Enhancing Genomic Prediction Accuracy in Beef Cattle Using WMGBLUP and SNP Pre-Selection

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Simulation
2.2. Population Grouping and Combination Strategy
2.3. GWAS Model
2.4. GBLUP Model
2.5. MGBLUP Model and WMGBLUP Model
2.6. Evaluation of Prediction Ability
3. Results
3.1. Results of Population Simulation
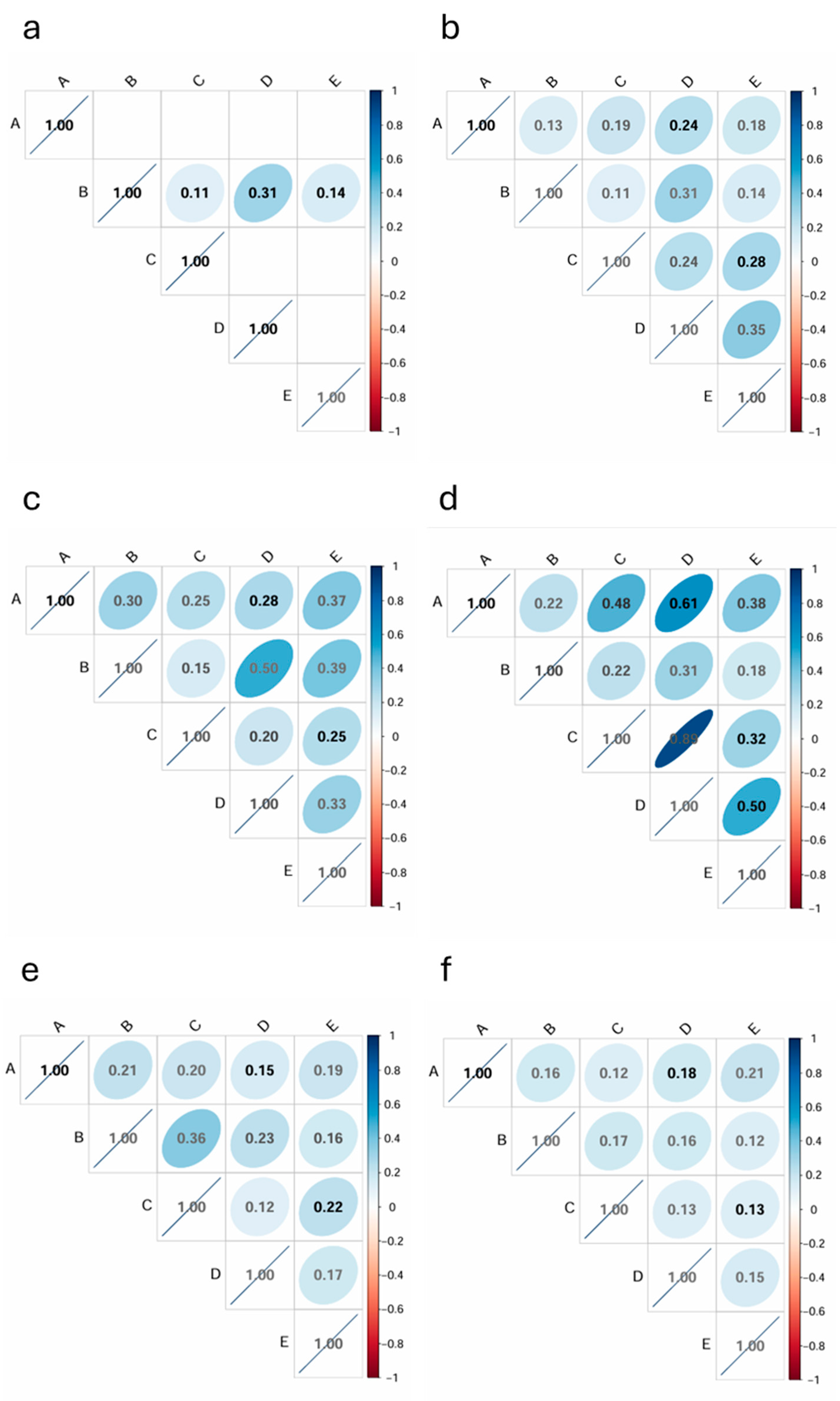
3.2. Genetic Correlation Between Populations
3.3. Genomic Prediction Accuracy
3.3.1. The Results of Genomic Prediction for Population A
3.3.2. The Results of Genomic Prediction for Population B
3.3.3. The Results of Genomic Prediction for Population E
4. Discussion
4.1. The Influence of Training Set Size and Population Composition on Prediction Accuracy
4.2. Influence of the Preselected SNP Proportion on Prediction Accuracy
4.3. The Influence of the Weighted Multiple G Matrix Model on Accuracy
4.4. Importance and Limitations of the Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Yin, L.; Ma, Y.; Xiang, T.; Zhu, M.; Yu, M.; Li, X.; Liu, X.; Zhao, S. The Progress and Prospect of Genomic Selection Models. Acta Vet. Zootech. Sin. 2019, 50, 233–242. [Google Scholar]
- Mcgowan, M.; Wang, J.; Dong, H.; Liu, X.; Zhang, Z. Ideas in Genomic Selection with the Potential to Transform Plant Molecular Breeding: A Review. Preprints 2021, 273–319. [Google Scholar]
- Sun, D.; Zhang, S.; Zhang, Q.; Li, J.; Zhang, G.; Liu, C.; Zheng, W. Application Progress on Genomic Selection Technology for Dairy Cattle in China. Acta Vet. Zootech. Sin. 2023, 54, 4028–4039. [Google Scholar]
- Hozé, C.; Fritz, S.; Phocas, F.; Boichard, D.; Ducrocq, V.; Croiseau, P. Efficiency of multi-breed genomic selection for dairy cattle breeds with different sizes of reference population. J. Dairy. Sci. 2014, 97, 3918–3929. [Google Scholar] [CrossRef]
- Toosi, A.; Fernando, R.L.; Dekkers, J.C. Genomic selection in admixed and crossbred populations. J. Anim. Sci. 2010, 88, 32–46. [Google Scholar] [CrossRef]
- Ibtisham, F.; Zhang, L.; Xiao, M.; An, L.; Ramzan, M.B.; Nawab, A.; Zhao, Y.; Li, G.; Xu, Y. Genomic selection and its application in animal breeding. Thai J. Vet. Med. 2017, 47, 301–310. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef]
- Misztal, I.; Steyn, Y.; Lourenco, D.A.L. Genomic evaluation with multibreed and crossbred data. JDS Commun. 2022, 3, 156–159. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.C.; Verbyla, K.; Goddard, M.E. Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet. Sel. Evol. 2009, 41, 51. [Google Scholar] [CrossRef]
