
TaqMan qPCR Detection and Quantification of Phytophthora cinnamomi in Soil and Plant Tissues for Walnut Disease Management


Abstract
1. Introduction
2. Materials and Methods
2.1. Phytophthora Isolates and Isolation
2.2. DNA Extraction from Pure Colony and Plant Tissue
2.3. Real-Time PCR Primer and Probe Design for P. cinnamomi
2.4. qPCR Conditions
2.5. Validation of the Real-Time PCR Method
2.6. Soil DNA Extraction and Sampling
3. Results
3.1. Primers and Probe Design
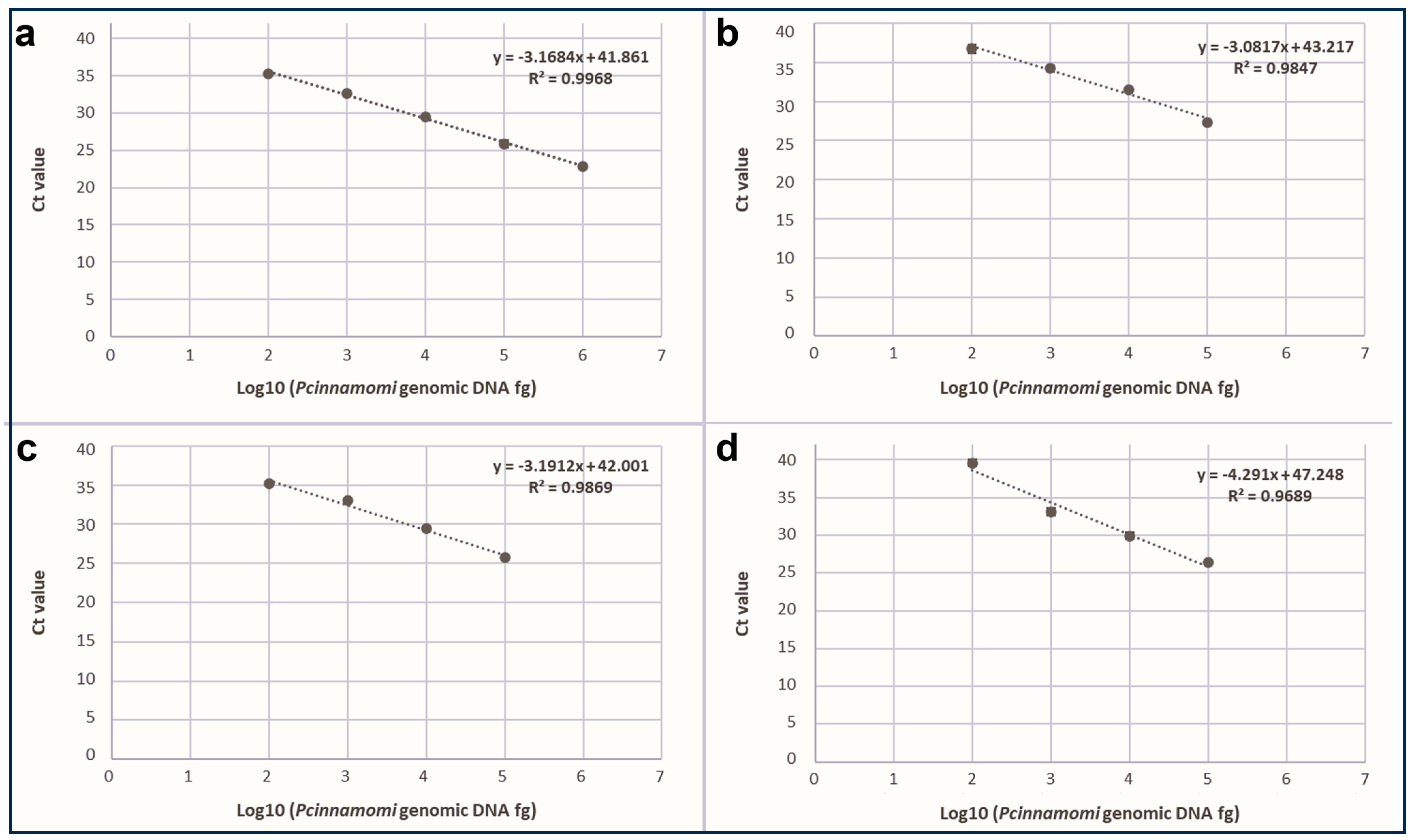
3.2. qPCR Sensitivity and Linearity
3.3. Specificity
3.4. Analysis of Naturally Infected Samples
3.4.1. Plant Material
3.4.2. Soil
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, X.; Tyler, B.M.; Hong, C. An expanded phylogeny for the genus Phytophthora. IMA Fungus 2017, 8, 355–384. [Google Scholar] [CrossRef]
- Hardham, A.R.; Blackman, L.M. Phytophthora cinnamomi. Mol. Plant. Pathol. 2018, 19, 260–285. [Google Scholar] [CrossRef] [PubMed]
- Erwin, D.C.; Ribeiro, O.K. Phytophthora Diseases Worldwide; APS Press: St. Paul, MN, USA, 1996; p. 562. ISBN 978-0-89054-212-5. [Google Scholar]
- Kamoun, S.; Furzer, O.; Jones, J.D.G.; Judelson, H.S.; Ali, G.S.; Dalio, R.J.D.; Roy, S.G.; Schena, L.; Zambounis, A.; Panabières, F.; et al. The Top 10 oomycete pathogens in molecular plant pathology. Mol. Plant Pathol. 2015, 16, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Belisario, A.; Maccaroni, M.; Vettorazzo, M. First Report of Phytophthora cambivora Causing Bleeding Cankers and Dieback on Beech (Fagus sylvatica) in Italy. Plant Dis. 2006, 90, 1362. [Google Scholar] [CrossRef]
- Burgess, T.I.; Scott, J.K.; Mcdougall, K.L.; Stukely, M.J.C.; Crane, C.; Dunstan, W.A.; Brigg, F.; Andjic, V.; White, D.; Rudman, T.; et al. Current and projected global distribution of Phytophthora cinnamomi, one of the world’s worst plant pathogens. Glob. Chang. Biol. 2017, 23, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Orlikowski, L.; Henricot, B.; Abad-Campos, P.; Aday, A.G.; Aguín Casal, O.; Bakonyi, J.; Cacciola, S.O.; Cech, T.; Chavarriaga, D.; et al. Widespread Phytophthora infestations in European nurseries put forest, semi-natural and horticultural ecosystems at high risk of Phytophthora diseases. For. Pathol. 2016, 46, 134–163. [Google Scholar] [CrossRef]
