
ddRAD Sequencing-Based Scanning of Genetic Variants in Sargassum fusiforme


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Double-Digest RAD Sequencing
2.2. Read Mapping and SNP Calling
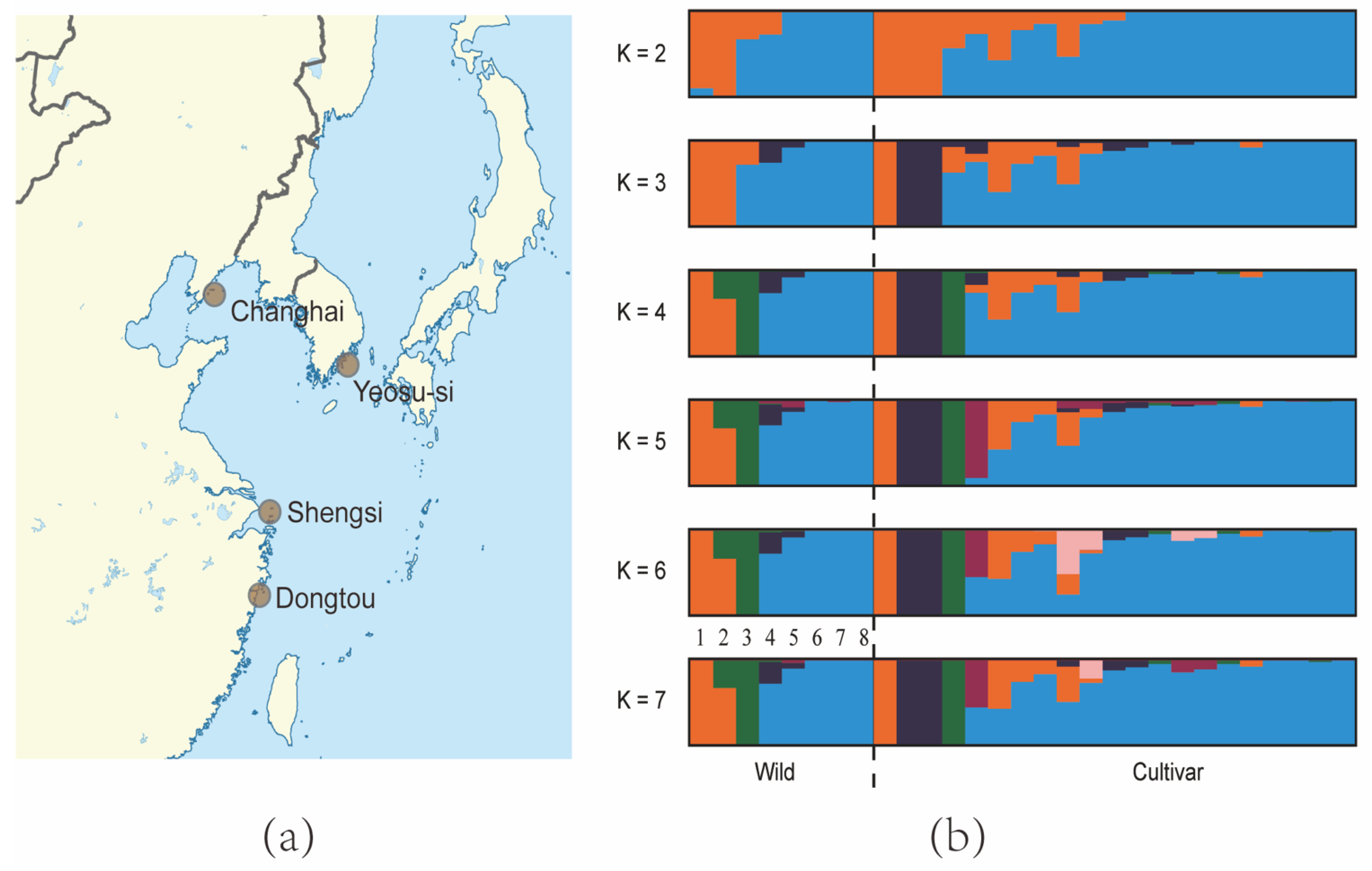
2.3. Genetic Diversity and Population Structure Analysis
2.4. Demographic Inference
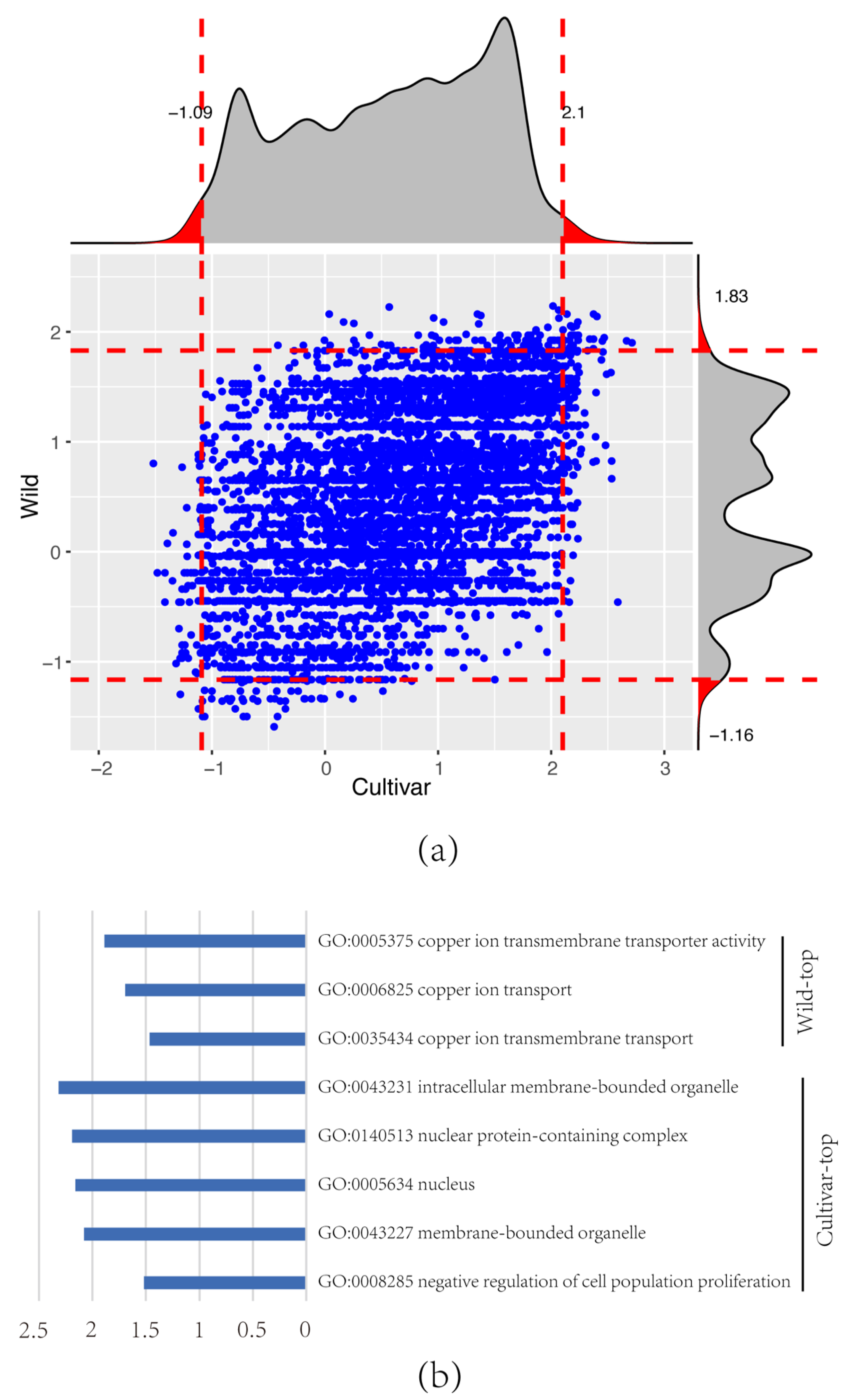
2.5. Genome-Wide Detection of Selection Signatures
3. Results
3.1. Processed Sequence Data
3.2. Genetic Diversity and Population Structure
3.3. Demographic History
3.4. Signature Selection
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, Z.; Wu, M.; Lin, L.; Thring, R.W.; Yu, H.; Zhang, X.; Zhao, M. Allelopathic interactions between the macroalga Hizikia fusiformis (Harvey) and the harmful blooms-forming dinoflagellate Karenia mikimotoi. Harmful Algae 2017, 65, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.; Hoeks, C.; Schepers, M.; Tiane, A.; Cuypers, A.; Leijten, F.; Chintapakorn, Y.; Suttiyut, T.; Pornpakakul, S.; Struik, D.; et al. Dietary Sargassum fusiforme improves memory and reduces amyloid plaque load in an Alzheimer’s disease mouse model. Sci. Rep. 2019, 9, 4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; He, D.; Zhang, Y.; Yang, S.; Chen, L.; Wang, S.; Zou, H.; Liao, Z.; Zhang, X.; Wu, M. Sargassum fusiforme polysaccharides activate antioxidant defense by promoting Nrf2-dependent cytoprotection and ameliorate stress insult during aging. Food Funct. 2016, 7, 4576–4588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, X.; Tang, Y.; Mao, J. Composition, isolation, purification and biological activities of Sargassum fusiforme polysaccharides: A review. Carbohydr. Polym. 2020, 228, 115381. [Google Scholar] [CrossRef]