- Pryce, J.E.; Gredler, B.; Bolormaa, S.; Bowman, P.J.; Egger-Danner, C.; Fuerst, C.; Emmerling, R.; Sölkner, J.; Goddard, M.E.; Hayes, B.J. Short communication: Genomic selection using a multi-breed, across-country reference population. J. Dairy. Sci. 2011, 94, 2625–2630. [Google Scholar] [CrossRef]
- Erbe, M.; Hayes, B.J.; Matukumalli, L.K.; Goswami, S.; Bowman, P.J.; Reich, C.M.; Mason, B.A.; Goddard, M.E. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy. Sci. 2012, 95, 4114–4129. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy. Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Goddard, M. Genomic selection: Prediction of accuracy and maximisation of long term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef]
- Calus, M.P.; Huang, H.; Vereijken, A.; Visscher, J.; Ten Napel, J.; Windig, J.J. Genomic prediction based on data from three layer lines: A comparison between linear methods. Genet. Sel. Evol. 2014, 46, 57. [Google Scholar] [CrossRef]
- Kachman, S.D.; Spangler, M.L.; Bennett, G.L.; Hanford, K.J.; Kuehn, L.A.; Snelling, W.M.; Thallman, R.M.; Saatchi, M.; Garrick, D.J.; Schnabel, R.D.; et al. Comparison of molecular breeding values based on within- and across-breed training in beef cattle. Genet. Sel. Evol. 2013, 45, 30. [Google Scholar] [CrossRef]
- Legarra, A.; Aguilar, I.; Misztal, I. A relationship matrix including full pedigree and genomic information. J. Dairy. Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef]
- Lourenco, D.A.; Tsuruta, S.; Fragomeni, B.O.; Masuda, Y.; Aguilar, I.; Legarra, A.; Bertrand, J.K.; Amen, T.S.; Wang, L.; Moser, D.W.; et al. Genetic evaluation using single-step genomic best linear unbiased predictor in American Angus. J. Anim. Sci. 2015, 93, 2653–2662. [Google Scholar] [CrossRef]
- Song, H.; Ye, S.; Jiang, Y.; Zhang, Z.; Zhang, Q.; Ding, X. Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs. Genet. Sel. Evol. 2019, 51, 58. [Google Scholar] [CrossRef]
- Goddard, M.E.; Kemper, K.E.; MacLeod, I.M.; Chamberlain, A.J.; Hayes, B.J. Genetics of complex traits: Prediction of phenotype, identification of causal polymorphisms and genetic architecture. Proc. Biol. Sci. 2016, 283, 1835. [Google Scholar] [CrossRef]
- Veerkamp, R.F.; Bouwman, A.C.; Schrooten, C.; Calus, M.P. Genomic prediction using preselected DNA variants from a GWAS with whole-genome sequence data in Holstein-Friesian cattle. Genet. Sel. Evol. 2016, 48, 95. [Google Scholar] [CrossRef]
- van den Berg, I.; Boichard, D.; Lund, M.S. Sequence variants selected from a multi-breed GWAS can improve the reliability of genomic predictions in dairy cattle. Genet. Sel. Evol. 2016, 48, 83. [Google Scholar] [CrossRef]
- Du, Y.; Huang, C.; Wang, Y.; Li, S.; Wen, J.; Chen, Z.; Zhao, G.; Zheng, M. Genomic Selection for RFI in Broiler Combining GWAS Prior Marker Information. Acta Vet. Zootech. Sin. 2022, 53, 3403–3411. [Google Scholar]
