entropy Energy Calculation
2.10. WaterSwap Absolute Energy Estimation
2.11. Secondary Structure Analysis
2.12. Principal Component Analysis (PCA)
2.13. Salt Bridges
3. Results
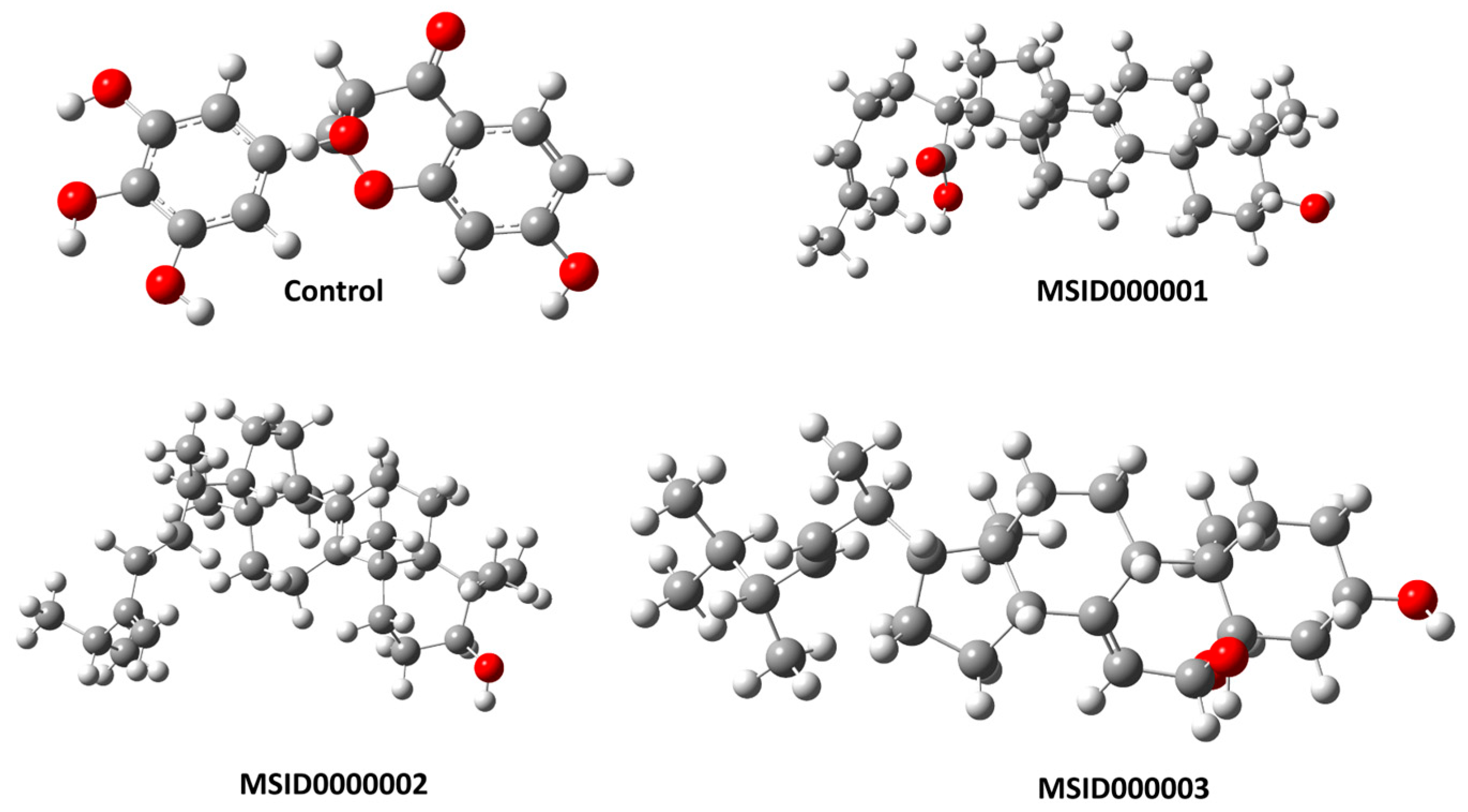
3.1. Structure Retrieval and Initial Preparation
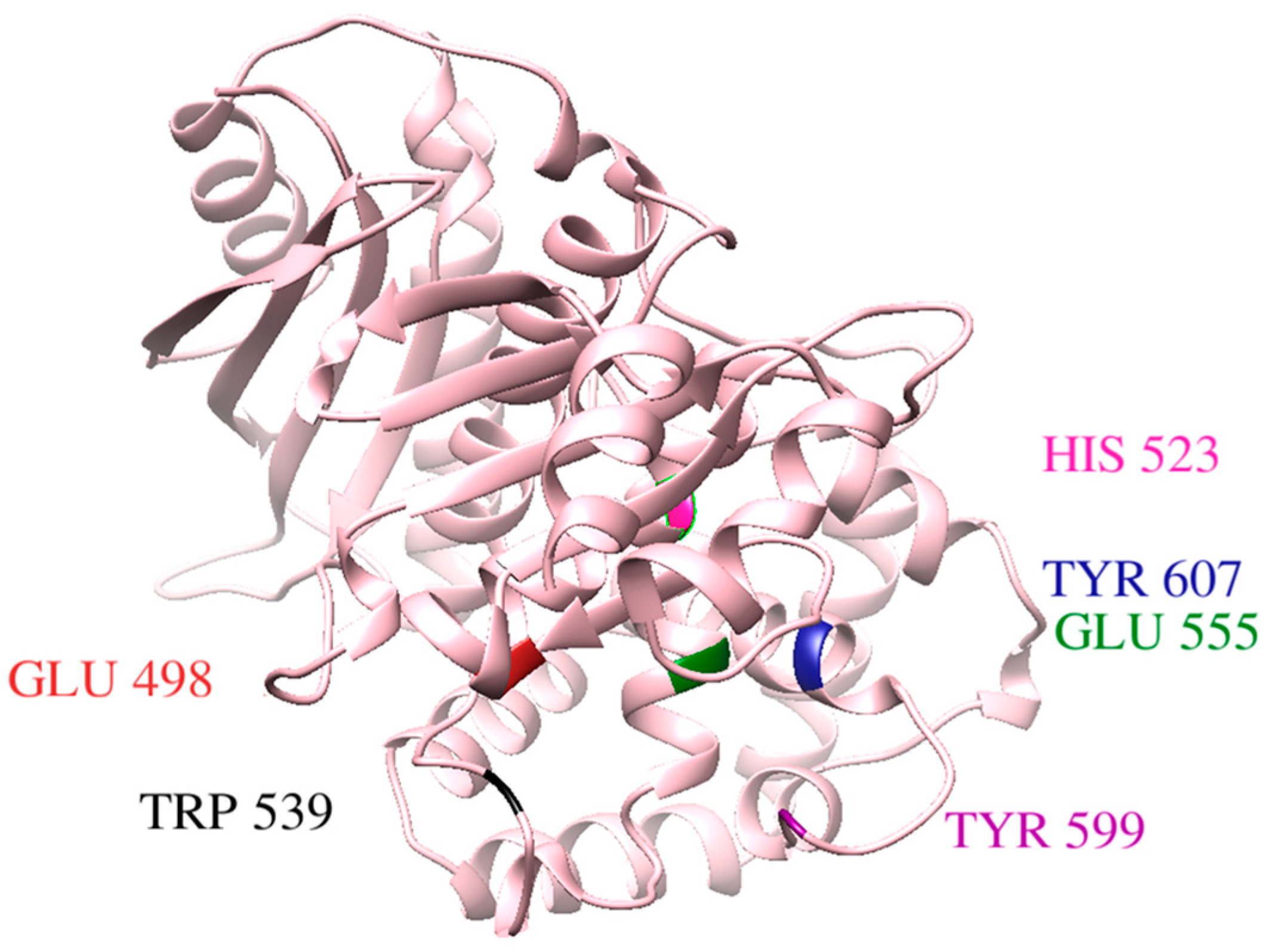
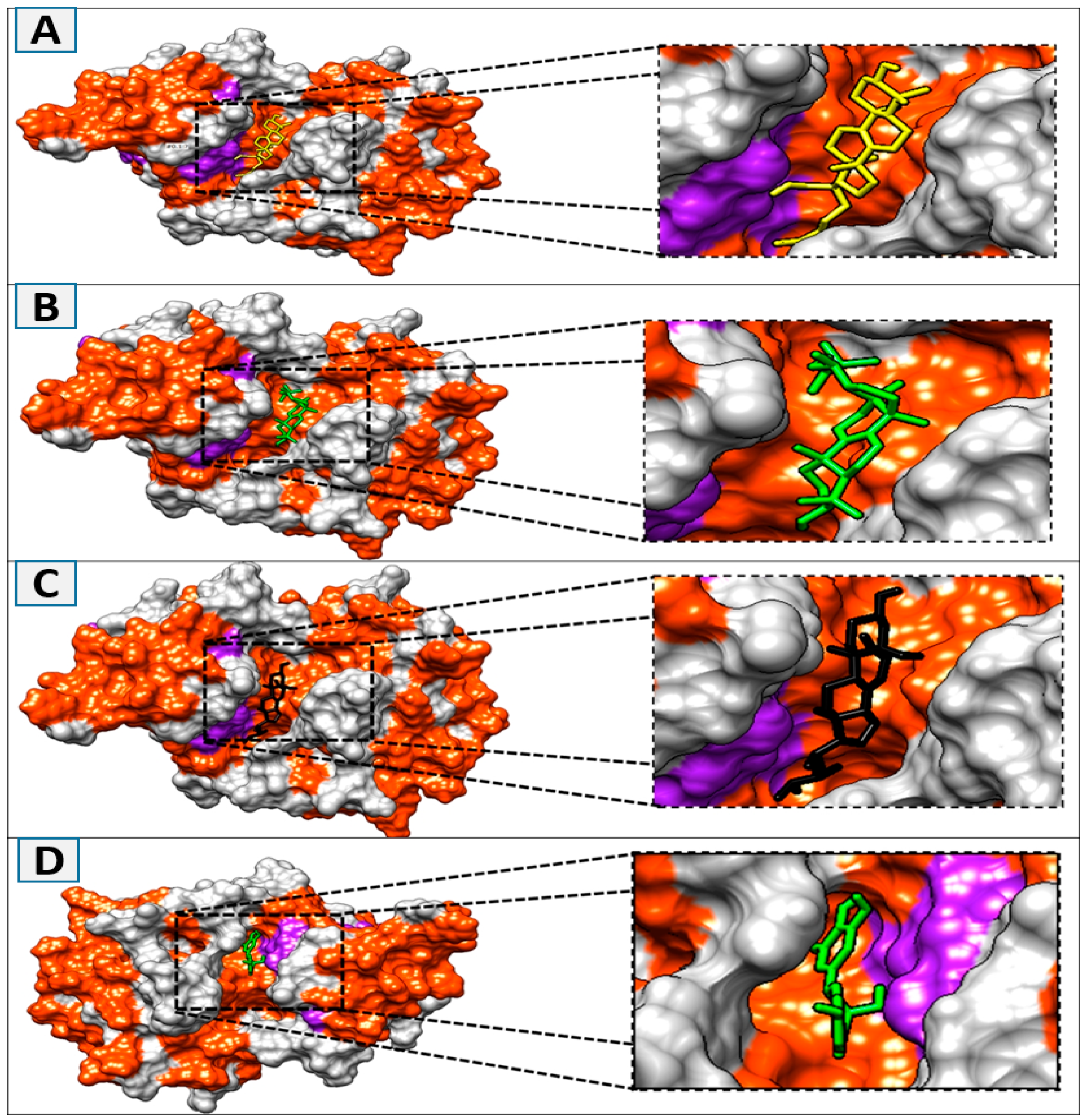
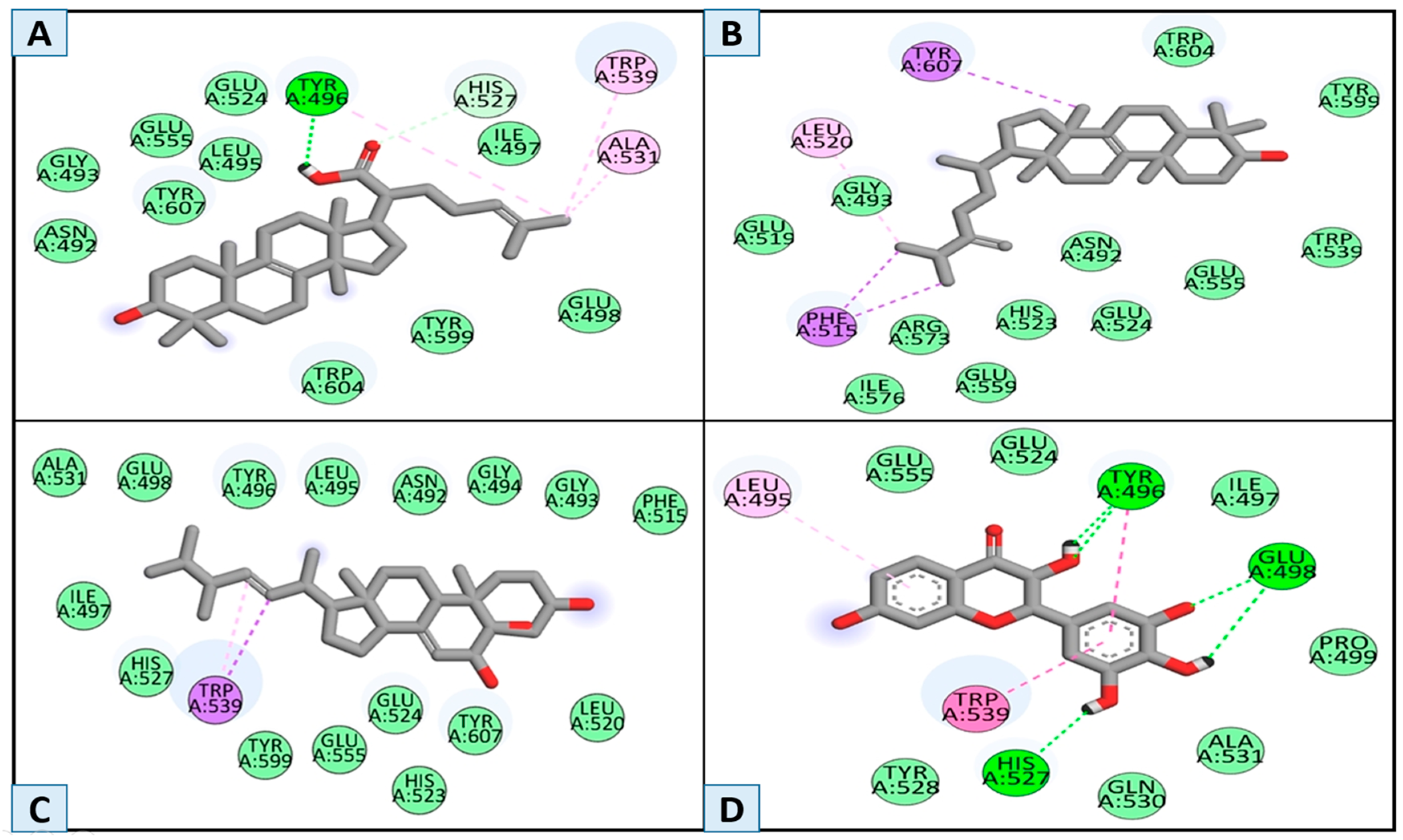
3.2. Molecular Docking and Binding Interaction/Poses Analysis
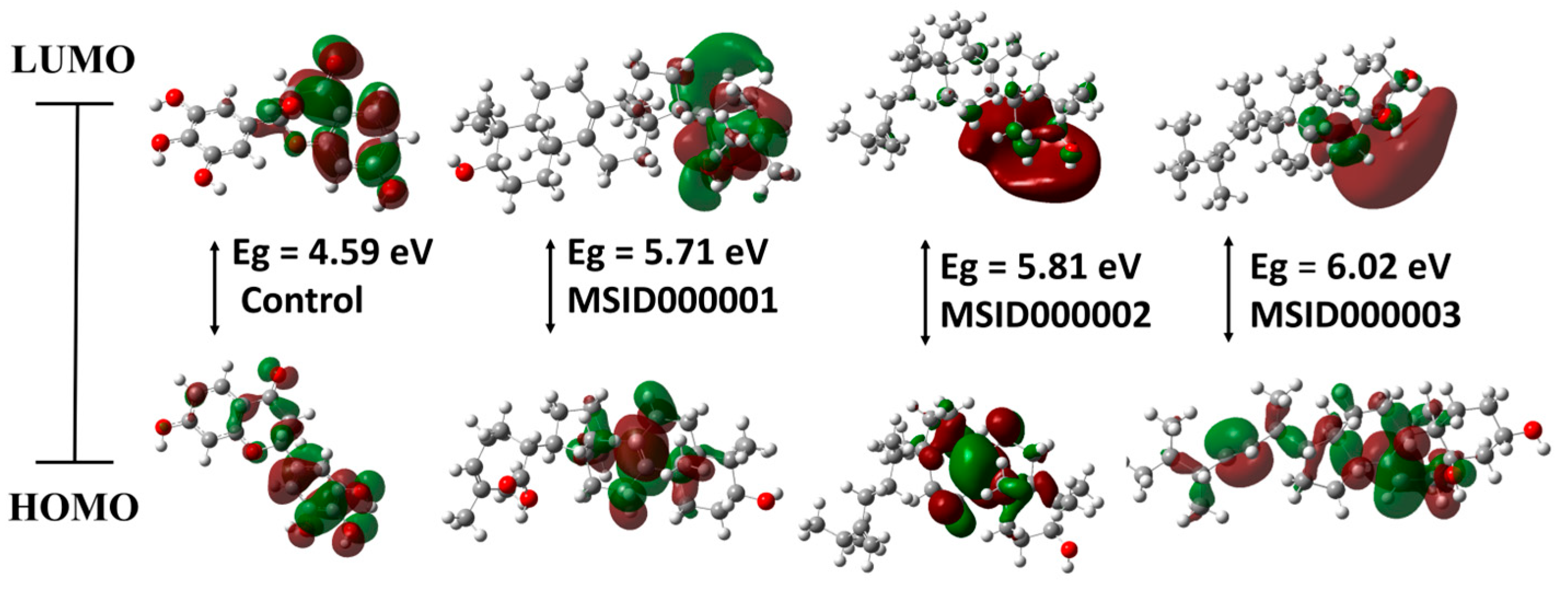
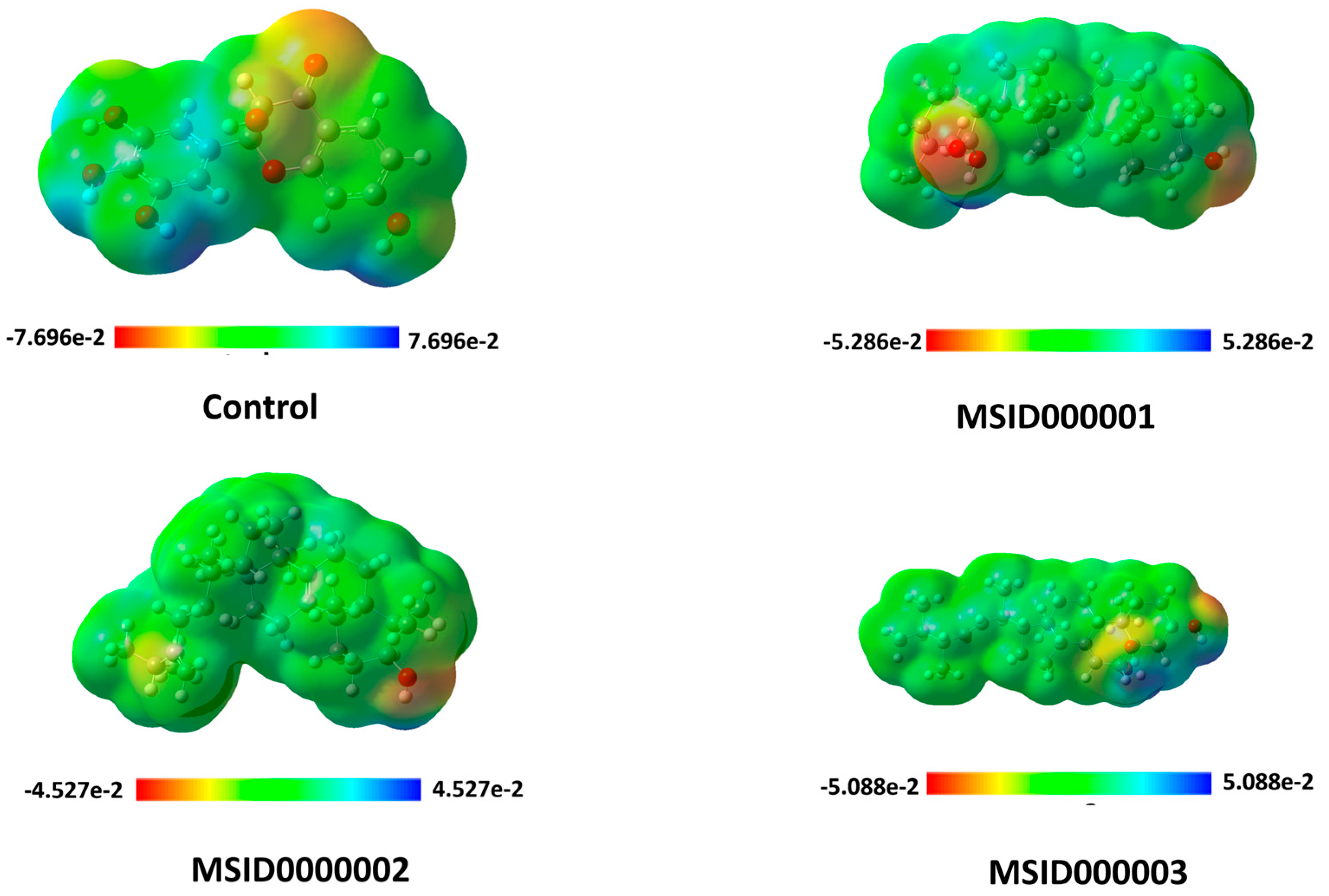
3.3. Density Functional Theory (DFT)
3.4. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Profiling
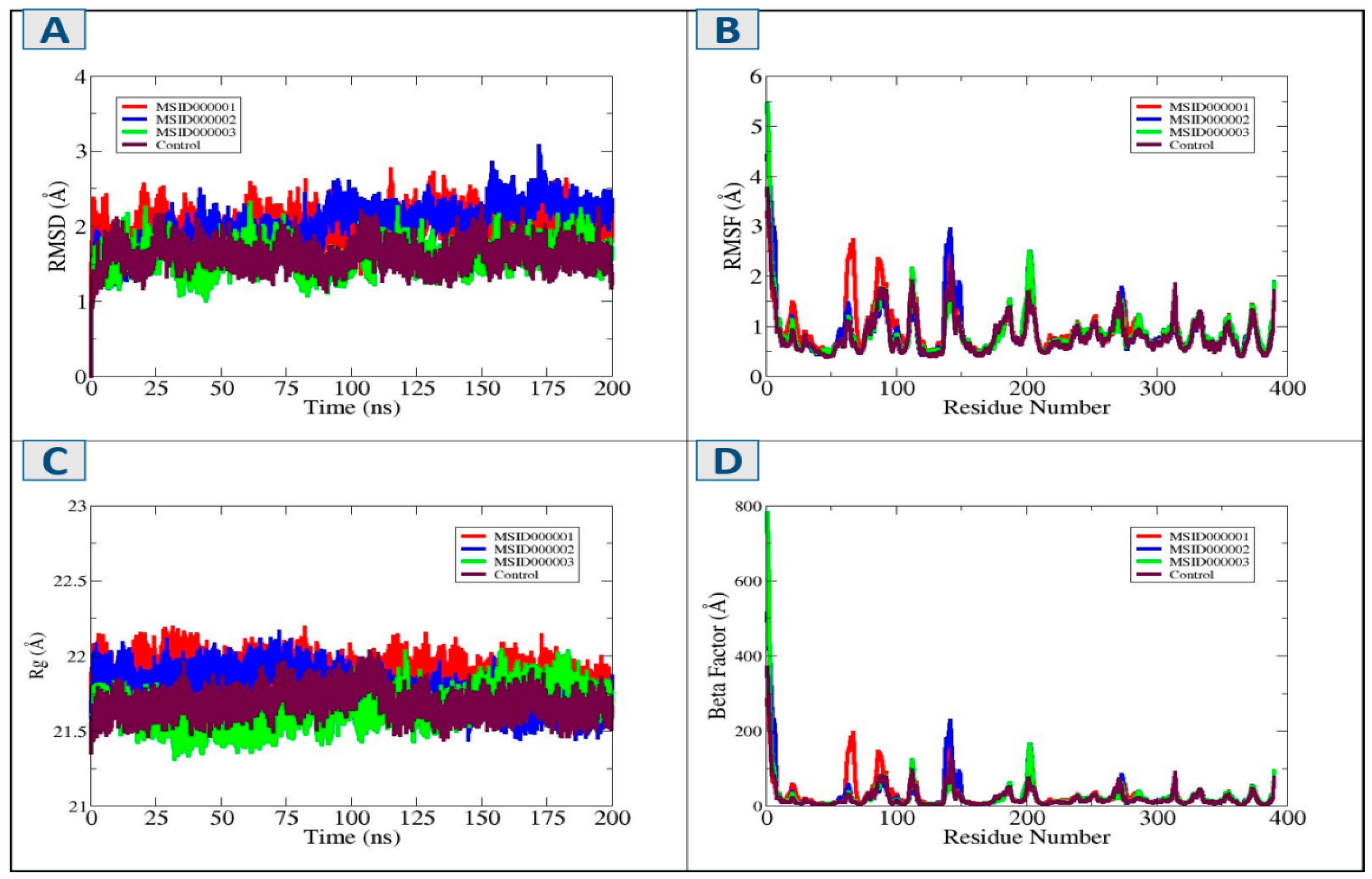
3.5. Molecular Dynamic Simulation
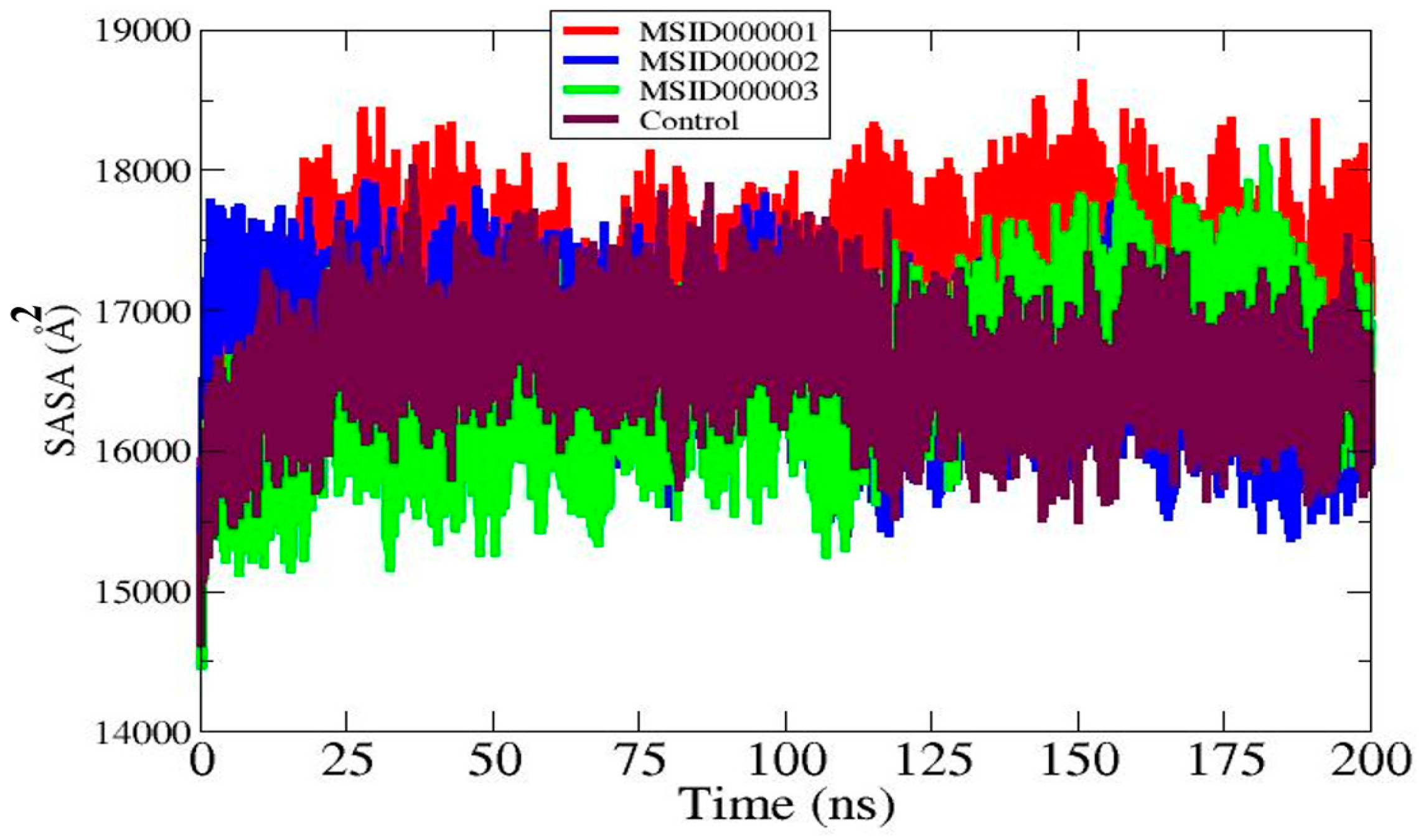
3.6. Solvent Accessible Surface Area (SASA)
3.7. H-Bonding Analysis
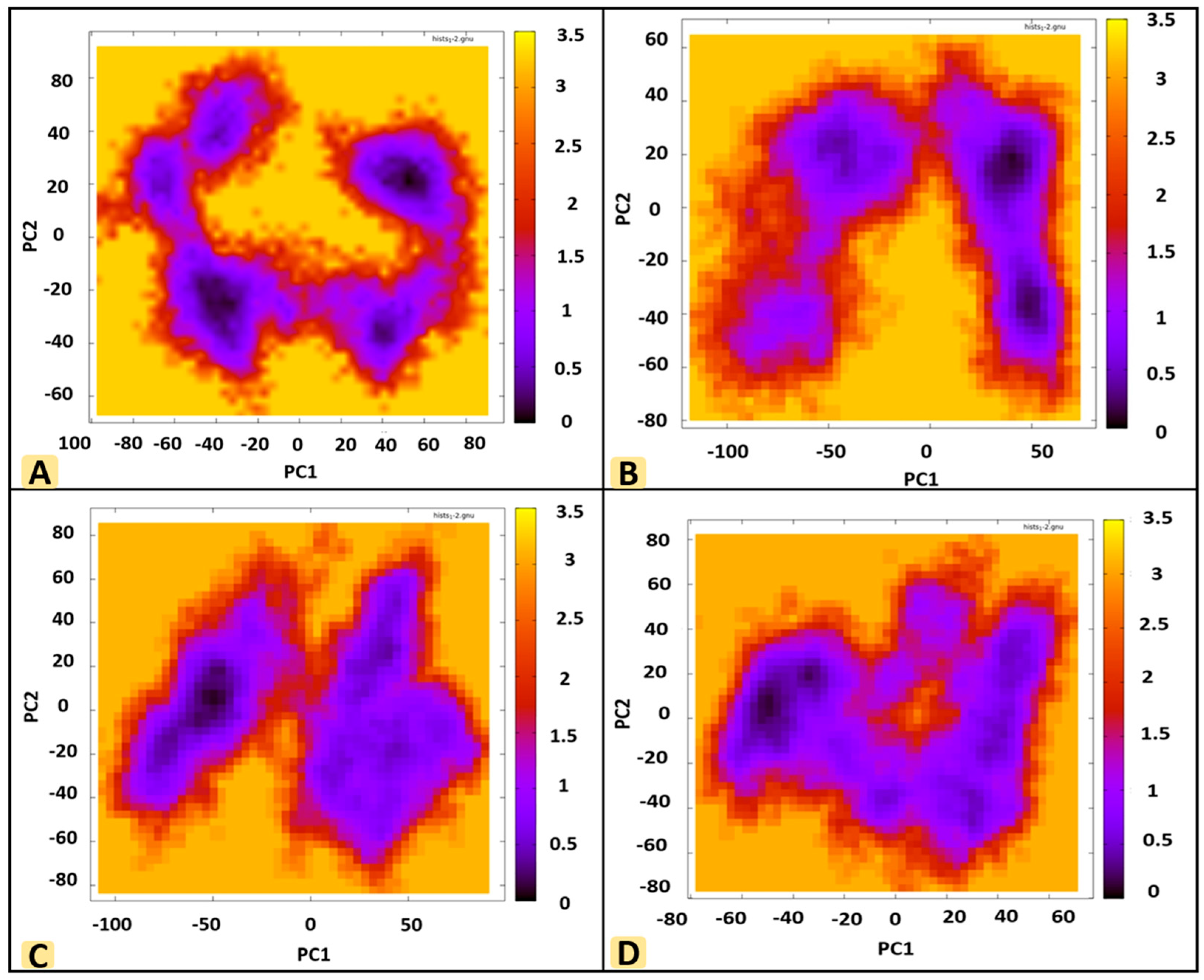
3.8. Principal Component Analysis (PCA)
3.9. Secondary Structure Analysis
3.10. MMPBSA/GSA Calculations
3.11. WaterSwap Energy Estimation
3.12. Entropy Energy Estimation
3.13. Salt Bridges Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alhayek, A.; Abdelsamie, A.S.; Schönauer, E.; Camberlein, V.; Hutterer, E.; Posselt, G.; Serwanja, J.; Blöchl, C.; Huber, C.G.; Haupenthal, J.; et al. Discovery and Characterization of Synthesized and FDA-Approved Inhibitors of Clostridial and Bacillary Collagenases. J. Med. Chem. 2022, 65, 12933–12955. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Rehman, M.T.; Shahwan, M.; Aatif, M. Origin of Antibiotics and Antibiotic Resistance, and Their Impacts on Drug Development: A Narrative Review. Pharmaceuticals 2023, 16, 1615. [Google Scholar] [CrossRef] [PubMed]
- Calvert, M.B.; Jumde, V.R.; Titz, A. Pathoblockers or Antivirulence Drugs as a New Option for the Treatment of Bacterial Infections. Beilstein J. Org. Chem. 2018, 14, 2607–2617. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Bouvet, P. Genetic Characteristics of Toxigenic Clostridia and Toxin Gene Evolution. Toxicon 2013, 75, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Larkinson, M.L.Y.; Clarke, T.B. Immunological Design of Commensal Communities to Treat Intestinal Infection and Inflammation. PLoS Pathog. 2021, 17, e1009191. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.S.; Hough, R.L.; West, H.M.; Avery, L.M. A Review of the Abundance, Behaviour and Detection of Clostridial Pathogens in Agricultural Soils. Eur. J. Soil Sci. 2019, 70, 911–929. [Google Scholar] [CrossRef]
- Gabrielson, A.T.; Spitz, J.T.; Hellstrom, W.J.G. Collagenase Clostridium Histolyticum in the Treatment of Urologic Disease: Current and Future Impact. Sex. Med. Rev. 2018, 6, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Overview of Bacterial Protein Toxins from Pathogenic Bacteria: Mode of Action and Insights into Evolution. Toxins 2024, 16, 182. [Google Scholar] [CrossRef] [PubMed]