- Scanu, B.; Hunter, G.C.; Linaldeddu, B.T.; Franceschini, A.; Maddau, L.; Jung, T.; Denman, S. A taxonomic re-evaluation reveals that Phytophthora cinnamomi and P. cinnamomi var. parvispora are separate species. For. Pathol. 2013, 44, 1–20. [Google Scholar] [CrossRef]
- Frisullo, S.; Lima, G.; Magnano di San Lio, G.; Camele, I.; Melissano, L.; Puglisi, I.; Pane, A.; Agosteo, G.E.; Prudente, L.; Cacciola, S.O. Phytophthora cinnamomi Involved in the Decline of Holm Oak (Quercus ilex) Stands in Southern Italy. For. Sci. 2018, 64, 290–298. [Google Scholar] [CrossRef]
- Vettraino, A.M.; Barzanti, P.; Bianco, M.C.; Ragazzi, A.; Capretti, P.; Paoletti, E.; Luisi, N.; Anselmi, N.; Vannini, A. Occurrence of Phytophthora species in oak stands in Italy and their association with declining oak trees. For Pathol. 2002, 32, 19–28. [Google Scholar] [CrossRef]
- Westbrook, J.W.; James, J.B.; Sisco, P.H.; Frampton, J.; Lucas, S.; Jeffers, S.N. Resistance to Phytophthora cinnamomi in American Chestnut (Castanea dentata) Backcross Populations that Descended from Two Chinese Chestnut (Castanea mollissima) Sources of Resistance. Plant Dis. 2019, 103, 1631–1641. [Google Scholar] [CrossRef]
- Belisario, A.; Galli, M.; Wajnberg, E. Evaluation of Juglans species for resistance to Phytophthora cinnamomi: Differences in isolate virulence and response to fosetyl-Al. For. Pathol. 2009, 39, 168–176. [Google Scholar] [CrossRef]
- Browne, G.T.; Leslie, C.A.; Grant, J.A.; Bhat, R.G.; SchmidtL, S.; Hackett, W.P.; Kluepfel, D.A.; Robinson, R.; McGranahan, G.H. Resistance to Species of Phytophthora Identified among Clones of Juglans macrocarpa × J. regia. HortScience 2015, 50, 1136–1142. [Google Scholar] [CrossRef]
- Guajardo, J.; Saa, S.; Camps, R.; Besoain, X. Outbreak of Crown and Root Rot of Walnut Caused by Phytophthora cinnamomi in Chile. Plant Dis. 2017, 101, 636. [Google Scholar] [CrossRef]
- Matheron, M.E.; Mircetich, S.M. Pathogenicity and relative virulence of Phytophthora spp. from walnut and other plants to rootstocks of English walnut trees. Phytopathology 1985, 75, 977–981. [Google Scholar] [CrossRef]
- Vitale, S.; Scotton, M.; Vettraino, A.M.; Vannini, A.; Haegi, A.; Luongo, L.; Scarpari, M.; Belisario, A. Characterization of Phytophthora cinnamomi from common walnut in Southern Europe environment. For. Pathol. 2018, 49, e12477. [Google Scholar] [CrossRef]