- Sargolzaei, M.; Schenkel, F.S. QMSim: A large-scale genome simulator for livestock. Bioinformatics 2009, 25, 680–681. [Google Scholar] [CrossRef]
- Snelling, W.M.; Chiu, R.; Schein, J.E.; Hobbs, M.; Abbey, C.A.; Adelson, D.L.; Aerts, J.; Bennett, G.L.; Bosdet, I.E.; Boussaha, M.; et al. A physical map of the bovine genome. Genome Biol. 2007, 8, R165. [Google Scholar] [CrossRef]
- Brown, B.C.; Ye, C.J.; Price, A.L.; Zaitlen, N. Transethnic Genetic-Correlation Estimates from Summary Statistics. Am. J. Hum. Genet. 2016, 99, 76–88. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Yin, C.; Zhou, P.; Wang, Y.; Yin, Z.; Liu, Y. Using genomic selection to improve the accuracy of genomic prediction for multi-populations in pigs. Animal 2024, 18, 101062. [Google Scholar] [CrossRef]
- Wientjes, Y.C.; Calus, M.P.; Goddard, M.E.; Hayes, B.J. Impact of QTL properties on the accuracy of multi-breed genomic prediction. Genet. Sel. Evol. 2015, 47, 42. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, I.; MacLeod, I.M.; Reich, C.M.; Breen, E.J.; Pryce, J.E. Optimizing genomic prediction for Australian Red dairy cattle. J. Dairy. Sci. 2020, 103, 6276–6298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Mitra, N.; Kanetsky, P.A.; Nathanson, K.L.; Rebbeck, T.R. A practical approach to adjusting for population stratification in genome-wide association studies: Principal components and propensity scores (PCAPS). Stat. Appl. Genet. Mol. Biol. 2018, 17, 6. [Google Scholar] [CrossRef]
- Wei, C.; Chang, C.; Zhang, W.; Ren, D.; Cai, X.; Zhou, T.; Shi, S.; Wu, X.; Si, J.; Yuan, X.; et al. Preselecting Variants from Large-Scale Genome-Wide Association Study Meta-Analyses Increases the Genomic Prediction Accuracy of Growth and Carcass Traits in Large White Pigs. Animals 2023, 13, 3746. [Google Scholar] [CrossRef]
- Raymond, B.; Wientjes, Y.C.J.; Bouwman, A.C.; Schrooten, C.; Veerkamp, R.F. A deterministic equation to predict the accuracy of multi-population genomic prediction with multiple genomic relationship matrices. Genet. Sel. Evol. 2020, 52, 21. [Google Scholar] [CrossRef]
- Su, G.; Christensen, O.F.; Janss, L.; Lund, M.S. Comparison of genomic predictions using genomic relationship matrices built with different weighting factors to account for locus-specific variances. J. Dairy. Sci. 2014, 97, 6547–6559. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Ding, X.; Bijma, P.; de Koning, D.J.; Zhang, Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE 2010, 5, e12648. [Google Scholar] [CrossRef]
