Impact of Exercise on Immunometabolism in Multiple Sclerosis




Abstract
:1. Introduction
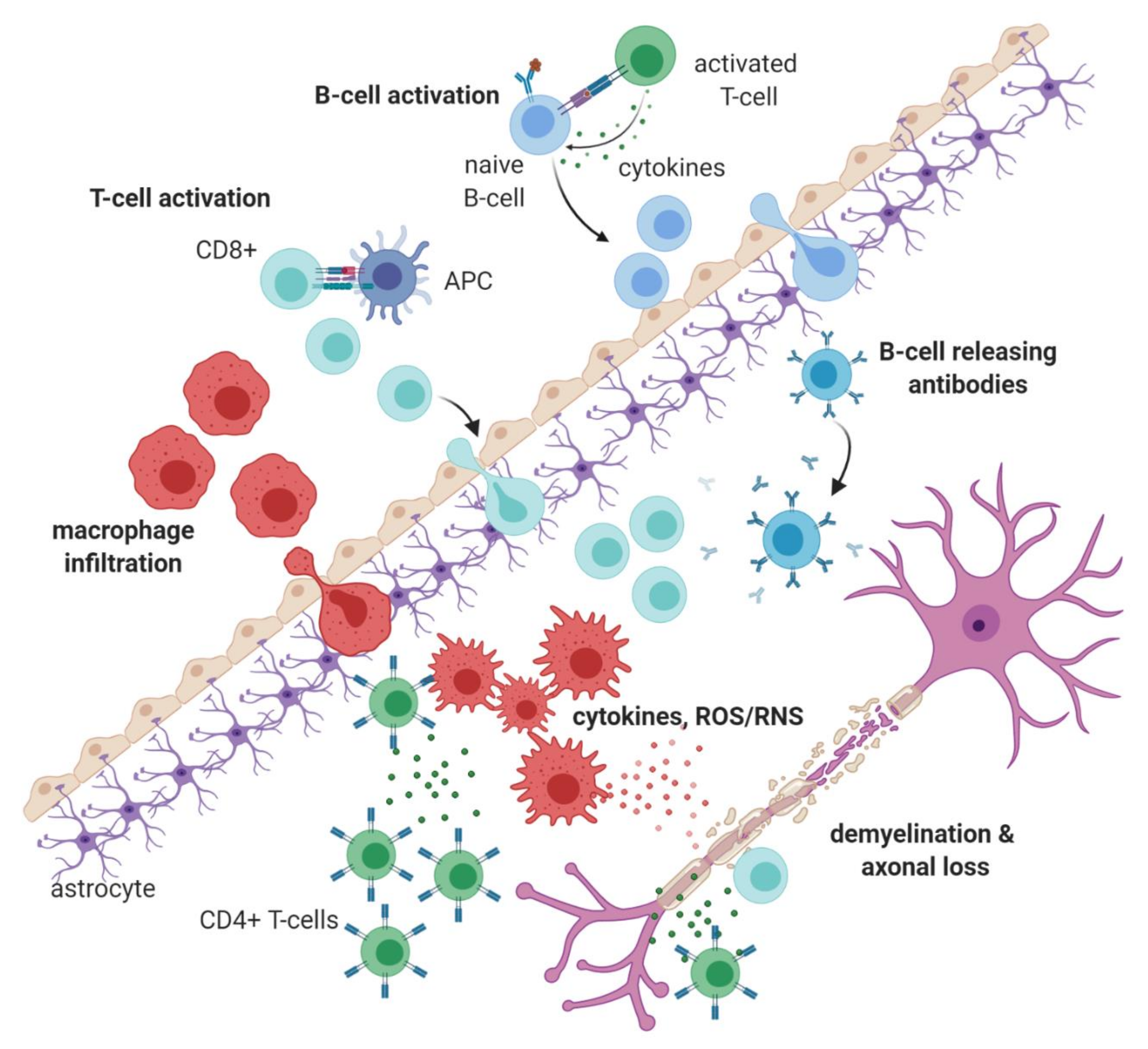
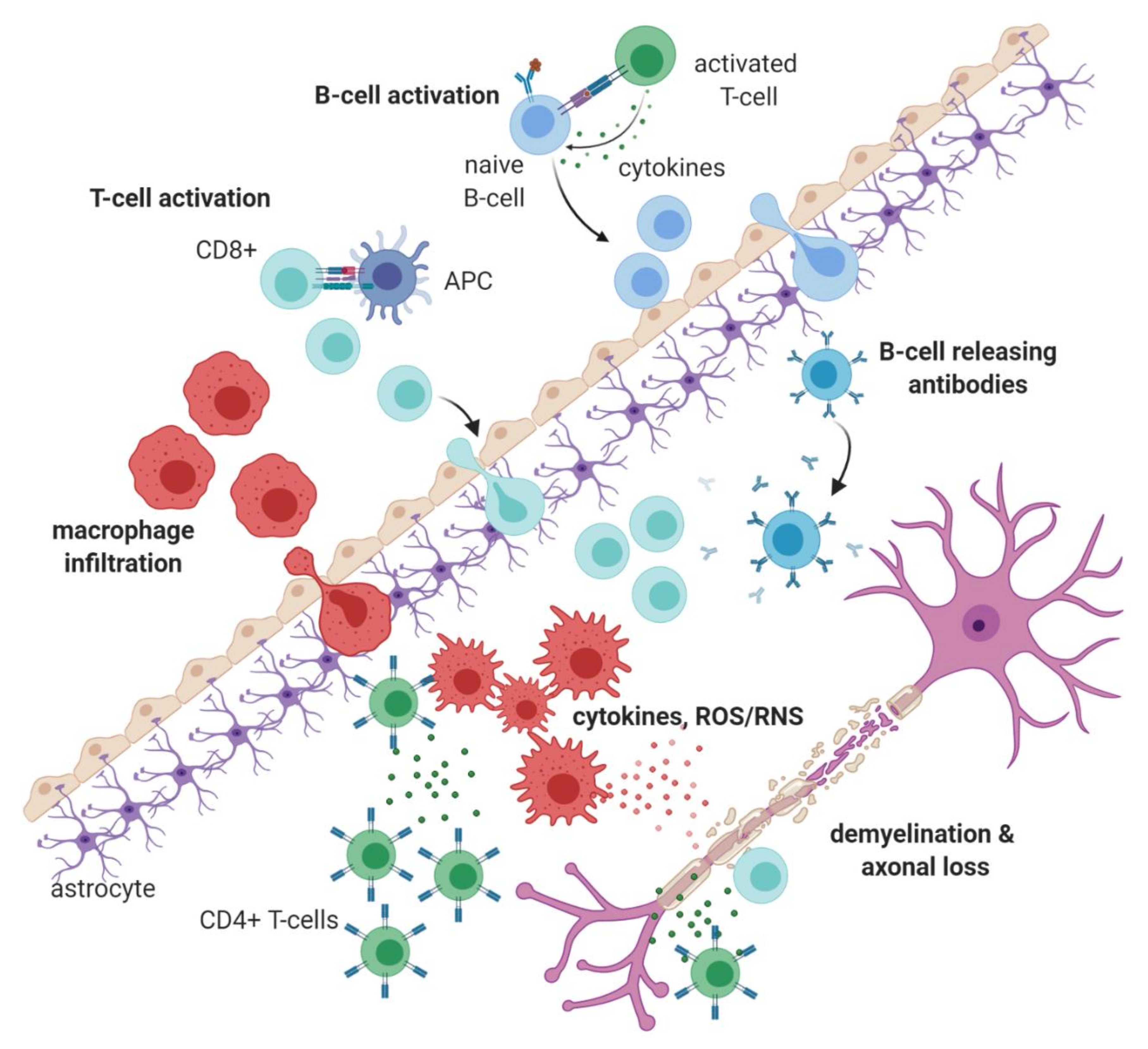
1.1. Immunopathology of Multiple Sclerosis
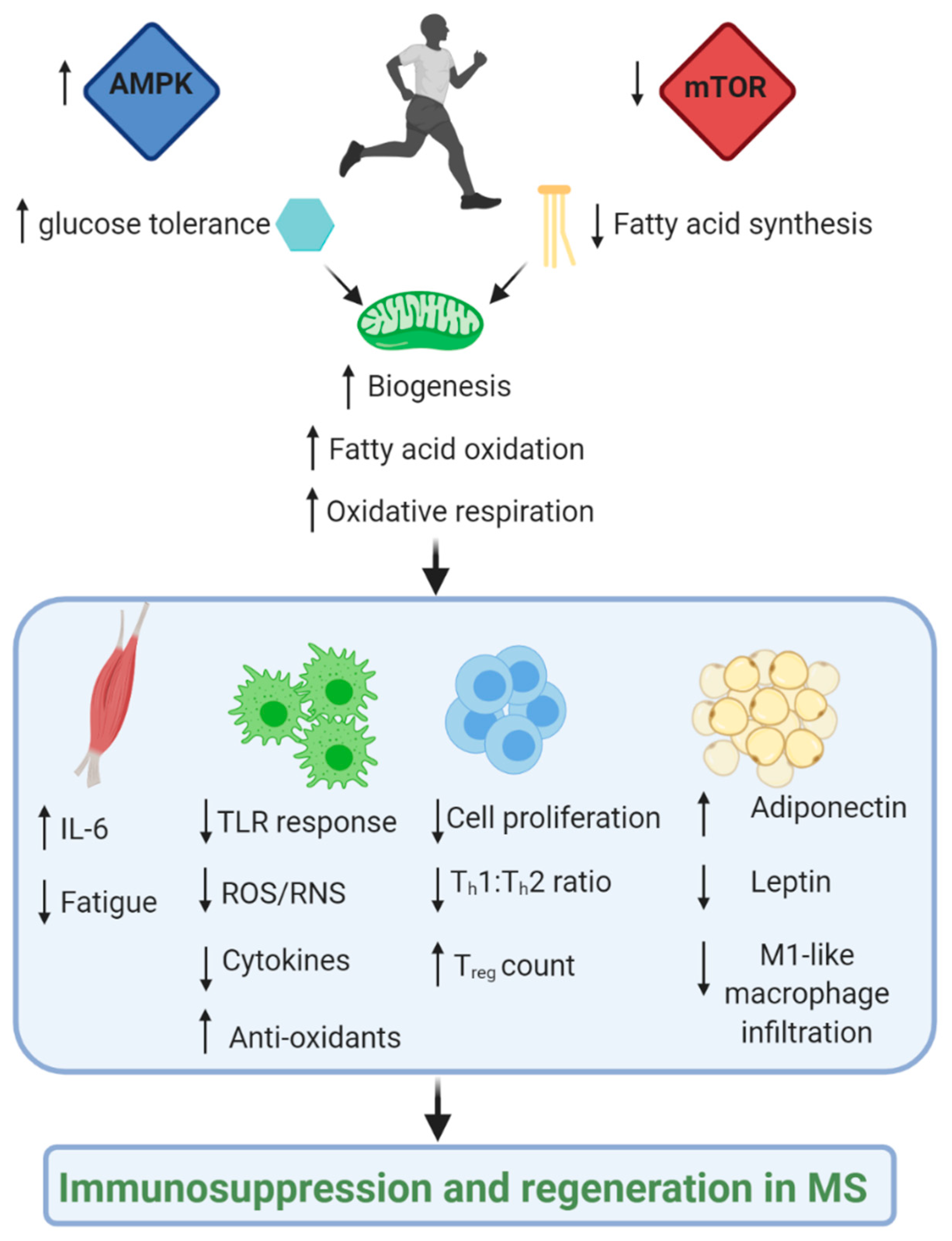
1.2. Exercise in Multiple Sclerosis
2. Exercise as a Regulator of Immunometabolism
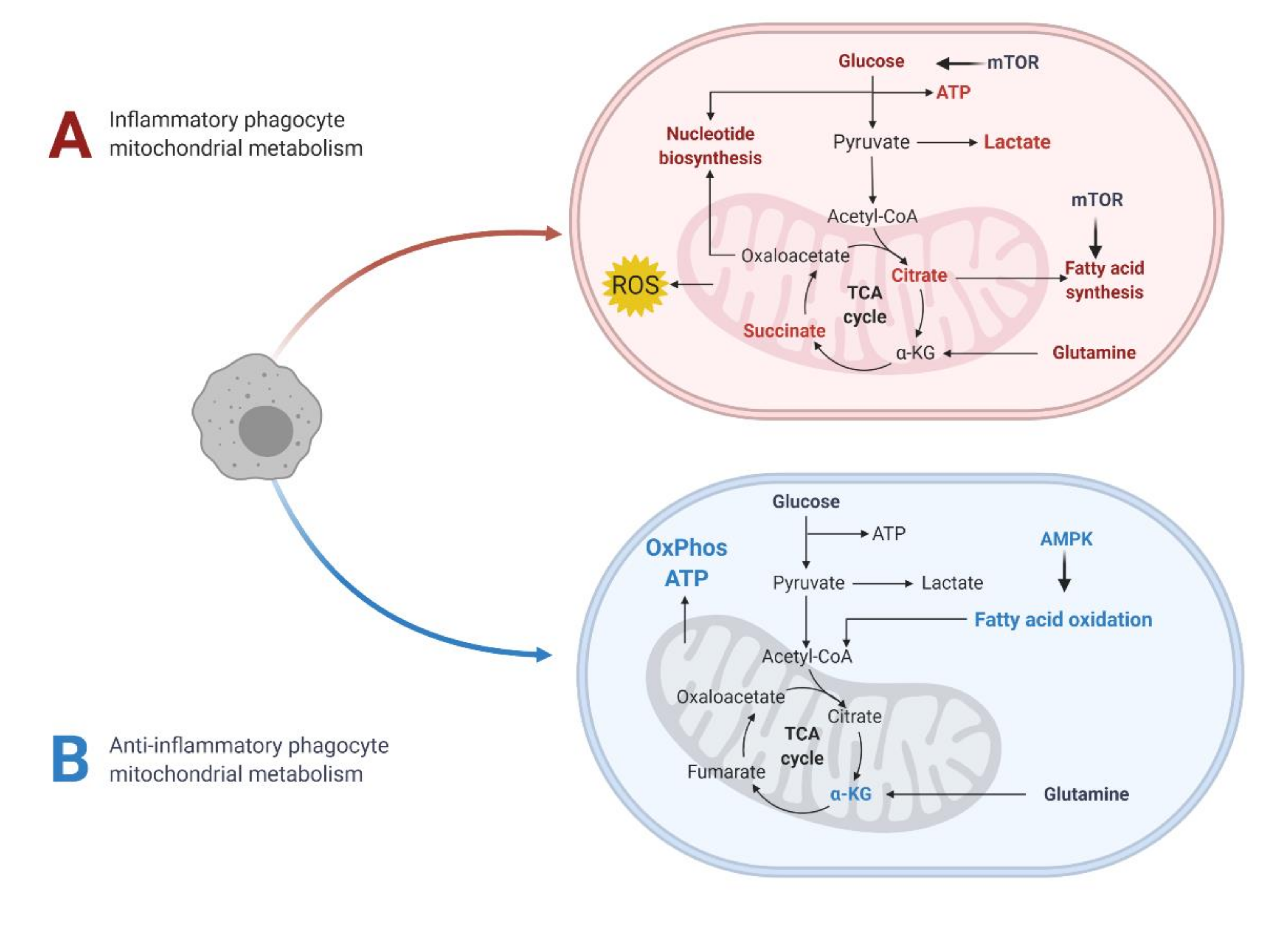
2.1. Overview
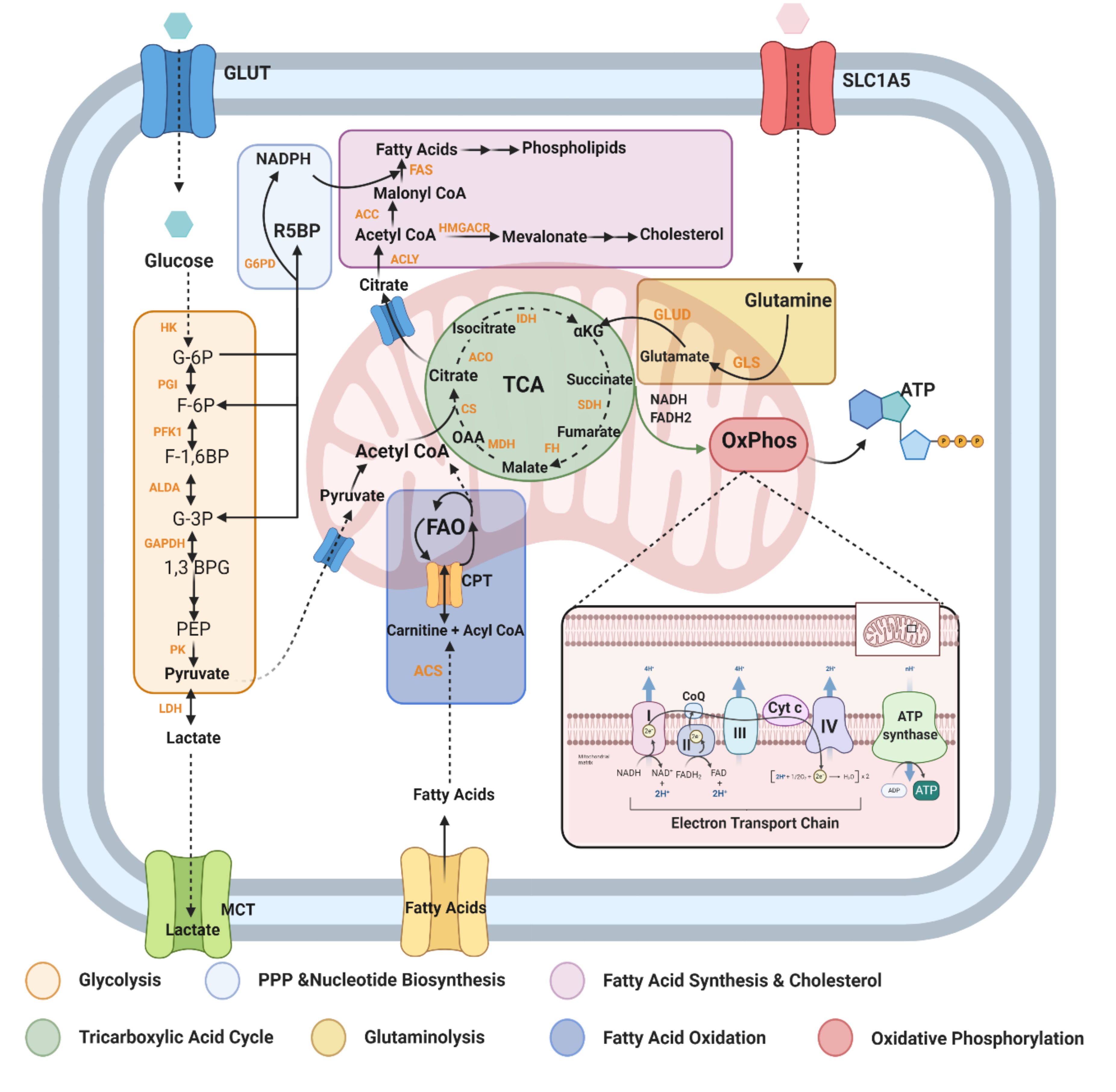
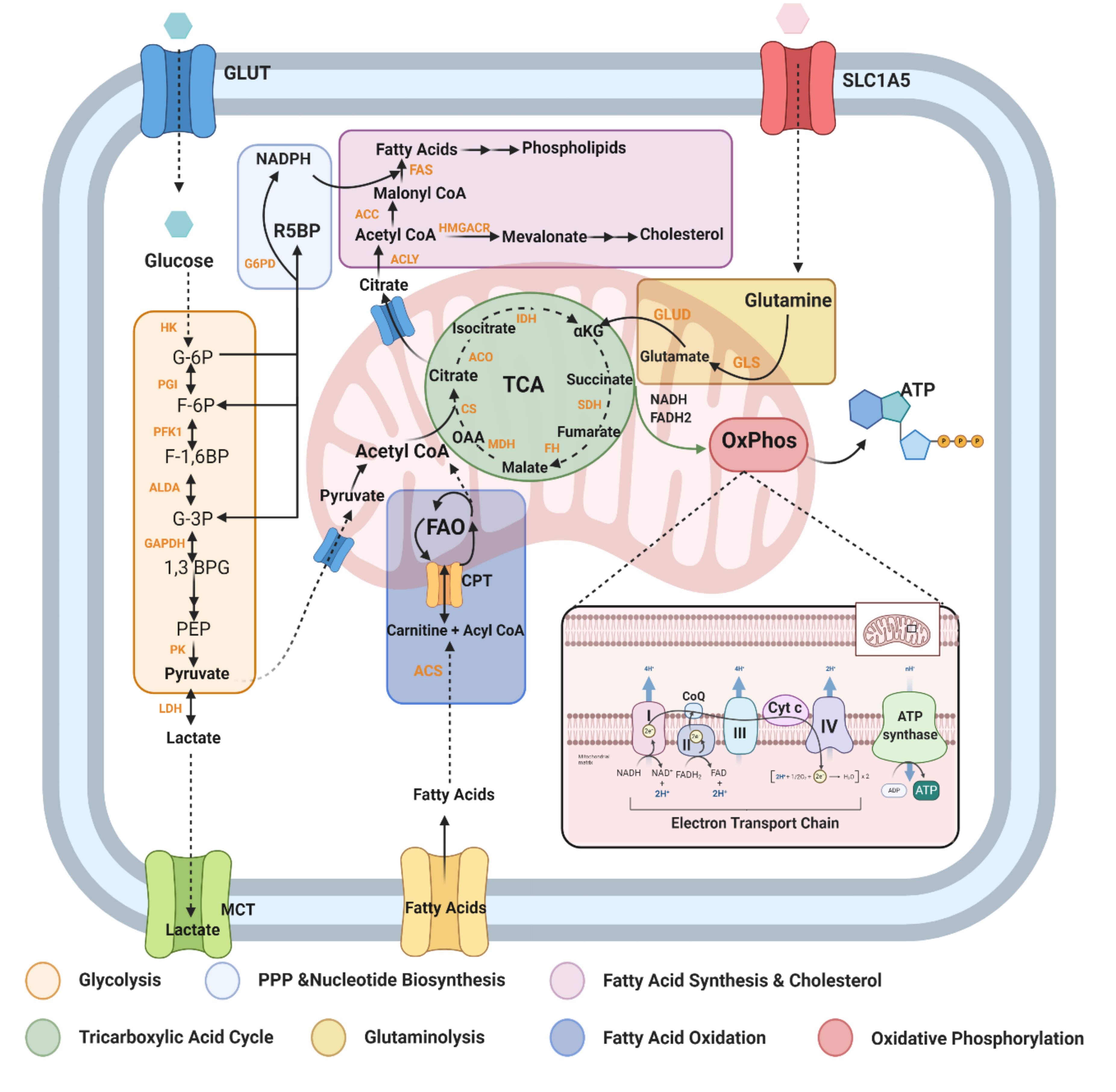
2.2. Metabolic Pathways and Modulators Disrupted in MS
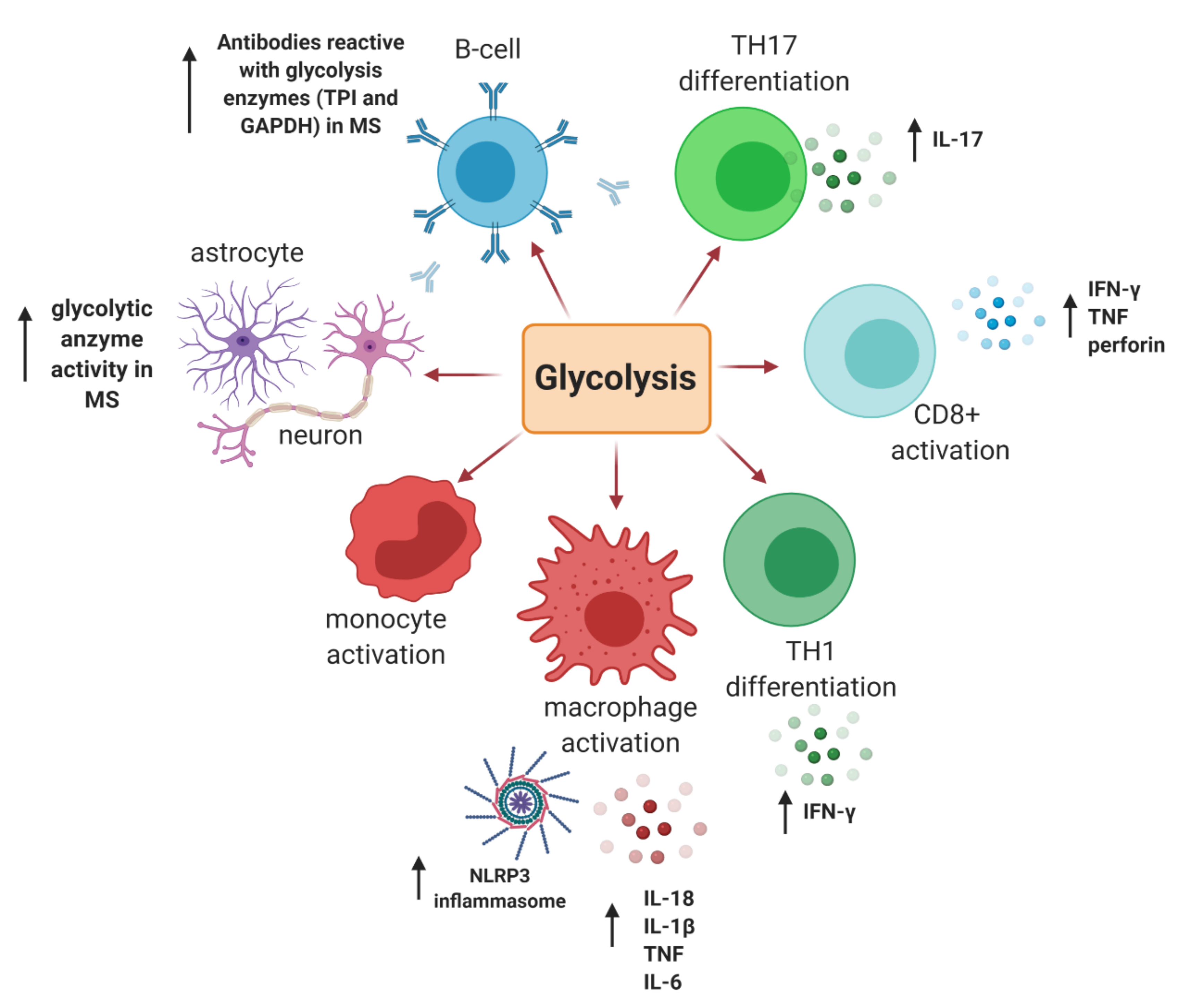
2.2.1. Glycolysis (Glucose Metabolism)
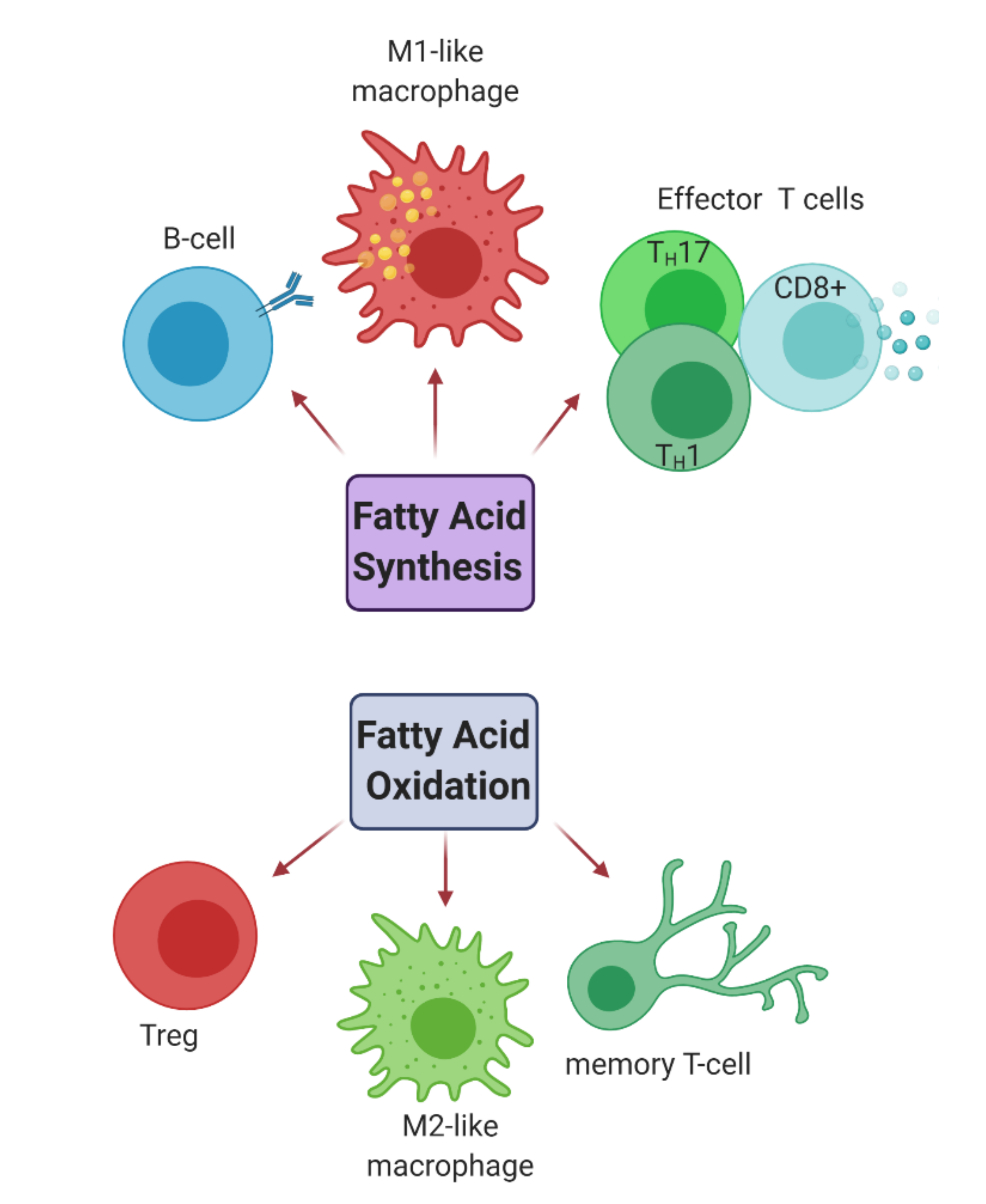
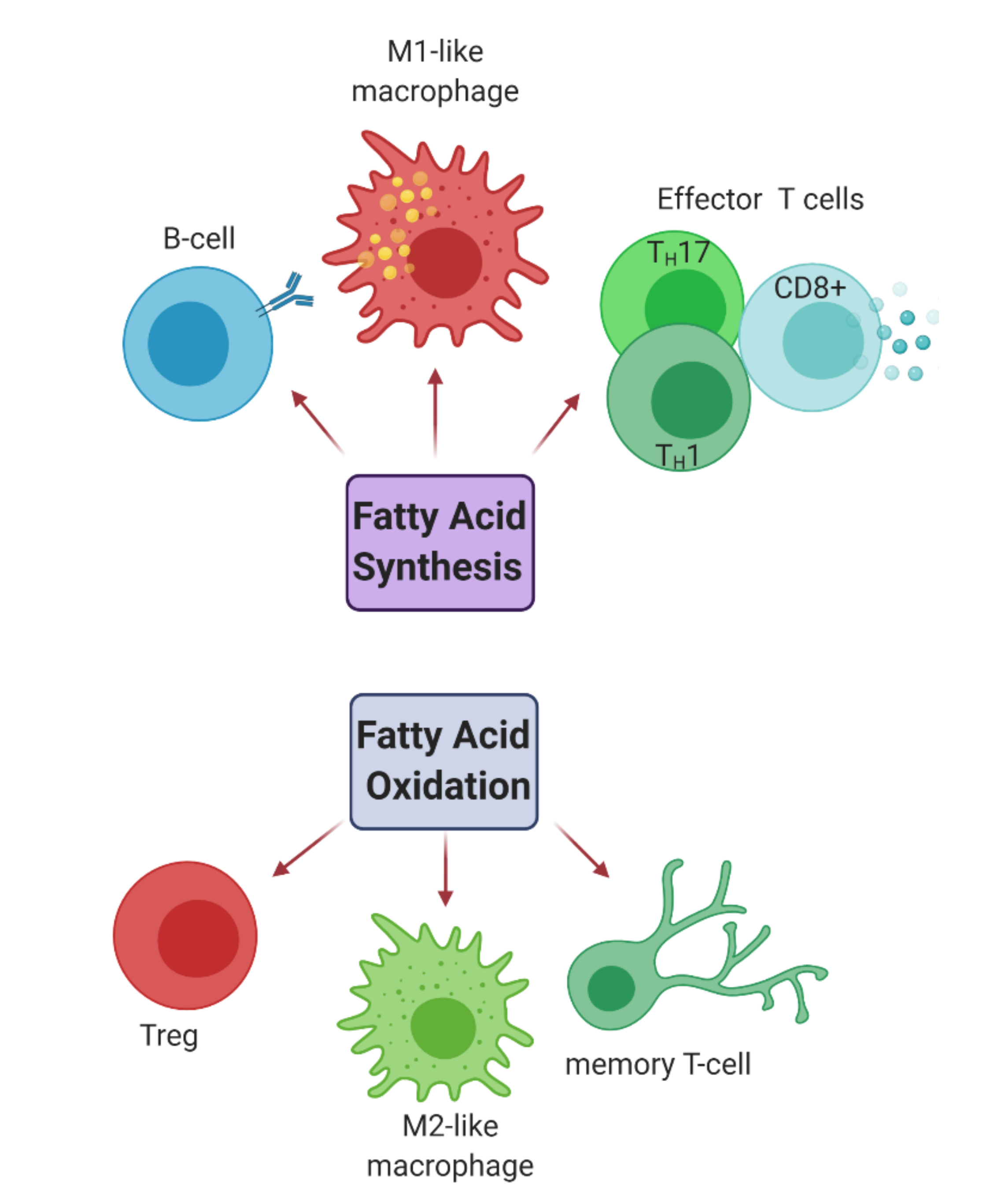
2.2.2. Fatty Acid Metabolism
2.2.3. Mitochondrial Oxidative Stress
2.2.4. Glutamine/Glutamate
2.2.5. mTOR and AMPK: Nutrient Sensing Pathways
2.2.6. Tryptophan/Kynurenine
3. Conclusions
4. Limitations
Author Contributions
Funding
Conflicts of Interest
References
- McCoy, C.E. miR-155 dysregulation and therapeutic intervention in Multiple Sclerosis. Adv. Exp. Med. Bio. 2017, 1024, 111–131. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- White, L.J.; Castellano, V. Exercise and brain health--implications for multiple sclerosis--Part II--immune factors and stress hormones. Sports Med. 2008, 38, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, B.; Rocca, M.R.; Caputo, D.; Zaffaroni, M.; Capra, R.; Bertolotto, A.; Martinelli, V.; Comi, G.; Filippi, M. In-vivo evidence for stable neuroaxonal damage in the brain of patients with benign multiple sclerosis. Mult. Scler. J. 2009, 15, 789–794. [Google Scholar] [CrossRef]
- Levin, M.; Gardner, L.; Douglas, J.; Meyers, L.; Lee, S.; Shin, Y. Neurodegeneration in multiple sclerosis involves multiple pathogenic mechanisms. Degener. Neurol. Neuromuscul. Dis. 2014, 49. [Google Scholar] [CrossRef] [Green Version]
- Ljubisavljevic, S.; Stojanovic, I.; Pavlovic, R.; Sokolovic, D.; Pavlovic, D.; Cvetkovic, T.; Stevanovic, I. Modulation of nitric oxide synthase by arginase and methylated arginines during the acute phase of experimental multiple sclerosis. J. Neurol. Sci. 2012, 318, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.; Lucchinetti, C.F. Pathology of demyelinating diseases. Annu. Rev. Pathol. 2012, 7, 185–217. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; De Rosa, V.; Carrieri, P.B.; Montella, S.; Bruzzese, D.; Porcellini, A.; Procaccini, C.; La Cava, A.; Matarese, G. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat. Med. 2014, 20, 69–74. [Google Scholar] [CrossRef]
- University of California, San Francisco MS-EPIC Team; Cree, B.A.C.; Gourraud, P.-A.; Oksenberg, J.R.; Bevan, C.; Crabtree-Hartman, E.; Gelfand, J.M.; Goodin, D.S.; Graves, J.; Green, A.J.; et al. Long-term evolution of multiple sclerosis disability in the treatment era. Ann. Neurol. 2016, 80, 499–510. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Duffy, C.P.; McCoy, C.E. The Role of MicroRNAs in Repair Processes in Multiple Sclerosis. Cells 2020, 9. [Google Scholar] [CrossRef]
- Nally, F.K.; De Santi, C.; McCoy, C.E. Nanomodulation of Macrophages in Multiple Sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspersen, C.J.; Powell, K.E.; Christenson, G.M. Physical activity, exercise, and physical fitness: Definitions and distinctions for health-related research. Public Health Rep. 1985, 100, 126–131. [Google Scholar]