- Kaya, C.; Hirsch, A.K.H. Targeting Extracellular Bacterial Proteases for the Development of Novel Antivirulence Agents. Chimia 2022, 76, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Eckhard, U.; Huesgen, P.F.; Brandstetter, H.; Overall, C.M. Proteomic Protease Specificity Profiling of Clostridial Collagenases Reveals Their Intrinsic Nature as Dedicated Degraders of Collagen. J. Proteom. 2014, 100, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Shekhter, A.B.; Balakireva, A.V.; Kuznetsova, N.V.; Vukolova, M.N.; Litvitsky, P.F.; Zamyatnin Jr, A.A. Collagenolytic Enzymes and Their Applications in Biomedicine. Curr. Med. Chem. 2019, 26, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, A.; Hakobyan, H.; Mardiyan, Z.; Jamharyan, S.; Dadayan, A.; Sargsyan, T.; Hovhannisyan, N. Modeling, Synthesis and in Vitro Screening of Unusual Amino Acids and Peptides As Protease Inhibitors. J. Chem. Technol. Metall. 2023, 58, 615–620. [Google Scholar] [CrossRef]
- María, R.R.; Arturo, C.J.; Alicia, J.; Paulina, M.G.; Gerardo, A. The Impact of Bioinformatics on Vaccine Design and Development. Vaccines 2017, 2, 3–6. [Google Scholar]
- dos Santos Nascimento, I.J.; da Silva Rodrigues, É.E.; da Silva, M.F.; de Araújo-Júnior, J.X.; de Moura, R.O. Advances in Computational Methods to Discover New NS2B-NS3 Inhibitors Useful against Dengue and Zika Viruses. Curr. Top. Med. Chem. 2022, 22, 2435–2462. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.; Sarkar, A. Review on Multiple Facets of Drug Resistance: A Rising Challenge in the 21st Century. J. Xenobiotics 2021, 11, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Kore, P.P.; Mutha, M.M.; Antre, R.V.; Oswal, R.J.; Kshirsagar, S.S. Computer-Aided Drug Design: An Innovative Tool for Modeling. Open J. Med. Chem. 2012, 2, 26238. [Google Scholar] [CrossRef]
- Sarkar, S.; Gulati, K.; Kairamkonda, M.; Mishra, A.; Poluri, K.M. Elucidating Protein-Protein Interactions through Computational Approaches and Designing Small Molecule Inhibitors against Them for Various Diseases. Curr. Top. Med. Chem. 2018, 18, 1719–1736. [Google Scholar] [CrossRef]
- Palchevskyi, S.; Czarnocki-Cieciura, M.; Vistoli, G.; Gervasoni, S.; Nowak, E.; Beccari, A.R.; Nowotny, M.; Talarico, C. Structure of Human TRPM8 Channel. Commun. Biol. 2023, 6, 1065. [Google Scholar] [CrossRef]
- Kaliappan, S.; Bombay, I.I.T. UCSF Chimera—Overview. 2018. Available online: https://www.cgl.ucsf.edu/chimera/ (accessed on 20 July 2024).
- Pencheva, T.; Lagorce, D.; Pajeva, I.; Villoutreix, B.O.; Miteva, M.A. AMMOS: Automated Molecular Mechanics Optimization Tool for in Silico Screening. BMC Bioinform. 2008, 9, 438. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; Saha, A.; Osman, S.M.; Alasmary, F.A.; Almutairi, T.M.; Islam, M.A. Structure-Based Identification of SARS-CoV-2 Main Protease Inhibitors from Anti-Viral Specific Chemical Libraries: An Exhaustive Computational Screening Approach. Mol. Divers. 2021, 25, 1979–1997. [Google Scholar] [CrossRef] [PubMed]
- Zothantluanga, J.; Aswin, S.K.; Rudrapal, M.; Chetia, D. Antimalarial Flavonoid-Glycoside from Acacia Pennata with Inhibitory Potential against PfDHFR-TS: An In-Silico Study. Biointerface Res. Appl. Chem 2021, 12, 4871–4887. [Google Scholar]
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and Other Widely Available Force Fields for Conformational Energies and for Intermolecular-interaction Energies and Geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Olawale, F.; Iwaloye, O.; Elekofehinti, O.O. Virtual Screening of Natural Compounds as Selective Inhibitors of Polo-like Kinase-1 at C-Terminal Polo Box and N-Terminal Catalytic Domain. J. Biomol. Struct. Dyn. 2022, 40, 13606–13624. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual Screening Strategies in Drug Discovery: A Critical Review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Kumar, S.P.; Girija, A.S.S.; Priyadharsini, J.V. Targeting NM23-H1-Mediated Inhibition of Tumour Metastasis in Viral Hepatitis with Bioactive Compounds from Ganoderma Lucidum: A Computational Study. Indian J. Pharm. Sci. 2020, 82, 300. [Google Scholar] [CrossRef]