- Zhang, Z.; Ober, U.; Erbe, M.; Zhang, H.; Gao, N.; He, J.; Li, J.; Simianer, H. Improving the accuracy of whole genome prediction for complex traits using the results of genome wide association studies. PLoS ONE 2014, 9, e93017. [Google Scholar] [CrossRef]
- Edwards, S.M.; Sørensen, I.F.; Sarup, P.; Mackay, T.F.; Sørensen, P. Genomic Prediction for Quantitative Traits Is Improved by Mapping Variants to Gene Ontology Categories in Drosophila melanogaster. Genetics 2016, 203, 1871–1883. [Google Scholar] [CrossRef]
- Meuwissen, T.; Eikje, L.S.; Gjuvsland, A.B. GWABLUP: Genome-wide association assisted best linear unbiased prediction of genetic values. Genet. Sel. Evol. 2024, 56, 17. [Google Scholar] [CrossRef]
- Raymond, B.; Bouwman, A.C.; Wientjes, Y.C.J.; Schrooten, C.; Houwing-Duistermaat, J.; Veerkamp, R.F. Genomic prediction for numerically small breeds, using models with pre-selected and differentially weighted markers. Genet. Sel. Evol. 2018, 50, 49. [Google Scholar] [CrossRef]
- Steyn, Y.; Lourenco, D.A.L.; Misztal, I. Genomic predictions in purebreds with a multibreed genomic relationship matrix. J. Anim. Sci. 2019, 97, 4418–4427. [Google Scholar] [CrossRef] [PubMed]
- Esrafili Taze Kand Mohammaddiyeh, M.; Rafat, S.A.; Shodja, J.; Javanmard, A.; Esfandyari, H. Selective genotyping to implement genomic selection in beef cattle breeding. Front. Genet. 2023, 14, 1083106. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Parameters | A | B | C | D | E |
|---|---|---|---|---|---|
| Initial males | 60 | 50 | 50 | 50 | 60 |
| Initial females | 2800 | 2600 | 3000 | 2400 | 2500 |
| Sire replacement | 0.50 | 0.50 | 0.60 | 0.50 | 0.60 |
| Sire growth rate | 0.07 | 0.06 | 0.1 | 0.07 | 0.11 |
| Dam replacement | 0.30 | 0.30 | 0.30 | 0.30 | 0.30 |
| Dam growth rate | 0.1 | 0.1 | 0.13 | 0.1 | 0.13 |
| Number of offspring per dam | 1 | ||||
| Mating design | Random | ||||
| GWAS Group | Training Set of the GP Group | Test Set of the GP Group |
|---|---|---|
| 800A 1 | 1000A | 800A |
| 1600A | 2000A | |
| 2400A | 3000A | |
| 3200A | 4000A | |
| 2400A_800B 2 | 3000A_1000B | |
| 2400A_800E | 3000A_1000E | |
| 1600A_1600B | 2000A_2000B | |
| 1600A_1600E | 2000A_2000E | |
| 800B | 1000B | 800B |
| 1600B | 2000B | |
| 3200B | 4000B | |
| 2400A_800B | 3000A_1000B | |
| 1600A_1600B | 2000A_2000B | |
| 800E | 1000E | 800E |
| 1600E | 2000E | |
| 3200E | 4000E | |
| 1600A_1600E | 2000A_2000E | |
| 2400A_800E | 3000A_1000E |
| Preselected SNP | Model | Training Set of GP Group | |||
|---|---|---|---|---|---|
| 1000A | 2000A | 3000A | 4000A | ||
| - | GBLUP | 0.246 (0.008) 1 | 0.320 (0.008) | 0.346 (0.007) | 0.420 (0.005) |
| top5% 2 | WMGBLUP | 0.278 (0.008) | 0.406 (0.011) | 0.450 (0.008) | 0.520 (0.005) |