- Guo, L.Y.; Lozinski, B.; Yong, V.W. Exercise in multiple sclerosis and its models: Focus on the central nervous system outcomes. J. Neurosci. Res. 2020, 98, 509–523. [Google Scholar] [CrossRef]
- van Praag, H. Exercise and the brain: Something to chew on. Trends Neurosci. 2009, 32, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K.; Saltin, B. Exercise as medicine—evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sports 2015, 25 (Suppl. 3), 1–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, D.Y.; Heo, J.W.; Ko, J.R.; Kwak, H.B. Exercise and Neuroinflammation in Health and Disease. Int. Neurourol. J. 2019, 23, S82–S92. [Google Scholar] [CrossRef]
- Sharif, K.; Watad, A.; Bragazzi, N.L.; Lichtbroun, M.; Amital, H.; Shoenfeld, Y. Physical activity and autoimmune diseases: Get moving and manage the disease. Autoimmun. Rev. 2018, 17, 53–72. [Google Scholar] [CrossRef]
- White, L.J.; Dressendorfer, R.H. Exercise and multiple sclerosis. Sports Med. (Auckl. N.Z.) 2004, 34, 1077–1100. [Google Scholar] [CrossRef]
- Klaren, R.E.; Motl, R.W.; Woods, J.A.; Miller, S.D. Effects of exercise in experimental autoimmune encephalomyelitis (an animal model of multiple sclerosis). J. Neuroimmunol. 2014, 274, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, W.M.; Freeman, K.G.; Larson, R.D.; Edwards, G.L.; White, L.J. Chronic exercise confers neuroprotection in experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2015, 93, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.S.; Gonçalves, E.D.; Pedroso, G.S.; Farias, H.R.; Junqueira, S.C.; Marcon, R.; Tuon, T.; Cola, M.; Silveira, P.C.L.; Santos, A.R.; et al. Physical Exercise Attenuates Experimental Autoimmune Encephalomyelitis by Inhibiting Peripheral Immune Response and Blood-Brain Barrier Disruption. Mol. Neurobiol. 2017, 54, 4723–4737. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Li, Z.; Wang, Y.; Xue, X.; Ma, W.; Zhang, Y.; Wang, J. Effects of moderate-versus high-intensity swimming training on inflammatory and CD4(+) T cell subset profiles in experimental autoimmune encephalomyelitis mice. J. Neuroimmunol. 2019, 328, 60–67. [Google Scholar] [CrossRef]
- Negaresh, R.; Motl, R.W.; Mokhtarzade, M.; Dalgas, U.; Patel, D.; Shamsi, M.M.; Majdinasab, N.; Ranjbar, R.; Zimmer, P.; Baker, J.S. Effects of exercise training on cytokines and adipokines in multiple Sclerosis: A systematic review. Mult. Scler. Relat. Disord. 2018, 24, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.J.; Kunz, H.; Agha, N.; Graff, R. Exercise and the Regulation of Immune Functions. Prog. Mol. Biol. Transl. Sci. 2015, 135, 355–380. [Google Scholar] [CrossRef]
- Fainstein, N.; Tyk, R.; Touloumi, O.; Lagoudaki, R.; Goldberg, Y.; Agranyoni, O.; Navon-Venezia, S.; Katz, A.; Grigoriadis, N.; Ben-Hur, T.; et al. Exercise intensity-dependent immunomodulatory effects on encephalomyelitis. Ann. Clin. Transl. Neurol. 2019, 6, 1647–1658. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-W.; Sung, Y.-H. Regular exercise promotes memory function and enhances hippocampal neuroplasticity in experimental autoimmune encephalomyelitis mice. Neuroscience 2017, 346, 173–181. [Google Scholar] [CrossRef]
- Dorans, K.S.; Massa, J.; Chitnis, T.; Ascherio, A.; Munger, K.L. Physical activity and the incidence of multiple sclerosis. Neurology 2016, 87, 1770. [Google Scholar] [CrossRef] [Green Version]