- Bilal, M.S.; Ejaz, S.A.; Zargar, S.; Akhtar, N.; Wani, T.A.; Riaz, N.; Aborode, A.T.; Siddique, F.; Altwaijry, N.; Alkahtani, H.M.; et al. Computational Investigation of 1, 3, 4 Oxadiazole Derivatives as Lead Inhibitors of VEGFR 2 in Comparison with EGFR: Density Functional Theory, Molecular Docking and Molecular Dynamics Simulation Studies. Biomolecules 2022, 12, 1612. [Google Scholar] [CrossRef] [PubMed]
- Masnabadi, N.; Thalji, M.R.; Alhasan, H.S.; Mahmoodi, Z.; Soldatov, A.V.; Ali, G.A.M. Structural, Electronic, Reactivity, and Conformational Features of 2, 5, 5-Trimethyl-1, 3, 2-Diheterophosphinane-2-Sulfide, and Its Derivatives: DFT, MEP, and NBO Calculations. Molecules 2022, 27, 4011. [Google Scholar] [CrossRef] [PubMed]
- Hashem, H.E.; Ahmad, S.; Kumer, A.; Bakri, Y. El In Silico and in Vitro Prediction of New Synthesized N-Heterocyclic Compounds as Anti-SARS-CoV-2. Sci. Rep. 2024, 14, 1152. [Google Scholar] [CrossRef]
- Fatima, A.; Pooja, K.; Savita, S.; Singh, M.; Verma, I.; Siddiqui, N.; Javed, S. Quantum Chemical, Experimental Spectroscopic, Hirshfeld Surface and Molecular Docking Studies of the Anti-Microbial Drug Sulfathiazole. J. Mol. Struct. 2021, 1245, 131118. [Google Scholar] [CrossRef]
- Bruno, A.; Costantino, G.; Sartori, L.; Radi, M. The in Silico Drug Discovery Toolbox: Applications in Lead Discovery and Optimization. Curr. Med. Chem. 2019, 26, 3838–3873. [Google Scholar] [CrossRef] [PubMed]
- Lohohola, P.O.; Mbala, B.M.; Bambi, S.-M.N.; Mawete, D.T.; Matondo, A.; Mvondo, J.G.M. In Silico ADME/T Properties of Quinine Derivatives Using SwissADME and PkCSM Webservers. Int. J. Trop. Dis. Health 2021, 42, 1–12. [Google Scholar]
- Makeneni, S.; Thieker, D.F.; Woods, R.J. Applying Pose Clustering and MD Simulations to Eliminate False Positives in Molecular Docking. J. Chem. Inf. Model. 2018, 58, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Tripathi, T. Molecular Dynamics Simulation of Protein and Protein–Ligand Complexes. In Computer-Aided Drug Design; Springer: Singapore, 2020; pp. 133–161. [Google Scholar]
- Pujadas, G.; Vaque, M.; Ardevol, A.; Blade, C.; Salvado, M.J.; Blay, M.; Fernandez-Larrea, J.; Arola, L. Protein-Ligand Docking: A Review of Recent Advances and Future Perspectives. Curr. Pharm. Anal. 2008, 4, 1–19. [Google Scholar] [CrossRef]
- Alamri, M.A.; Mirza, M.U.; Adeel, M.M.; Ashfaq, U.A.; Tahir Ul Qamar, M.; Shahid, F.; Ahmad, S.; Alatawi, E.A.; Albalawi, G.M.; Allemailem, K.S.; et al. Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals 2022, 15, 659. [Google Scholar] [CrossRef] [PubMed]
- Miandad, K.; Ullah, A.; Bashir, K.; Khan, S.; Abideen, S.A.; Shaker, B.; Alharbi, M.; Alshammari, A.; Ali, M.; Haleem, A.; et al. Virtual Screening of Artemisia Annua Phytochemicals as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Molecules 2022, 27, 8103. [Google Scholar] [CrossRef] [PubMed]