| MGBLUP | 0.275 (0.009) | 0.389 (0.010) | 0.432 (0.009) | 0.510 (0.005) | |
| top10% | WMGBLUP | 0.269 (0.008) | 0.377 (0.010) | 0.428 (0.007) | 0.497 (0.004) |
| MGBLUP | 0.268 (0.009) | 0.365 (0.009) | 0.414 (0.012) | 0.485 (0.004) | |
| top15% | WMGBLUP | 0.263 (0.008) | 0.360 (0.009) | 0.403 (0.008) | 0.478 (0.004) |
| MGBLUP | 0.262 (0.009) | 0.353 (0.009) | 0.394 (0.006) | 0.467 (0.005) | |
| top20% | WMGBLUP | 0.267 (0.009) | 0.348 (0.009) | 0.391 (0.008) | 0.455 (0.006) |
| MGBLUP | 0.258 (0.008) | 0.344 (0.009) | 0.383 (0.013) | 0.454 (0.005) | |
| top25% | WMGBLUP | 0.262 (0.008) | 0.346 (0.008) | 0.385 (0.008) | 0.473 (0.004) |
| MGBLUP | 0.255 (0.008) | 0.338 (0.008) | 0.368 (0.005) | 0.446 (0.005) | |
| Preselected SNP | Model | Training Set of GP Group | |||
|---|---|---|---|---|---|
| 3000A_1000B | 3000A_1000E | 2000A_2000B | 2000A_2000E | ||
| - | GBLUP | 0.366 (0.008) | 0.398 (0.009) | 0.360 (0.006) | 0.341 (0.010) |
| top5% | WMGBLUP | 0.513 (0.008) | 0.514 (0.006) | 0.433 (0.007) | 0.461 (0.006) |
| MGBLUP | 0.490 (0.008) | 0.496 (0.007) | 0.441 (0.007) | 0.460 (0.006) | |
| top10% | WMGBLUP | 0.478 (0.008) | 0.479 (0.007) | 0.423 (0.007) | 0.441 (0.007) |
| MGBLUP | 0.449 (0.008) | 0.470 (0.008) | 0.422 (0.007) | 0.431 (0.007) | |
| top15% | WMGBLUP | 0.448 (0.008) | 0.460 (0.008) | 0.409 (0.008) | 0.422 (0.008) |
| MGBLUP | 0.424 (0.008) | 0.452 (0.008) | 0.406 (0.008) | 0.411 (0.008) | |
| top20% | WMGBLUP | 0.426 (0.008) | 0.447 (0.008) | 0.397 (0.008) | 0.406 (0.008) |
| MGBLUP | 0.409 (0.008) | 0.440 (0.008) | 0.394 (0.008) | 0.395 (0.008) | |
| top25% | WMGBLUP | 0.409 (0.008) | 0.434 (0.008) | 0.388 (0.008) | 0.391 (0.009) |
| MGBLUP | 0.398 (0.008) | 0.429 (0.009) | 0.385 (0.008) | 0.382 (0.009) | |
| Preselected SNP | Model | Training Set of GP Group | ||||
|---|---|---|---|---|---|---|
| 1000B | 2000B | 4000B | 3000A_1000B | 2000A_2000B | ||
| - | GBLUP | 0.207 (0.013) | 0.309 (0.007) | 0.423 (0.007) | 0.109 (0.010) | 0.293 (0.013) |
| top5% | WMGBLUP | 0.250 (0.012) | 0.375 (0.006) | 0.534 (0.006) | 0.253 (0.009) | 0.432 (0.008) |
| MGBLUP | 0.237 (0.013) | 0.352 (0.008) | 0.509 (0.007) | 0.227 (0.010) | 0.417 (0.010) | |
| top10% | WMGBLUP | 0.249 (0.011) | 0.347 (0.011) | 0.518 (0.006) | 0.224 (0.010) | 0.414 (0.009) |
| MGBLUP | 0.229 (0.013) | 0.333 (0.006) | 0.487 (0.007) | 0.190 (0.010) | 0.387 (0.011) | |
| top15% | WMGBLUP | 0.247 (0.010) | 0.344 (0.007) | 0.506 (0.006) | 0.202 (0.010) | 0.392 (0.011) |
| MGBLUP | 0.222 (0.013) | 0.323 (0.004) | 0.473 (0.007) | 0.169 (0.010) | 0.362 (0.012) | |
| top20% | WMGBLUP | 0.245 (0.012) | 0.322 (0.008) | 0.520 (0.006) | 0.184 (0.011) | 0.377 (0.011) |