- Kraszula, L.; Jasińska, A.; Eusebio, M.; Kuna, P.; Głąbiński, A.; Pietruczuk, M. Evaluation of the relationship between leptin, resistin, adiponectin and natural regulatory T cells in relapsing-remitting multiple sclerosis. Neurol. I Neurochir. Pol. 2012, 46, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Matarese, G.; Carrieri, P.B.; La Cava, A.; Perna, F.; Sanna, V.; De Rosa, V.; Aufiero, D.; Fontana, S.; Zappacosta, S. Leptin increase in multiple sclerosis associates with reduced number of CD4(+)CD25+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5150–5155. [Google Scholar] [CrossRef] [Green Version]
- Mokhtarzade, M.; Ranjbar, R.; Majdinasab, N.; Patel, D.; Molanouri Shamsi, M. Effect of aerobic interval training on serum IL-10, TNFalpha, and adipokines levels in women with multiple sclerosis: Possible relations with fatigue and quality of life. Endocrine 2017, 57, 262–271. [Google Scholar] [CrossRef]
- Barry, A.; Cronin, O.; Ryan, A.M.; Sweeney, B.; Yap, S.M.; O’Toole, O.; Allen, A.P.; Clarke, G.; O’Halloran, K.D.; Downer, E.J. Impact of Exercise on Innate Immunity in Multiple Sclerosis Progression and Symptomatology. Front. Physiol. 2016, 7, 194. [Google Scholar] [CrossRef] [PubMed]
- Castellano, V.; Patel, D.I.; White, L.J. Cytokine responses to acute and chronic exercise in multiple sclerosis. J. Appl. Physiol. (1985) 2008, 104, 1697–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjolhede, T.; Dalgas, U.; Gade, A.B.; Bjerre, M.; Stenager, E.; Petersen, T.; Vissing, K. Acute and chronic cytokine responses to resistance exercise and training in people with multiple sclerosis. Scand. J. Med. Sci. Sports 2016, 26, 824–834. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Gaber, T.; Strehl, C.; Sawitzki, B.; Hoff, P.; Buttgereit, F. Cellular energy metabolism in T-lymphocytes. Int. Rev. Immunol. 2015, 34, 34–49. [Google Scholar] [CrossRef]
- Tänzer, A. Molecular Mechanisms of Immunometabolic Dysfunction in Multiple Sclerosis. Ph.D. Thesis, Humboldt-Universität zu Berlin, Berlin, Germany, 2019. [Google Scholar]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Olenchock, B.A.; Rathmell, J.C.; Vander Heiden, M.G. Biochemical Underpinnings of Immune Cell Metabolic Phenotypes. Immunity 2017, 46, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanely, R.A.; Nieman, D.C.; Henson, D.A.; Jin, F.; Knab, A.M.; Sha, W. Inflammation and oxidative stress are lower in physically fit and active adults. Scand. J. Med. Sci. Sports 2013, 23, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Wedell-Neergaard, A.S.; Lang Lehrskov, L.; Christensen, R.H.; Legaard, G.E.; Dorph, E.; Larsen, M.K.; Launbo, N.; Fagerlind, S.R.; Seide, S.K.; Nymand, S.; et al. Exercise-Induced Changes in Visceral Adipose Tissue Mass Are Regulated by IL-6 Signaling: A Randomized Controlled Trial. Cell Metab. 2019, 29, 844–855.e843. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.D.; Farrell, L.; Wood, M.J.; Martinovic, M.; Arany, Z.; Rowe, G.C.; Souza, A.; Cheng, S.; McCabe, E.L.; Yang, E.; et al. Metabolic signatures of exercise in human plasma. Sci. Transl. Med. 2010, 2, 33ra37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, R.; Zhao, X.; Weigert, C.; Simon, P.; Fehrenbach, E.; Fritsche, J.; Machann, J.; Schick, F.; Wang, J.; Hoene, M.; et al. Medium chain acylcarnitines dominate the metabolite pattern in humans under moderate intensity exercise and support lipid oxidation. PLoS ONE 2010, 5, e11519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, R.J.; McFarlin, B.K.; McSporran, C.; Spielmann, G.; ó Hartaigh, B.; Guy, K. Toll-like receptor expression on classic and pro-inflammatory blood monocytes after acute exercise in humans. Brain Behav. Immun. 