- Li, X.; Wang, W. Chinese Materia Medica: Combinations and Applications; Donica Publishing: Potters Bar, UK, 2002. [Google Scholar]
- Zhang, Q.; Liu, G. The development and utilization on Sargassum fusiforme resources of Rongcheng coastal waters. Mar. Sci. 2009, 33, 45–48. [Google Scholar]
- Hu, Z.; Li, J.; Sun, Z.; Gao, X.; Yao, J.; Choi, H.; Endo, H.; Duan, D. Hidden diversity and phylogeographic history provide conservation insights for the edible seaweed Sargassum fusiforme in the Northwest Pacific. Evol. Appl. 2017, 10, 366–378. [Google Scholar] [CrossRef]
- Lin, L.; Chen, B.; Wang, S.; Li, J.; Ma, Z. Comparative transcriptome and cross-section analyses provide insights into the mechanisms underlying Sargassum fusiforme responses to the harmful algal bloom-forming dinoflagellate Karenia mikimotoi. Aquat. Bot. 2021, 172, 103383. [Google Scholar] [CrossRef]
- Unamba, C.I.N.; Nag, A.; Sharma, R.K. Next Generation Sequencing Technologies: The Doorway to the Unexplored Genomics of Non-Model Plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S.; Cardi, T.; Campanelli, G.; Sestili, S.; Díez, M.J.; Soler, S.; Prohens, J.; Tripodi, P. ddRAD sequencing-based genotyping for population structure analysis in cultivated tomato provides new insights into the genomic diversity of Mediterranean ‘da serbo’ type long shelf-life germplasm. Hortic. Res. 2020, 7, 134. [Google Scholar] [CrossRef]
- Avia, K.; Coelho, S.M.; Montecinos, G.J.; Cormier, A.; Lerck, F.; Mauger, S.; Faugeron, S.; Valero, M.; Cock, J.M.; Boudry, P. High-density genetic map and identification of QTLs for responses to temperature and salinity stresses in the model brown alga Ectocarpus. Sci. Rep. 2017, 7, 