- Vitale, S.; Luongo, L.; Scarpari, M.; Tizzani, L.; Garaguso, I.; Galli, M.; Belisario, A.; Scotton, M.; Mughini, G.; Gras, M.; et al. Sources of resistance to Phytophthora cinnamomi in Juglans spp. for potential rootstocks. Hortscience 2021, 56, 667–671. [Google Scholar] [CrossRef]
- Akinsanmi, O.A.; Drenth, A. Phosphite and metalaxyl rejuvenate macadamia trees in decline caused by Phytophthora cinnamomi. Crop Prot. 2013, 53, 29–36. [Google Scholar] [CrossRef]
- Dunstan, W.A.; Rudman, T.; Shearer, B.L.; Moore, N.A.; Paap, T.; Calver, M.C.; Dell, B.; Hardy, G.E.S.J. Containment and spot eradication of a highly destructive, invasive plant pathogen (Phytophthora cinnamomi) in natural ecosystems. Biol. Invasions 2010, 12, 913–925. [Google Scholar] [CrossRef]
- Carter, Z.T.; McNaughton, E.J.; Fea, M.P.; Horner, I.; Johnson, K.; Killick, S.; Glen, A.S. Evaluating scent detection dogs as a tool to detect pathogenic Phytophthora species. Conserv. Sci. Pract. 2023, 5, e12997. [Google Scholar] [CrossRef]
- Schena, L.; Duncan, J.M.; Cooke, D.E.L. Development and application of a PCR-based ‘molecular toolbox’ for the identification of Phytophthora species damaging forests and natural ecosystems. Plant Pathol. 2008, 57, 64–75. [Google Scholar] [CrossRef]
- Langrell, S.R.H.; Morel, O.; Robin, C. Touchdown nested multiplex PCR detection of Phytophthora cinnamomi and P. cambivora from French and English chestnut grove soils. Fungal Biol. 2011, 115, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Engelbrecht, J.; Duong, T.A.; van den Berg, N. Development of a Nested Quantitative Real-Time PCR for Detecting Phytophthora cinnamomi in Persea americana Rootstocks. Plant Dis. 2013, 97, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, G.J.; Martin, F.N.; Coffey, M.D.; Blomquist, C.L. Development of a Multiplex Assay for Genus- and Species-Specific Detection of Phytophthora Based on Differences in Mitochondrial Gene Order. Phytopathology 2014, 104, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Kunadiya, M.; Dunstan, W.A.D.; White, D.; Hardy, G.E.S.J.; Grigg, A.H.; Burgess, T.I. Assay for the Detection of Phytophthora cinnamomi Including an mRNA Protocol Designed to Establish Propagule Viability in Environmental Samples. Plant Dis. 2019, 103, 2443–2450. [Google Scholar] [CrossRef]