2009, 23, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.; McFarlin, B.; Flynn, M. Exercise and Toll-like receptors. Exerc. Immunol. Rev. 2006, 12, 34–53. [Google Scholar] [PubMed]
- Timmerman, K.L.; Flynn, M.G.; Coen, P.M.; Markofski, M.M.; Pence, B.D. Exercise training-induced lowering of inflammatory (CD14+CD16+) monocytes: A role in the anti-inflammatory influence of exercise? J. Leukoc. Biol. 2008, 84, 1271–1278. [Google Scholar] [CrossRef]
- Kawanishi, N.; Yano, H.; Yokogawa, Y.; Suzuki, K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc. Immunol. Rev. 2010, 16, 105–118. [Google Scholar]
- Kawanishi, N.; Mizokami, T.; Yano, H.; Suzuki, K. Exercise attenuates M1 macrophages and CD8+ T cells in the adipose tissue of obese mice. Med. Sci. Sports Exerc. 2013, 45, 1684–1693. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Kiernan, K.; MacIver, N.J. Changes in Nutritional Status Impact Immune Cell Metabolism and Function. Front. Immunol. 2018, 9, 1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsholme, P.; Curi, R.; Gordon, S.; Newsholme, E.A. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J. 1986, 239, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Diskin, C.; Palsson-McDermott, E.M. Metabolic Modulation in Macrophage Effector Function. Front. Immunol. 2018, 9, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langston, P.K.; Shibata, M.; Horng, T. Metabolism Supports Macrophage Activation. Front. Immunol. 2017, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.S.; Hisata, S.; Park, M.A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.H.; Jones, H.H., Jr.; Bunch, L.D. Biochemical studies in multiple sclerosis. Ann. Intern. Med. 1950, 33, 831–840. [Google Scholar] [CrossRef]
- Royds, J.A.; Timperley, W.R.; Taylor, C.B. Levels of enolase and other enzymes in the cerebrospinal fluid as indices of pathological change. J. Neurol. Neurosurg. Psychiatry 1981, 44, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Nijland, P.G.; Molenaar, R.J.; van der Pol, S.M.A.; van der Valk, P.; van Noorden, C.J.F.; de Vries, H.E.; van Horssen, J. Differential expression of glucose-metabolizing enzymes in multiple sclerosis lesions. Acta Neuropathol. Commun. 2015, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Radu, C.G.; Shu, C.J.; Shelly, S.M.; Phelps, M.E.; Witte, O.N. Positron emission tomography with computed tomography imaging of neuroinflammation in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2007, 104, 1937. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.R.; Simão, A.N.; Kallaur, A.P.; de Almeida, E.R.; Morimoto, H.K.; Lopes, J.; Dichi, I.; Kaimen-Maciel, D.R.; Reiche, E.M. Disability in patients with multiple sclerosis: influence of insulin resistance, adiposity, and oxidative stress. Nutrition 2014, 30, 268–273. [Google Scholar] [CrossRef]