43241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzinski, J.; Ballenghien, M.; Daguin-Thiébaut, C.; Lévêque, L.; Viard, F. Population genomics of the introduced and cultivated Pacific kelp Undaria pinnatifida: Marinas—Not farms—Drive regional connectivity and establishment in natural rocky reefs. Evol. Appl. 2018, 11, 1582–1597. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-D.; Wu, M.-J. The diversity of cultivated Sargassum fusiforme strains in Dongtou, Zhejiang province by the combination of air-bladder phenotype and simplified genome. Oceanol. Limnol. Sin. 2021, 52, 1224–1235. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lin, L.; Shi, Y.; Qian, W.; Li, N.; Yan, X.; Zou, H.; Wu, M. First Draft Genome Assembly of the Seaweed Sargassum fusiforme. Front. Genet. 2020, 11, 590065. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Liu, X.; Fu, Y.-X. Stairway Plot 2: Demographic history inference with folded SNP frequency spectra. Genome Biol. 2020, 21, 280. [Google Scholar] [CrossRef]
- Chan, S.W.; Cheang, C.C.; Chirapart, A.; Gerung, G.; Tharith, C.; Ang, P. Homogeneous Population of the Brown Alga Sargassum polycystum in Southeast Asia: Possible Role of Recent Expansion and Asexual Propagation. PLoS ONE 2013, 8, e77662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.R.; Arrigo, K.R.; Alderkamp, A.-C.; Allen, A.E. Massive difference in synonymous substitution rates among mitochondrial, plastid, and nuclear genes of Phaeocystis algae. Mol. Phylogenet. Evol. 2014, 71, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. TopGo: Enrichment Analysis for Gene Ontology; Bioconductor Version; Release (3.15). 2022. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 5 May 2022).