- Rieder, S.R.; Ries, B.; Schaller, K.; Champion, C.; Barros, E.P.; Hünenberger, P.H.; Riniker, S. Replica-Exchange Enveloping Distribution Sampling Using Generalized AMBER Force-Field Topologies: Application to Relative Hydration Free-Energy Calculations for Large Sets of Molecules. J. Chem. Inf. Model. 2022, 62, 3043–3056. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F.; Sirimulla, S. An Improved Free Energy Perturbation FEP+ Sampling Protocol for Flexible Ligand-Binding Domains. Sci. Rep. 2019, 9, 16829. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Sharma, P.; Kumar, M.; Kapil, A.; Abdul Samath, E.; Kaur, P. Identification of Novel Natural MurD Ligase Inhibitors as Potential Antimicrobial Agents Targeting Acinetobacter Baumannii: In Silico Screening and Biological Evaluation. J. Biomol. Struct. Dyn. 2022, 40, 14051–14066. [Google Scholar] [CrossRef] [PubMed]
- Honarparvar, B.; Govender, T.; Maguire, G.E.M.; Soliman, M.E.S.; Kruger, H.G. Integrated Approach to Structure-Based Enzymatic Drug Design: Molecular Modeling, Spectroscopy, and Experimental Bioactivity. Chem. Rev. 2014, 114, 493–537. [Google Scholar] [CrossRef] [PubMed]
- Zschau, R.L.; Zacharias, M. Mechanism of Β-hairpin Formation in AzoChignolin and Chignolin. J. Comput. Chem. 2023, 44, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Justino, G.C.; Nascimento, C.P.; Justino, M.C. Molecular Dynamics Simulations and Analysis for Bioinformatics Undergraduate Students. Biochem. Mol. Biol. Educ. 2021, 49, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Brandsdal, B.O.; Österberg, F.; Almlöf, M.; Feierberg, I.; Luzhkov, V.B.; Åqvist, J. Free Energy Calculations and Ligand Binding. Adv. Protein Chem. 2003, 66, 123–158. [Google Scholar] [PubMed]
- Gul, S.; Ozcan, O.; Asar, S.; Okyar, A.; Barıs, I.; Kavakli, I.H. In Silico Identification of Widely Used and Well-Tolerated Drugs as Potential SARS-CoV-2 3C-like Protease and Viral RNA-Dependent RNA Polymerase Inhibitors for Direct Use in Clinical Trials. J. Biomol. Struct. Dyn. 2021, 39, 6772–6791. [Google Scholar] [CrossRef] [PubMed]
- Miller III, B.R.; McGee Jr, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.J.; Malaisree, M.; Michel, J.; Long, B.; McIntosh-Smith, S.; Mulholland, A.J. Rapid Decomposition and Visualisation of Protein–Ligand Binding Free Energies by Residue and by Water. Faraday Discuss. 2014, 169, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Bergström, C.A.S.; Larsson, P. Computational Prediction of Drug Solubility in Water-Based Systems: Qualitative and Quantitative Approaches Used in the Current Drug Discovery and Development Setting. Int. J. Pharm. 2018, 540, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Montgomerie, S.; Sundararaj, S.; Gallin, W.J.; Wishart, D.S. Improving the Accuracy of Protein Secondary Structure Prediction Using Structural Alignment. BMC Bioinform. 2006, 7, 301. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, H.; Kumar, D.T.; Doss, C.G.P.; Zayed, H. Bioinformatics Classification of Mutations in Patients with Mucopolysaccharidosis IIIA. Metab. Brain Dis. 2019, 34, 1577–1594. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, W.; Zhao, L.; Wei, B.; Chen, J. Binding Mechanism of Inhibitors to BRD4 and BRD9 Decoded by Multiple Independent Molecular Dynamics Simulations and Deep Learning. Molecules 2024, 29, 1857. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Anasti, K.; Manne, K.; Stalls, V.; Saunders, C.; Bililign, Y.; Williams, A.; Bubphamala, P.; Montani, M.; Kachhap, S. Engineering Immunogens That Select for Specific Mutations in HIV Broadly Neutralizing Antibodies. bioRxiv 2023. [Google Scholar] [CrossRef]