- Ruiz-Argüelles, A.; Méndez-Huerta, M.A.; Lozano, C.D.; Ruiz-Argüelles, G.J. Metabolomic profile of insulin resistance in patients with multiple sclerosis is associated to the severity of the disease. Mult. Scler. Relat. Disord. 2018, 25, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Wens, I.; Dalgas, U.; Deckx, N.; Cools, N.; Eijnde, B.O. Does multiple sclerosis affect glucose tolerance? Mult. Scler. 2014, 20, 1273–1276. [Google Scholar] [CrossRef] [PubMed]
- Kolln, J.; Ren, H.M.; Da, R.R.; Zhang, Y.; Spillner, E.; Olek, M.; Hermanowicz, N.; Hilgenberg, L.G.; Smith, M.A.; van den Noort, S.; et al. Triosephosphate isomerase- and glyceraldehyde-3-phosphate dehydrogenase-reactive autoantibodies in the cerebrospinal fluid of patients with multiple sclerosis. J. Immunol. 2006, 177, 5652–5658. [Google Scholar] [CrossRef] [Green Version]
- La Rocca, C.; Carbone, F.; De Rosa, V.; Colamatteo, A.; Galgani, M.; Perna, F.; Lanzillo, R.; Brescia Morra, V.; Orefice, G.; Cerillo, I.; et al. Immunometabolic profiling of T cells from patients with relapsing-remitting multiple sclerosis reveals an impairment in glycolysis and mitochondrial respiration. Metabolism 2017, 77, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Riccardis, L.; Rizzello, A.; Ferramosca, A.; Urso, E.; De Robertis, F.; Danieli, A.; Giudetti, A.M.; Trianni, G.; Zara, V.; Maffia, M. Bioenergetics profile of CD4(+) T cells in relapsing remitting multiple sclerosis subjects. J. Biotechnol. 2015, 202, 31–39. [Google Scholar] [CrossRef]
- Kaushik, D.K.; Bhattacharya, A.; Mirzaei, R.; Rawji, K.S.; Ahn, Y.; Rho, J.M.; Yong, V.W. Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis. J. Clin. Investig. 2019, 129, 3277–3292. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Nagasaki, M.; Nakai, N.; Fushimi, T. Physical Exercise Improves Glucose Metabolism in Lifestyle-Related Diseases. Exp. Biol. Med. 2003, 228, 1208–1212. [Google Scholar] [CrossRef]
- Batatinha, H.A.P.; Biondo, L.A.; Lira, F.S.; Castell, L.M.; Rosa-Neto, J.C. Nutrients, immune system, and exercise: Where will it take us? Nutrition 2019, 61, 151–156. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Toft, A.D. Effects of exercise on lymphocytes and cytokines. Br. J. Sports Med. 2000, 34, 246. [Google Scholar] [CrossRef] [Green Version]
- Wens, I.; Dalgas, U.; Vandenabeele, F.; Verboven, K.; Hansen, D.; Deckx, N.; Cools, N.; Eijnde, B.O. High Intensity Aerobic and Resistance Exercise Can Improve Glucose Tolerance in Persons With Multiple Sclerosis: A Randomized Controlled Trial. Am. J. Phys. Med. Rehabil. 2017, 96, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.J.; Haidar, M.; Kooij, G.; Hendriks, J.J.A. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Micioni Di Bonaventura, M.V.; Gaetani, S.; Giudetti, A.M. Fats for thoughts: An update on brain fatty acid metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Houdebine, L.; Gallelli, C.A.; Rastelli, M.; Sampathkumar, N.K.; Grenier, J. Effect of physical exercise on brain and lipid metabolism in mouse models of multiple sclerosis. Chem. Phys. Lipids 2017, 207, 127–134. [Google Scholar] [CrossRef]
- Duc, D.; Vigne, S.; Pot, C. Oxysterols in Autoimmunity. Int. J. Mol. Sci. 2019, 20, 4522. [Google Scholar] [CrossRef] [Green Version]
- Teunissen, C.E.; Dijkstra, C.D.; Polman, C.H.; Hoogervorst, E.L.; von Bergmann, K.; Lütjohann, D. Decreased levels of the brain specific 24S-hydroxycholesterol and cholesterol precursors in serum of multiple sclerosis patients. Neurosci. Lett. 2003, 347, 159–162. [Google Scholar] [CrossRef]