| MGBLUP | 0.218 (0.013) | 0.314 (0.011) | 0.461 (0.007) | 0.154 (0.010) | 0.346 (0.012) | |
| top25% | WMGBLUP | 0.241 (0.012) | 0.312 (0.009) | 0.511 (0.006) | 0.171 (0.011) | 0.364 (0.011) |
| MGBLUP | 0.215 (0.013) | 0.311 (0.007) | 0.452 (0.007) | 0.142 (0.010) | 0.333 (0.012) | |
| Preselected SNP | Model | Training Set of GP Group | ||||
|---|---|---|---|---|---|---|
| 1000E | 2000E | 4000E | 3000A_1000E | 2000A_2000E | ||
| - | GBLUP | 0.254 (0.010) | 0.321 (0.015) | 0.429 (0.007) | 0.120 (0.012) | 0.334 (0.012) |
| top5% | WMGBLUP | 0.287 (0.009) | 0.389 (0.012) | 0.537 (0.007) | 0.285 (0.011) | 0.438 (0.010) |
| MGBLUP | 0.271 (0.009) | 0.381 (0.012) | 0.525 (0.006) | 0.252 (0.013) | 0.430 (0.011) | |
| top10% | WMGBLUP | 0.285 (0.009) | 0.356 (0.005) | 0.523 (0.006) | 0.255 (0.012) | 0.428 (0.010) |
| MGBLUP | 0.266 (0.009) | 0.350 (0.009) | 0.504 (0.006) | 0.213 (0.013) | 0.408 (0.011) | |
| top15% | WMGBLUP | 0.280 (0.009) | 0.351 (0.006) | 0.510 (0.006) | 0.230 (0.012) | 0.414 (0.010) |
| MGBLUP | 0.263 (0.009) | 0.349 (0.011) | 0.488 (0.006) | 0.188 (0.013) | 0.391 (0.011) | |
| top20% | WMGBLUP | 0.277 (0.009) | 0.349 (0.013) | 0.501 (0.006) | 0.211 (0.012) | 0.401 (0.009) |
| MGBLUP | 0.261 (0.009) | 0.342 (0.006) | 0.476 (0.006) | 0.171 (0.013) | 0.377 (0.011) | |
| top25% | WMGBLUP | 0.275 (0.009) | 0.344 (0.007) | 0.498 (0.006) | 0.196 (0.011) | 0.392 (0.010) |
| MGBLUP | 0.259 (0.009) | 0.324 (0.012) | 0.466 (0.007) | 0.159 (0.013) | 0.348 (0.012) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Xie, X.; Ma, H.; Zhou, P.; Xu, B.; Zhang, Y.; Xu, L.; Gao, H.; Li, J.; Wang, Z.; et al. Enhancing Genomic Prediction Accuracy in Beef Cattle Using WMGBLUP and SNP Pre-Selection. Agriculture 2025, 15, 1094. https://doi.org/10.3390/agriculture15101094
Zhao H, Xie X, Ma H, Zhou P, Xu B, Zhang Y, Xu L, Gao H, Li J, Wang Z, et al. Enhancing Genomic Prediction Accuracy in Beef Cattle Using WMGBLUP and SNP Pre-Selection. Agriculture. 2025; 15(10):1094. https://doi.org/10.3390/agriculture15101094
Chicago/Turabian StyleZhao, Huqiong, Xueyuan Xie, Haoran Ma, Peinuo Zhou, Boran Xu, Yuanqing Zhang, Lingyang Xu, Huijiang Gao, Junya Li, Zezhao Wang, and et al. 2025. "Enhancing Genomic Prediction Accuracy in Beef Cattle Using WMGBLUP and SNP Pre-Selection" Agriculture 15, no. 10: 1094. https://doi.org/10.3390/agriculture15101094
APA StyleZhao, H., Xie, X., Ma, H., Zhou, P., Xu, B., Zhang, Y., Xu, L., Gao, H., Li, J., Wang, Z., & Niu, X. (2025). Enhancing Genomic Prediction Accuracy in Beef Cattle Using WMGBLUP and SNP Pre-Selection. Agriculture, 15(10), 1094. https://doi.org/10.3390/agriculture15101094