- Mortier, J.; Rakers, C.; Bermudez, M.; Murgueitio, M.S.; Riniker, S.; Wolber, G. The Impact of Molecular Dynamics on Drug Design: Applications for the Characterization of Ligand–Macromolecule Complexes. Drug Discov. Today 2015, 20, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.A.; Ali, N.; Qureshi, U.; Khalil, R.; Qasmi, Z.-U.H. Molecular Dynamics Simulation of Human Pancreatic Lipase and Lipase-Colipase Complex: Insight into the Structural Fluctuations and Conformational Changes. Int. J. Comput. Theor. Chem. 2020, 8, 19. [Google Scholar] [CrossRef]
- Elmaaty, A.A.; Darwish, K.M.; Khattab, M.; Elhady, S.S.; Salah, M.; Hamed, M.I.A.; Al-Karmalawy, A.A.; Saleh, M.M. In a Search for Potential Drug Candidates for Combating COVID-19: Computational Study Revealed Salvianolic Acid B as a Potential Therapeutic Targeting 3CLpro and Spike Proteins. J. Biomol. Struct. Dyn. 2022, 40, 8866–8893. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef] [PubMed]
- Gewers, F.L.; Ferreira, G.R.; De Arruda, H.F.; Silva, F.N.; Comin, C.H.; Amancio, D.R.; Costa, L.d.F. Principal Component Analysis: A Natural Approach to Data Exploration. ACM Comput. Surv. 2021, 54, 1–34. [Google Scholar] [CrossRef]
- Yoo, C.; Shahlaei, M. The Applications of PCA in QSAR Studies: A Case Study on CCR5 Antagonists. Chem. Biol. Drug Des. 2018, 91, 137–152. [Google Scholar] [CrossRef]
- Karnati, K.R.; Wang, Y. Structural and Binding Insights into HIV-1 Protease and P2-Ligand Interactions through Molecular Dynamics Simulations, Binding Free Energy and Principal Component Analysis. J. Mol. Graph. Model. 2019, 92, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Kumar, K.; Venkatesu, P.; Masram, D.T. Protein Immobilization on Graphene Oxide or Reduced Graphene Oxide Surface and Their Applications: Influence over Activity, Structural and Thermal Stability of Protein. Adv. Colloid Interface Sci. 2021, 289, 102367. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.B.; Ismail, S.; Yousaf, R.; Qazi, A.S.; Iftkhar, S.; Abbasi, S.W. Exploring Novel 1-Hydroxynaphthalene-2-Carboxanilides Based Inhibitors Against C-Jun N-Terminal Kinases Through Molecular Dynamic Simulation and WaterSwap Analysis. Appl. Biochem. Biotechnol. 2023, 196, 1803–1819. [Google Scholar] [CrossRef] [PubMed]
- Panday, S.K.; Ghosh, I. In Silico Structure-Based Prediction of Receptor–Ligand Binding Affinity: Current Progress and Challenges. In Structural Bioinformatics: Applications in Preclinical Drug Discovery Process. Challenges and Advances in Computational Chemistry and Physics; Springer: Cham, Switzerland, 2019; pp. 109–175. [Google Scholar]
- Kirby, J.P. The Amidinium-Carboxylate Salt Bridge and Electron Transfer Reactions; Michigan State University: East Lansing, MI, USA, 1997; ISBN 0591744015. [Google Scholar]
- Gupta, M.N.; Uversky, V.N. Biological Importance of Arginine: A Comprehensive Review of the Roles in Structure, Disorder, and Functionality of Peptides and Proteins. Int. J. Biol. Macromol. 2023, 257, 128646. [Google Scholar] [CrossRef] [PubMed]