- Moutinho, M.; Nunes, M.J.; Rodrigues, E. Cholesterol 24-hydroxylase: Brain cholesterol metabolism and beyond. Biochim. Biophys. Acta 2016, 1861, 1911–1920. [Google Scholar] [CrossRef]
- Novakova, L.; Axelsson, M.; Malmeström, C.; Zetterberg, H.; Björkhem, I.; Karrenbauer, V.D.; Lycke, J. Reduced cerebrospinal fluid concentrations of oxysterols in response to natalizumab treatment of relapsing remitting multiple sclerosis. J. Neurol. Sci. 2015, 358, 201–206. [Google Scholar] [CrossRef]
- Diestel, A.; Aktas, O.; Hackel, D.; Hake, I.; Meier, S.; Raine, C.S.; Nitsch, R.; Zipp, F.; Ullrich, O. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: A link between demyelination and neuronal damage. J. Exp. Med. 2003, 198, 1729–1740. [Google Scholar] [CrossRef] [Green Version]
- Rosklint, T.; Ohlsson, B.G.; Wiklund, O.; Norén, K.; Hultén, L.M. Oxysterols induce interleukin-1beta production in human macrophages. Eur. J. Clin. Investig. 2002, 32, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bähre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Torres-Gonzalez, M.; Olson, L.K. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem. Pharmacol. 2011, 81, 649–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidan, A.; Hedya, S.E.; Elfeky, D.M.; Abdin, A.A. The possible anti-apoptotic and antioxidant effects of acetyl l-carnitine as an add-on therapy on a relapsing-remitting model of experimental autoimmune encephalomyelitis in rats. Biomed. Pharmacother. 2018, 103, 1302–1311. [Google Scholar] [CrossRef]
- Tomassini, V.; Pozzilli, C.; Onesti, E.; Pasqualetti, P.; Marinelli, F.; Pisani, A.; Fieschi, C. Comparison of the effects of acetyl L-carnitine and amantadine for the treatment of fatigue in multiple sclerosis: Results of a pilot, randomised, double-blind, crossover trial. J. Neurol. Sci. 2004, 218, 103–108. [Google Scholar] [CrossRef]
- Athanassakis, I.; Mouratidou, M.; Sakka, P.; Evangeliou, A.; Spilioti, M.; Vassiliadis, S. l-carnitine modifies the humoral immune response in mice after in vitro or in vivo treatment. Int. Immunopharmacol. 2001, 1, 1813–1822. [Google Scholar] [CrossRef]
- Powell, B.R.; Kennaway, N.G.; Rhead, W.J.; Reece, C.J.; Burlingame, T.G.; Buist, N.R. Juvenile multiple sclerosis-like episodes associated with a defect of mitochondrial beta oxidation. Neurology 1990, 40, 487–491. [Google Scholar] [CrossRef]
- Youssef, S.; Stüve, O.; Patarroyo, J.C.; Ruiz, P.J.; Radosevich, J.L.; Hur, E.M.; Bravo, M.; Mitchell, D.J.; Sobel, R.A.; Steinman, L.; et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 2002, 420, 78–84. [Google Scholar] [CrossRef]
- Howie, D.; Ten Bokum, A.; Necula, A.S.; Cobbold, S.P.; Waldmann, H. The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front. Immunol. 2018, 8, 1949. [Google Scholar] [CrossRef]
- Melanson, E.L.; MacLean, P.S.; Hill, J.O. Exercise improves fat metabolism in muscle but does not increase 24-h fat oxidation. Exerc. Sport Sci. Rev. 2009, 37, 93–101. [Google Scholar] [CrossRef]
- Achten, J.; Jeukendrup, A.E. Optimizing fat oxidation through exercise and diet. Nutrition 2004, 20, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Jeukendrup, A.E. Regulation of fat metabolism in skeletal muscle. Ann. New York Acad. Sci. 2002, 967, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, H.M.; Holloway, G.P.; Steinberg, G.R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 2013, 366, 135–151. [Google Scholar] [CrossRef]