- Dai, T.; Yang, X.; Hu, T.; Li, Z.Y.; Xu, Y.; Lu, C.C. A novel LAMP assay for the detection of Phytophthora cinnamomi utilizing a new target gene identified from genome sequences. Plant Dis. 2019, 103, 3101–3107. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Wang, A.; Yang, X.; Yu, X.; Tian, W.; Xu, Y.; Hu, T. PHYCI_587572: An RxLR Effector Gene and New Biomarker in A Recombinase Polymerase Amplification Assay for Rapid Detection of Phytophthora cinnamomi. Forests 2020, 11, 306. [Google Scholar] [CrossRef]
- Kunadiya, M.; White, D.; Dunstan, W.A.D.; Hardy, G.E.S.J.; Grigg, A.H.; Andjic, V.; Burgess, T.I. Pathways to false-positive diagnoses using molecular genetic detection methods; Phytophthora cinnamomi a case study. FEMS Microbiol. Lett. 2017, 364, fnx009. [Google Scholar] [CrossRef]
- Ioos, R.; Laugustin, L.; Schenck, N.; Rose, S.; Husson, C.; Frey, P. Usefulness of single copy genes containing introns in Phytophthora for the development of detection tools for the regulated species P. ramorum and P. fragariae. Eur. J. Plant Pathol. 2006, 116, 171–176. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—Occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Jeffers, S.N.; Martin, S.B. Comparison of two media selective for Phytophthora and Pythium species. Plant Dis. 1986, 70, 1038–1043. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H.J. Bioinformatics Methods and Protocols: Methods in Molecular Biology. In Primer 3 on the WWW for General Users and for Biologist Programmers; Krawetzand, S., Misener, S., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. ISSN 1064-3745. [Google Scholar]
- Cooke, D.E.L.; Drenth, A.; Duncan, J.M.; Wagels, G.; Brasier, C.M. A molecular phylogeny of Phytophthora and related oomycetes. Fungal Genet. Biol. 2000, 30, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Riolo, M.; Aloi, F.; La Spada, F.; Sciandrello, S.; Moricca, S.; Santilli, E.; Pane, A.; Cacciola, S.O. Diversity of Phytophthora communities across different types of mediterranean vegetation in a nature reserve area. Forests 2020, 11, 853. [Google Scholar] [CrossRef]