- The Gene Ontology Consortium; Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Bemmels, J.B.; Dick, C.W.; Lohmann, L.G. Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 2017, 17, 1136–1147. [Google Scholar] [CrossRef]
- Graf, L.; Shin, Y.; Yang, J.H.; Choi, J.W.; Hwang, I.K.; Nelson, W.; Bhattacharya, D.; Viard, F.; Yoon, H.S. A genome-wide investigation of the effect of farming and human-mediated introduction on the ubiquitous seaweed Undaria pinnatifida. Nat. Ecol. Evol. 2021, 5, 360–368. [Google Scholar] [CrossRef]
- Yao, J.; Shuai, L.; Li, S.; Xu, C.; Wang, X. Genetic analysis of selected Sargassum fusiforme (Harvey) Setchell (Sargassaceae, Phaeophyta) strains with RAPD and ISSR markers. J. Oceanol. Limnol. 2019, 37, 783–789. [Google Scholar] [CrossRef]
- Li, S.; Xu, C.; Li, J.; Yao, J.; Liu, J.; Duan, D. Selection, artificial breeding and cultivation of Hizikia “Lu Feng No. 1”. Prog. Fish. Sci. 2010, 31, 88–94. [Google Scholar]
- Marandel, F.; Charrier, G.; Lamy, J.; Le Cam, S.; Lorance, P.; Trenkel, V.M. Estimating effective population size using RADseq: Effects of SNP selection and sample size. Ecol. Evol. 2020, 10, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Patton, A.H.; Margres, M.J.; Stahlke, A.R.; Hendricks, S.; Lewallen, K.; Hamede, R.K.; Ruiz-Aravena, M.; Ryder, O.; McCallum, H.I.; Jones, M.E.; et al. Contemporary Demographic Reconstruction Methods Are Robust to Genome Assembly Quality: A Case Study in Tasmanian Devils. Mol. Biol. Evol. 2019, 36, 2906–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giantsis, I.A.; Kravva, N.; Apostolidis, A.P. Genetic characterization and evaluation of anthropogenic impacts on genetic patterns in cultured and wild populations of mussels (Mytilus galloprovincialis) from Greece. Genet. Mol. Res. 2012, 11, 3814–3823. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Z.; Ning, X.R.; Le, F.F.; Chen, W.D.; Zhuang, D.G. Long term changes of biodiversity of benthic macroalgae in the intertidal zone of the Nanji Islands. Acta Ecol. Sin. 2010, 30, 106–112. [Google Scholar] [CrossRef]
- Nin, C.J.; Rodgher, S. Effect of a temperature rise on metal toxicity for the aquatic biota: A systematic review. Braz. J. Environ. Sci. Online 2021, 56, 710–720. [Google Scholar] [CrossRef]
- Leal, P.P.; Hurd, C.L.; Sander, S.G.; Armstrong, E.; Fernández, P.A.; Suhrhoff, T.J.; Roleda, M.Y. Copper pollution exacerbates the effects of ocean acidification and warming on kelp microscopic early life stages. Sci. Rep. 2018, 8, 14763. [Google Scholar] [CrossRef]
- Markert, J.A.; Champlin, D.M.; Gutjahr-Gobell, R.; Grear, J.S.; Kuhn, A.; McGreevy, T.J.; Roth, A.; Bagley, M.J.; Nacci, D.E. Population genetic diversity and fitness in multiple environments. BMC Evol. Biol. 2010, 10, 205. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Location Name | Type | Sample Size |
|---|---|---|
| Dongtou, Zhejiang, China | Cultivar | 21 |
| Shengsi, Zhejiang, China | Wild | 1 |
| Changhai, Liaoning, China | Wild | 2 |
| Yeosu-si, Jeollanam-do, South Korea | Wild | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, L.; Wang, F.; Wu, M.; Wang, S. ddRAD Sequencing-Based Scanning of Genetic Variants in Sargassum fusiforme. J. Mar. Sci. Eng. 2022, 10, 958. https://doi.org/10.3390/jmse10070958
Lin L, Wang F, Wu M, Wang S. ddRAD Sequencing-Based Scanning of Genetic Variants in Sargassum fusiforme. Journal of Marine Science and Engineering. 2022; 10(7):958. https://doi.org/10.3390/jmse10070958
Chicago/Turabian StyleLin, Lidong, Fei Wang, Mingjiang Wu, and Shengqin Wang. 2022. "ddRAD Sequencing-Based Scanning of Genetic Variants in Sargassum fusiforme" Journal of Marine Science and Engineering 10, no. 7: 958. https://doi.org/10.3390/jmse10070958
APA StyleLin, L., Wang, F., Wu, M., & Wang, S. (2022). ddRAD Sequencing-Based Scanning of Genetic Variants in Sargassum fusiforme. Journal of Marine Science and Engineering, 10(7), 958. https://doi.org/10.3390/jmse10070958