- Nieman, D.C.; Gillitt, N.D.; Sha, W.; Esposito, D.; Ramamoorthy, S. Metabolic recovery from heavy exertion following banana compared to sugar beverage or water only ingestion: A randomized, crossover trial. PLoS ONE 2018, 13, e0194843. [Google Scholar] [CrossRef] [Green Version]
- Nieman, D.C.; Lila, M.A.; Gillitt, N.D. Immunometabolism: A Multi-Omics Approach to Interpreting the Influence of Exercise and Diet on the Immune System. Annu. Rev. Food Sci. Technol. 2019, 10, 341–363. [Google Scholar] [CrossRef]
- Liepinsh, E.; Makarova, E.; Plakane, L.; Konrade, I.; Liepins, K.; Videja, M.; Sevostjanovs, E.; Grinberga, S.; Makrecka-Kuka, M.; Dambrova, M. Low-intensity exercise stimulates bioenergetics and increases fat oxidation in mitochondria of blood mononuclear cells from sedentary adults. Physiol. Rep. 2020, 8, e14489. [Google Scholar] [CrossRef]
- Mähler, A.; Steiniger, J.; Bock, M.; Brandt, A.U.; Haas, V.; Boschmann, M.; Paul, F. Is Metabolic Flexibility Altered in Multiple Sclerosis Patients? PLoS ONE 2012, 7, e43675. [Google Scholar] [CrossRef]
- Hansen, D.; Dendale, P.; van Loon, L.J.; Meeusen, R. The impact of training modalities on the clinical benefits of exercise intervention in patients with cardiovascular disease risk or type 2 diabetes mellitus. Sports Med. (Auckl. N.Z.) 2010, 40, 921–940. [Google Scholar] [CrossRef]
- Jorissen, W.; Vanmierlo, T.; Wens, I.; Somers, V.; Van Wijmeersch, B.; Bogie, J.F.; Remaley, A.T.; Eijnde, B.O.; Hendriks, J.J.A. Twelve Weeks of Medium-Intensity Exercise Therapy Affects the Lipoprotein Profile of Multiple Sclerosis Patients. Int. J. Mol. Sci. 2018, 19, 193. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Correa-da-Silva, F.; Pereira, J.A.S.; de Aguiar, C.F.; de Moraes-Vieira, P.M.M. Mitoimmunity-when mitochondria dictates macrophage function. Cell Biol. Int. 2018, 42, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Selak, M.; O’Connor, J.; Croul, S.; Lorenzana, C.; Butunoi, C.; Kalman, B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J. Neurol. Sci. 2000, 177, 95–103. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Recks, M.S.; Stormanns, E.R.; Bader, J.; Arnhold, S.; Addicks, K.; Kuerten, S. Early axonal damage and progressive myelin pathology define the kinetics of CNS histopathology in a mouse model of multiple sclerosis. Clin. Immunol. 2013, 149, 32–45. [Google Scholar] [CrossRef]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef]
- Kent-Braun, J.A.; Ng, A.V.; Castro, M.; Weiner, M.W.; Gelinas, D.; Dudley, G.A.; Miller, R.G. Strength, skeletal muscle composition, and enzyme activity in multiple sclerosis. J. Appl. Physiol. 1997, 83, 1998–2004. [Google Scholar] [CrossRef]
- di Penta, A.; Moreno, B.; Reix, S.; Fernandez-Diez, B.; Villanueva, M.; Errea, O.; Escala, N.; Vandenbroeck, K.; Comella, J.X.; Villoslada, P. Oxidative stress and proinflammatory cytokines contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation. PLoS ONE 2013, 8, e54722. [Google Scholar] [CrossRef] [Green Version]
- Malkiewicz, M.A.; Szarmach, A.; Sabisz, A.; Cubala, W.J.; Szurowska, E.; Winklewski, P.J. Blood-brain barrier permeability and physical exercise. J. Neuroinflammation. 2019, 16, 15. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Ng, X.; Sadeghian, M.; Heales, S.; Hargreaves, I.P. Assessment of Mitochondrial Dysfunction in Experimental Autoimmune Encephalomyelitis (EAE) Models of Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 4975. [Google Scholar] [CrossRef] [Green Version]