- Schena, L.; Hughes, J.D.; Cooke, D.E.L. Detection and quantification of Phytophthora ramorum, P. kernoviae, P. citricola, and P. quercina in symptomatic leaves by multiplex real-time PCR. Mol. Plant Pathol. 2006, 7, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, E.; Ceustermans, A.; Baets, D.; Ferreira, J.; Bonants, P.; Melis, P.; Van Hemelrijck, W.; Bylemans, S.; Rediers, H.; Lievens, B. Quantitative PCR for detection and quantification of Phytophthora cactorum in the cultivation of strawberry. Eur J Plant Pathol 2021, 160, 867–882. [Google Scholar] [CrossRef]
- Jung, T.; Colquhoun, I.J.; Hardy, G.E.S.J. New insights into the survival strategy of the invasive soilborne pathogen Phytophthora cinnamomi in different natural ecosystems in Western Australia. For. Pathol. 2013, 43, 266–288. [Google Scholar] [CrossRef]
- Osawa, H.; Suzuki, N.; Akino, S.; Araki, H.; Asano, K.; Akai, K.; Kondo, N. Quantification of Phytophthora infestans population densities and their changes in potato field soil using real-time PCR. Sci. Rep. 2021, 11, 6266. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | CREADC Isolate Number | Clade a | Host | Country | Real-Time PCR Result |
|---|---|---|---|---|---|
| P. x alni | Om293 | 7a | Alnus glutinosa | Germany | - |
| P. x multiformis | Om294 | 7a | Alnus glutinosa | The Netherlands | - |
| P. uniformis | Om295 b | 7a | Alnus glutinosa | Germany | - |
| P. cactorum | Om60 | 1a | Juglans regia | Italy | - |
| Om61 | Juglans regia | Italy | - | ||
| P. cambivora | Om133 | 7a | Fagus | Italy | - |
| Om134 | Fagus | Italy | - | ||
| P. capsici | Om246 | 2b | Capsicum annum | Italy | - |
| P. cinnamomi | Om69 | 7c | Quercus rubra | France | + |
| Om70 | Juglans regia | Italy | + | ||
| Om74 | Juglans regia | Italy | + | ||
| Om76 | Juglans regia | Italy | + | ||
| Om119 | Juglans regia | Italy | + | ||
| Om194 | Juglans regia | Italy | + | ||
| Om202 | Juglans regia | Italy | + | ||
| Om274 | Juglans regia | Italy | + | ||
| Om281 | Juglans regia | Italy | + | ||
| Om283 | Juglans regia | Italy | + | ||
| Om139 | Castanea sativa | Spain | + | ||
| Om141 | Castanea sativa | Spain | + | ||
| Om142 | Castanea sativa | Spain | + | ||
| Om144 | Quercus sp. | Italy | + | ||
| Om145 | Quercus sp. | Italy | + | ||
| P. citricola | Om161 | 2c | Juglans regia | Italy | - |
| P. cryptogea | Om26 | 8a | Actinidia deliciosa | Italy | - |