- Pakhrin, S.C.; Shrestha, B.; Adhikari, B.; Kc, D.B. Deep Learning-Based Advances in Protein Structure Prediction. Int. J. Mol. Sci. 2021, 22, 5553. [Google Scholar] [CrossRef] [PubMed]
- Gomes, D.; Silvestre, S.; Duarte, A.P.; Venuti, A.; Soares, C.P.; Passarinha, L.; Sousa, Â. In Silico Approaches: A Way to Unveil Novel Therapeutic Drugs for Cervical Cancer Management. Pharmaceuticals 2021, 14, 741. [Google Scholar] [CrossRef] [PubMed]
- Al-Thakafi, S.; Al-Hathal, N. Peyronie’s Disease: A Literature Review on Epidemiology, Genetics, Pathophysiology, Diagnosis and Work-Up. Transl. Androl. Urol. 2016, 5, 280. [Google Scholar] [CrossRef] [PubMed]
- Alipour, H.; Raz, A.; Zakeri, S.; Djadid, N.D. Therapeutic Applications of Collagenase (Metalloproteases): A Review. Asian Pac. J. Trop. Biomed. 2016, 6, 975–981. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A.; Mastrolorenzo, A. Bacterial Proteases: Current Therapeutic Use and Future Prospects for the Development of New Antibiotics. Expert Opin. Ther. Pat. 2001, 11, 221–259. [Google Scholar] [CrossRef]
- Nitulescu, G.; Nitulescu, G.M.; Zanfirescu, A.; Mihai, D.P.; Gradinaru, D. Candidates for Repurposing as Anti-Virulence Agents Based on the Structural Profile Analysis of Microbial Collagenase Inhibitors. Pharmaceutics 2021, 14, 62. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | Compounds | Structure | Binding Affinity |
|---|---|---|---|
| 1 | MSID000001 2-(3-hydroxy-4,4,10,13,14-pentamethyl2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-6-methylhept-6-enoic acid | 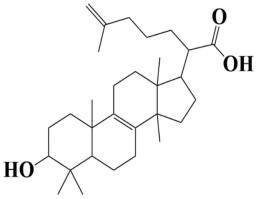 | −10.7 kcal/mol |
| 2 | MSID000002 4,4,10,13,14-pentamethyl-17-(6-methyl-5-methyleneheptan-2-yl)-2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol | 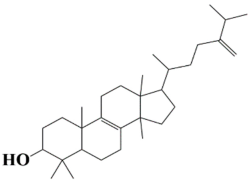 | −9.8 kcal/mol |
| 3 | MSID000003 (E)-17-(5,6-dimethylhept-3-en-2-yl)-10,13-dimethyl-2,3,4,5,6,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthrene-3,5,6-triol | 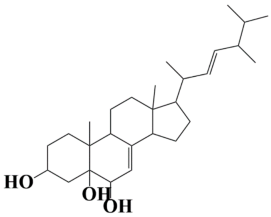 | −9.5 kcal/mol |
| 4 | MSID000004 2-(3-acetoxy-4,4,10,13,14-pentamethyl-2,3,4,5,6,7,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-6-methyl-3-oxohept-5-enoic acid | 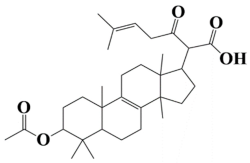 | −9.3 kcal/mol |
| 5 | MSID000006 2,8-dimethyl-1,2,4,5,6,7,8,8a-octahydroazulene-2,4,5-triyl)trimethanol | 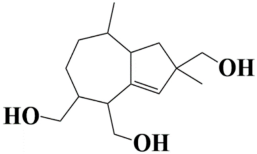 | −9.2 kcal/mol |
| 6 | MSID000009 (6-hydroxy-2,2,8-trimethyl-1,2,4,5,6,7,8,8a-octahydroazulene-4,5-diyl)dimethanol | 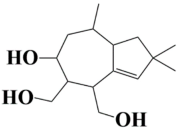 | −9 kcal/mol |
| 7 | MSID000010 3a-(2-(2,5-dihydroxyphenyl)-2-oxoethyl)-8-hydroxytetrahydrocyclopenta[1,2-b:2,3-c′]difuran-3,7(1H,8H)-dione | 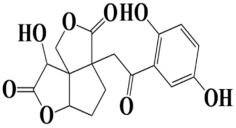 | −8.9 kcal/mol |
| 8 | MSID000016 6a-(2-(2,5-dihydroxyphenyl)-2-oxoethyl)-3a-(dimethoxymethyl)-4-ethoxyhexahydro-1H-cyclopenta[c]furan-1-one | 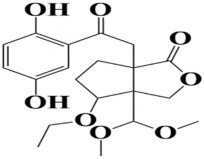 | −8.6 kcal/mol |
| 9 | MSID000020 methyl 1-(2-(2,5-dihydroxyphenyl)-2-oxoethyl)-2,4-dihydroxy-3-methylenecyclohexanecarboxylate | 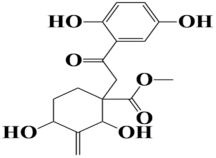 | −8.5 kcal/mol |
| 10 | MSID000022 4-(2-(2,5-dihydroxyphenyl)-2-oxoethyl)-5-hydroxy-6-methylene-2-oxabicyclo[2.2.2]octan-3-one | 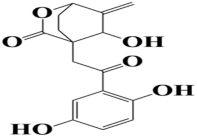 | −8.4 kcal/mol |
| 11 | Control 3,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)chroman-4-one | 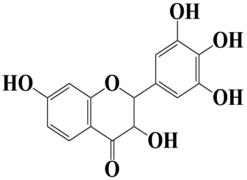 | −8 kcal/mol |