| Om28 | Actinidia deliciosa | Italy | - | ||
| P. drechsleri | Om41 c | 8a | Unknown | Unknown | - |
| Om220 | Unknown | Germany | - | ||
| P. gonapodyides | Om261 | 6b | Juglans regia | Italy | - |
| P. hedraiandra | Om68 | 1a | Viburnum tinus | Italy | - |
| P. hydropathica | Om234 | 9a1 | Viburnum tinus | Italy | - |
| Om236 | Viburnum tinus | Italy | - | ||
| P. kernoviae | Om273 d | 10 | England | - | |
| P. megasperma | Om198 | 6b | Juglans regia | Italy | - |
| Om199 | Juglans regia | Italy | - | ||
| Om239 | Celtis australis | Italy | - | ||
| Om284 | Juglans regia | Italy | - | ||
| P. nicotianae | Om263 | 1c | Vinca | Italy | - |
| Om265 | Capsicum annuum | Italy | - | ||
| P. niederhauserii | Om153 | 7b | Hedera helix | Italy | - |
| Om154 | Hedera helix | Italy | - | ||
| Om242 | Heuchera sp. | Germany | - | ||
| Om404 | Hedera helix | Norway | - | ||
| P. palmivora | Om19 | 4 | Pittosporum tobira | Italy | - |
| Om22 | Pittosporum tobira | Italy | - | ||
| P. parvispora | Om298 e | 7c | Beaucameare curvata | Mexico | - |
| P. ramorum | Om229 | 8c | Viburnus tinus | Italy | - |
| P. tropicalis | Om210 | 2b | Rhododendron sp. | Italy | - |
| Om212 | Rhododendron sp. | Italy | - | ||
| Om237 | 2b | Albizia julibrissin | Italy | - | |
| Pythium chamaehyphon | Om162 | Juglans regia | Italy | - | |
| Pythium sterilum | Om164 | Juglans regia | Italy | - |
| Primers or Probe | Sequences (5′-3′) | Tm (°C) | DNA Region | Position a (bp) | Product Size (bp) | Reference |
|---|---|---|---|---|---|---|
| P. cinn 3.4F | TTTGTGAGTGCCGAGACAAG | 58.42 | Intron3/Ypt1 | 4–23 | 75 | This study |
| P. cinn 3.78R | GCACAGAAACAACAACGACG | 58.55 | Intron3/Ypt1 | 31–52 | 75 | This study |
| P. cinn 3.31Probe b | [FAM]-CCTGTCTGCCCCATTCAACAGA-[BHQ] | 63.48 | Intron3/Ypt1 | 59–78 | -- | This study |
| YPh1F | CGACCATKGGTGTGGACTTT | ~450 | [21] | |||
| YPh2R | ACGTTCTCMCAGGCGTATCT | ~450 | [21] |
| Host | Tree Condition | qPCR | Mean Quantity * pg of Pathogen DNA/mg Host Tissue |
|---|---|---|---|
| Walnut Zn 3/18 | Symptomatic | Positive | 24.1 |
| Walnut Zn 8/19 | Symptomatic | Positive | 19.6 |
| Walnut Tas10/8 | Asymptomatic | Negative | UD |
| Walnut Tas 3/9 | Asymptomatic | Negative | UD |
| Walnut Tas 7/12 | Asymptomatic | Negative | UD |
| Walnut Tas 9/20 | Symptomatic | Positive | 18.1 |
| Walnut Tas11/18 | Symptomatic | Positive | 14.5 |
| Walnut Tas13/13 | Symptomatic | Positive | 9.4 |
| Walnut BD5/6 | Symptomatic | Positive | 12.75 |
| Chestnut | Symptomatic | Positive | 15.4 |
| Chestnut | Symptomatic | Positive | 2.3 |
| Chestnut | Symptomatic | Positive | 10.1 |
| Oak | Symptomatic | Positive | 1.6 |
| Oak | Symptomatic | Positive | 17.3 |
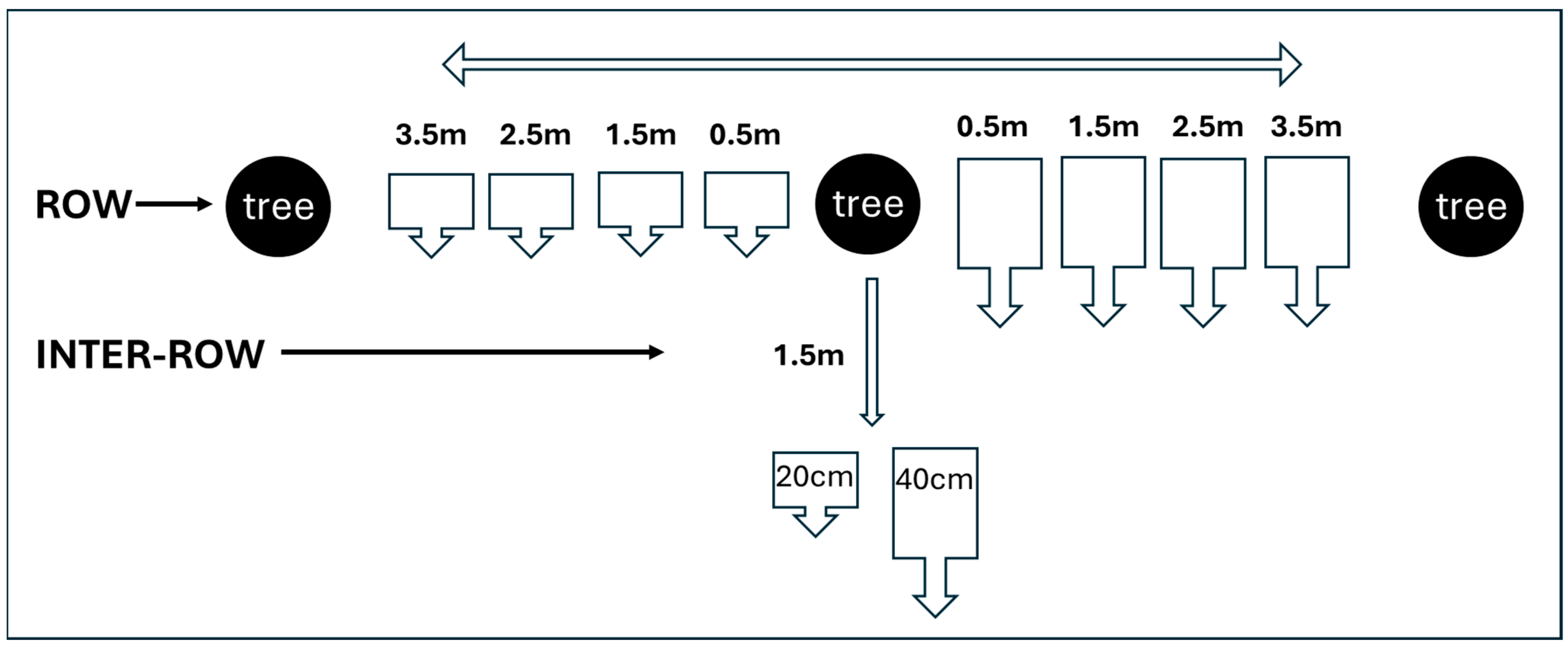
| Walnut Tree | Distance from the Tree | Depth | Sample I | qPCR Result | Sample II | qPCR Result |
|---|---|---|---|---|---|---|
| Zn 3/18 | ||||||
| 0.5 m | 20 | 3A | P1 P2 | 3.1 | P1 P2 | |
| 1.5 m | 20 | 3B | P1 P2 | 3.2 | P1 X2 | |
| 2.5 m | 20 | 3C | X1 X2 | 3.3 | X1 X2 | |
| 3.5 m | 20 | 3D | P1 X2 | 3.4 | X1 P2 | |
| inter-row 1.5 m | 20 | 3E | X1 X2 | 3.5 | X1 P2 | |
| 0.5 m | 40 | 3F | P1 P2 | 3.6 | P1 P2 | |
| 1.5 m | 40 | 3G | X1 P2 | 3.7 | P1 P2 | |
| 2.5 m | 40 | 3H | X1 X2 | 3.8 | X1 X2 | |
| 3.5 m | 40 | 3I | X1 X2 | 3.9 | X1 X2 | |
| Inter-row 1.5 m | 40 | 3L | P1 P2 | 3.10 | P1 P2 | |
| Zn 8/19 | ||||||
| 0.5 m | 20 | 8A | P1 P2 | 8.1 | P1 P2 | |
| 1.5 m | 20 | 8B | X1 X2 | 8.2 | P1 P2 | |
| 2.5 m | 20 | 8C | P1 X2 | 8.3 | P1 X2 | |
| 3.5 m | 20 | 8D | P1 X2 | 8.4 | P1 X2 | |
| Inter-row 1.5 m | 20 | 8E | P1 X2 | 8.5 | X1 X2 | |
| 0.5 m | 40 | 8F | P1 P2 | 8.6 | P1 P2 | |
| 1.5 m | 40 | 8G | P1 X2 | 8.7 | P1 X2 | |
| 2.5 m | 40 | 8H | X1 P2 | 8.8 | X1 P2 | |
| 3.5 m | 40 | 8I | X1 P2 | 8.9 | P1 X2 | |
| Inter-row 1.5 m | 40 | 8L | X1 P2 | 8.10 | X1 X2 |
| Plant for Soil Samples | Tree Condition | Soil Sample and Depth | qPCR Results | Mean Ct and Standard Deviation | Mean Quantity * pg of Pathogen DNA /g of Soil |
|---|---|---|---|---|---|
| Rhododendron | Symptomatic | Potted soil sample | positive | 29.95 ± 0.08 | 140.4 |
| Rhododendron | Symptomatic | Potted soil sample | positive | 30.18 ± 0.03 | 123.5 |
| Rhododendron | Symptomatic | Potted soil sample | positive | 28.47 ± 0.18 | 390.1 |
| Walnut Zn 7/18 | Symptomatic | 20 cm | positive | 37.98 ± 0.51 a | <0.25 |
| 40 cm | positive | Pos nested qPCR | <0.25 | ||
| Walnut Tas 10/8 | Asymptomatic | 20 cm | negative | UD | - |
| 40 cm | negative | UD | - | ||
| Walnut Tas 3/9 | Asymptomatic | 20 cm | negative | UD | - |
| 40 cm | negative | UD | - | ||