| Compounds | H-Bond | Van der Waals | Pi–Alkyl | Alkyl | Carbon–Hydrogen Bond | Pi-Sigma | Pi–Pi Stacked and Pi–Pi T-Shaped |
|---|---|---|---|---|---|---|---|
| MSID000001 | Tyr496 | Glu498, Tyr599, Trp604, Ile497, Glu524, Leu495, Glu555,Gly493, Asn492 | Trp530 | Ala531 | His527 | - | - |
| MSID000002 | - | Ile576, Glu559, Arg573. His523, Glu524, Asn 492, Glu555, Trp539, Tyr599, Trp604, Glu519, Gly 493 | - | Leu520 | - | Phe515, Tyr607 | - |
| MSID000003 | - | Leu520, Tyr607, Glu524, Tyr599, His527, Ile497, Ala531, Glu498, Tyr496, Leu495, Asn492, Gly494, Phe515 | - | - | - | Trp539 | |
| Control | His527, Glu498, Tyr496 | Pro499, Ala531, Gln530,Tyr528, Glu555, Ile497,Glu524 | Leu495 | - | - | - | Trp539, Leu495 |
| Ligand Code | Optimization Energy (a.u.) | Dipole Moment (debye) | Polarizability (α) (a.u.) | EH (eV) | EL (eV) | Eg (eV) |
|---|---|---|---|---|---|---|
| Control | −1105.73 | 6.10 | 207.35 | −6.45 | −1.86 | 4.59 |
| MSID000001 | −1398.26 | 3.08 | 347.85 | −6.13 | −0.43 | 5.71 |
| MSID000002 | −1288.26 | 1.80 | 352.91 | −6.01 | −0.21 | 5.81 |
| MSID000003 | −1320.80 | 2.30 | 338.32 | −6.53 | −0.51 | 6.02 |
| Ligand Code | Chemical Potential µ (eV) | Electronegativity χ (eV) | Hardness η (eV) | Softness S (eV) | Electrophilicity ω (eV) | Ionization Potential (I) | Electron Affinity (A) |
|---|---|---|---|---|---|---|---|
| Control | 4.15 | −4.15 | 1.37 | 0.68 | 11.76 | 6.45 | 1.86 |
| MSID000001 | 3.28 | −3.28 | 2.64 | 1.32 | 14.20 | 6.13 | 0.43 |
| MSID000002 | 3.11 | −3.11 | 2.80 | 1.40 | 13.53 | 6.01 | 0.21 |
| MSID000003 | 3.52 | −3.52 | 2.75 | 1.38 | 17.08 | 6.53 | 0.51 |
| MSID000001 | ||
|---|---|---|
| Donor | Acceptor | Occupancy |
| HIE128-Side | LIG391-Main | 0.30% |
| HIE128-Side | LIG391-Main | 0.10% |
| LIG391-Side | GLU156-Side | 0.10% |
| MSID000002 | ||
| LIG391-Main | ASP184-Side | 29.80% |
| MSID000003 | ||
| LIG391-Main | TYR208-Side | 1–10% |
| LIG391-Side | GLU156-Side | 5.40% |
| LIG391-Main | TYR200-Side | 0.30% |
| LIG391-Main | GLU156-Side | 0.10% |
| LIG391-Side | TYR208-Side | 1.50% |
| TYR208-Side | LIG391-Side | 0.10% |
| LIG391-Side | TYR200-Side | 0.10% |
| Control | ||
| LIG391-Side | GLU125-Side | 70.00% |
| LIG391-Side | GLU99-Side | 2.90% |
| Energy Parameter | MSID000001 | MSID000002 | MSID000003 | Control |
|---|---|---|---|---|
| MMGBSA | ||||
| van der Waals energy | −65.14 | −61.23 | −55.91 | −60.99 |
| Energy electrostatic | −24.01 | −20.81 | −15.49 | −17.67 |
| Total gas phase energy | −89.15 | −82.04 | −71.4 | −78.66 |
| Total salvation energy | 10.57 | 11.60 | 12.08 | 10.46 |
| Net energy | −78.58 | −70.44 | −59.32 | −68.2 |
| MMPBSA | ||||
| Energy van der Waals | −65.14 | −61.23 | −55.91 | −60.99 |
| Energy electrostatics | −24.01 | −20.81 | −15.49 | −17.67 |
| Total gas phase energy | −89.15 | −82.04 | −71.4 | −78.66 |
| Total energy salvation | 9.61 | 8.05 | 9.14 | 8.00 |
| Net energy | −79.54 | −73.99 | −62.26 | −70.66 |
| Complex | Translational | Vibrational | Rotational | ΔS Total |
|---|---|---|---|---|
| MSID000001 | 5.01 | 7.86 | 1147.09 | −5.96 |
| MSID000002 | 10.85 | 12.66 | 1269.48 | −2.85 |
| MSID000003 | 15.96 | 13.05 | 1566.12 | −1.36 |
| Control | 10.53 | 12.04 | 1428.64 | −2.60 |
| Complexes | Salt Bridges Interaction |
|---|---|