| Walnut Tas 7/12 | Asymptomatic | 20 cm | negative | UD | - |
| 40 cm | negative | UD | - | ||
| Walnut Tas 9/20 | Symptomatic | 20 cm | positive | 36.99 ± 1.47 | 0.61 |
| 40 cm | positive | Pos nested qPCR | <0.25 | ||
| Walnut Tas 11/18 | Symptomatic | 20 cm | positive | 30.47 ±0.07 | 134 |
| 40 cm | positive | 29.21 ±0.16 | 267 | ||
| Walnut Tas 13/13 | Symptomatic | 20 cm | positive | 34.47 ±1.028 | 15 |
| 40 cm | positive | 34.99 ±1.57 | 13.9 | ||
| Walnut Tas 16/12 | Symptomatic | 20 cm | positive | 35.47 ± 1.19 | 8.7 |
| 40 cm | positive | 31.21 ± 0.14 | 89.8 | ||
| Walnut Dossei 1 | Symptomatic | 20 cm | Positive | 38.06 ± 1.05 a | <0.25 |
| 40 cm | positive | Pos nested qPCR | <0.25 | ||
| Walnut Dossei 2 | Asymptomatic | 20 cm | positive | Pos nested qPCR | <0.25 |
| 40 cm | positive | Pos nested qPCR | <0.25 | ||
| Walnut Dossei 3 | Symptomatic | 20 cm | positive | Pos nested qPCR | <0.25 |
| 40 cm | positive | 35.32 ± 1.56 | 2.8 | ||
| Walnut BD 5/6 | Symptomatic | 20 cm | positive | 35.47 ± 0.27 | 2.49 |
| Walnut BD 9/10 | Symptomatic | 20 cm | positive | 33.99 ± 0.12 | 6.67 |
| 40cm | positive | 35.16 ± 0.81 | 3.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haegi, A.; Luongo, L.; Vitale, S.; Tizzani, L.; Belisario, A. TaqMan qPCR Detection and Quantification of Phytophthora cinnamomi in Soil and Plant Tissues for Walnut Disease Management. Agriculture 2024, 14, 999. https://doi.org/10.3390/agriculture14070999
Haegi A, Luongo L, Vitale S, Tizzani L, Belisario A. TaqMan qPCR Detection and Quantification of Phytophthora cinnamomi in Soil and Plant Tissues for Walnut Disease Management. Agriculture. 2024; 14(7):999. https://doi.org/10.3390/agriculture14070999
Chicago/Turabian StyleHaegi, Anita, Laura Luongo, Salvatore Vitale, Lorenza Tizzani, and Alessandra Belisario. 2024. "TaqMan qPCR Detection and Quantification of Phytophthora cinnamomi in Soil and Plant Tissues for Walnut Disease Management" Agriculture 14, no. 7: 999. https://doi.org/10.3390/agriculture14070999
APA StyleHaegi, A., Luongo, L., Vitale, S., Tizzani, L., & Belisario, A. (2024). TaqMan qPCR Detection and Quantification of Phytophthora cinnamomi in Soil and Plant Tissues for Walnut Disease Management. Agriculture, 14(7), 999. https://doi.org/10.3390/agriculture14070999