| MSID000001 | Glu34-Lys37, Glu147-Lys148, Glu220-Lys221, Asp1-Arg101, Asp279-Arg38, Asp222-Lys254, Glu120-Arg174, Glu61-Arg133, Asp338-Lys335, Asp345-Arg52, Asp67-Arg133, Asp187-Lus185, Glu8-Arg26, Glu45-Arg123, Asp6-Lys2, Glu33-Lys14, Glu292-Lys289, Asp345-Arg52, Asp204-Lys194, Asp280-Lys360, Glu339-Lys335, Asp6-Lys9, Glu33-Lys37, Glu78-Lys81, Asp19-Arg44, Glu264-Lys268, Asp92-Lys81, Glu339-Lys331, Glu-Arg44 |
| MSID000002 | Glu349-Arg167, Asp6-Lys9, Glu328-Lys331, Asp380-Arg370, Asp222-Lys254, Glu34-Arg38, Asp187-Lys185, Glu108-Lys35, Glu339-Lys342, Asp5-Lys72, Glu8-Lys72, Glu8-Lys72, Glu34-Arg38, Glu45-Arg123, Asp66-Arg133, Glu220-Lys268, Glu160-Arg174, Glu33-Lys14, Glu292-Lys289, Glu160-Arg174,Glu119-Arg123, Glu264-Lys221, Asp338-Lys342, Asp5-Arg101, Asp184-Lys183, Asp19-Arg44, Asp57-Arg322, Asp242-Arg150, Asp184-Lys183, Asp280-Lys283, Glu108-Lys35, Glu339-Lys331, Asp220-Lys254 |
| MSID000003 | Glu34-Lys37, Asp6-Lys9, Asp57-Lys58, Glu8-Arg26, Asp237-Lys239, Asp380-Arg370, Asp29-Lys30, Asp374-Lys376, Glu292-Lys299, Glu339-Lys342, Glu45-Arg123, Asp66-Arg133, Glu220-Lys268,Asp5-Lys9, Asp267-Lys268, Asp345-Arg52, Glu339-Lys343, Asp250-Lys264, Asp5-Lys9, Asp338-Lys342, Asp57-Arg322, Asp242-Arg150, Glu34-Lys37, Asp280-Lys283, Asp242-Arg150, Glu257-Lys254, Asp29-Lys30, Glu349-Arg167, Asp242-Arg150, Asp5-Lys2, Asp222-Lys254 |
| Control | Asp6-Lys2, Glu264-Lys268, Glu34-Arg38, Glu328-Lys331, Glu243-Lys264, Asp279-Arg38, Glu333-Lys231, Asp187-Lus185, Glu33-Lys14, Glu311-Arg44, Glu45-Arg123, Asp242-Lys148, Asp338-Lys335, Asp279-Arg38, Asp66-Arg133, Glu34-Arg38, Glu333-Lys228, Glu243-Lys239, Glu292-Lys289, Asp250-Lys193, Glu119-Arg44, Asp338-Lys342, Asp250-Lys264, Asp242-Arg150, Asp57-Arg322, Asp57-Arg322, Glu339-Lys331, Glu257-Lys254, Asp336-Lys331, Glu108-Lys35, Glu243-Lys246, Glu311-Arg44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anjum, F.; Hazazi, A.; Alsaeedi, F.A.; Bakhuraysah, M.; Shafie, A.; Alshehri, N.A.; Hawsawi, N.; Ashour, A.A.; Banjer, H.J.; Alharthi, A.; et al. Identification and Dynamics Understanding of Novel Inhibitors of Peptidase Domain of Collagenase G from Clostridium histolyticum. Computation 2024, 12, 153. https://doi.org/10.3390/computation12080153
Anjum F, Hazazi A, Alsaeedi FA, Bakhuraysah M, Shafie A, Alshehri NA, Hawsawi N, Ashour AA, Banjer HJ, Alharthi A, et al. Identification and Dynamics Understanding of Novel Inhibitors of Peptidase Domain of Collagenase G from Clostridium histolyticum. Computation. 2024; 12(8):153. https://doi.org/10.3390/computation12080153
Chicago/Turabian StyleAnjum, Farah, Ali Hazazi, Fouzeyyah Ali Alsaeedi, Maha Bakhuraysah, Alaa Shafie, Norah Ali Alshehri, Nahed Hawsawi, Amal Adnan Ashour, Hamsa Jameel Banjer, Afaf Alharthi, and et al. 2024. "Identification and Dynamics Understanding of Novel Inhibitors of Peptidase Domain of Collagenase G from Clostridium histolyticum" Computation 12, no. 8: 153. https://doi.org/10.3390/computation12080153
APA StyleAnjum, F., Hazazi, A., Alsaeedi, F. A., Bakhuraysah, M., Shafie, A., Alshehri, N. A., Hawsawi, N., Ashour, A. A., Banjer, H. J., Alharthi, A., & Niaz, M. I. (2024). Identification and Dynamics Understanding of Novel Inhibitors of Peptidase Domain of Collagenase G from Clostridium histolyticum. Computation, 12(8), 153. https://doi.org/10.3390/computation12080153