- Radak, Z.; Suzuki, K.; Higuchi, M.; Balogh, L.; Boldogh, I.; Koltai, E. Physical exercise, reactive oxygen species and neuroprotection. Free Radic. Biol. Med. 2016, 98, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.; Vadnal, J.; Houston, S.; Freeman, E.; McDonough, J. Impaired regulation of electron transport chain subunit genes by nuclear respiratory factor 2 in multiple sclerosis. J. Neurol. Sci. 2009, 279, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Nijland, P.G.; Drexhage, J.A.R.; Gerritsen, W.; Geerts, D.; van het Hof, B.; Reijerkerk, A.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Reduced expression of PGC-1α partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol. 2013, 125, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Malfroy, B.; Doctrow, S.R.; Orr, P.L.; Tocco, G.; Fedoseyeva, E.V.; Benichou, G. Prevention and suppression of autoimmune encephalomyelitis by EUK-8, a synthetic catalytic scavenger of oxygen-reactive metabolites. Cell. Immunol. 1997, 177, 62–68. [Google Scholar] [CrossRef]
- Guy, J.; McGorray, S.; Fitzsimmons, J.; Beck, B.; Mancuso, A.; Rao, N.A.; Hamed, L. Reversals of blood-brain barrier disruption by catalase: A serial magnetic resonance imaging study of experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3456–3465. [Google Scholar]
- Qi, X.; Hauswirth, W.W.; Guy, J. Dual gene therapy with extracellular superoxide dismutase and catalase attenuates experimental optic neuritis. Mol. Vis. 2007, 13, 1–11. [Google Scholar]
- McGuire, V.A.; Ruiz-Zorrilla Diez, T.; Emmerich, C.H.; Strickson, S.; Ritorto, M.S.; Sutavani, R.V.; Weiβ, A.; Houslay, K.F.; Knebel, A.; Meakin, P.J.; et al. Dimethyl fumarate blocks pro-inflammatory cytokine production via inhibition of TLR induced M1 and K63 ubiquitin chain formation. Sci. Rep. 2016, 6, 31159. [Google Scholar] [CrossRef] [Green Version]
- Boveris, A.; Navarro, A. Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free Radic. Biol. Med. 2008, 44, 224–229. [Google Scholar] [CrossRef]
- E, L.; Burns, J.M.; Swerdlow, R.H. Effect of high-intensity exercise on aged mouse brain mitochondria, neurogenesis, and inflammation. Neurobiol. Aging. 2014, 35, 2574–2583. [Google Scholar] [CrossRef] [Green Version]
- Leick, L.; Lyngby, S.S.; Wojtasewski, J.F.P.; Pilegaard, H. PGC-1α is required for training-induced prevention of age-associated decline in mitochondrial enzymes in mouse skeletal muscle. Exp. Gerontol. 2010, 45, 336–342. [Google Scholar] [CrossRef]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.P.; Blannin, A.K.; Robson, P.J.; Gleeson, M. Glutamine, Exercise, and Immune Function. Sports Med. 1998, 26, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Manev, H.; Favaron, M.; Guidotti, A.; Costa, E. Delayed increase of Ca2+ influx elicited by glutamate: Role in neuronal death. Mol. Pharmacol. 1989, 36, 106–112. [Google Scholar] [PubMed]
- Smaili, S.S.; Ureshino, R.P.; Rodrigues, L.; Rocha, K.K.; Carvalho, J.T.; Oseki, K.T.; Bincoletto, C.; Lopes, G.S.; Hirata, H. The role of mitochondrial function in glutamate-dependent metabolism in neuronal cells. Curr. Pharm. Des. 2011, 17, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Tisell, A.; Leinhard, O.D.; Warntjes, J.B.; Aalto, A.; Smedby, Ö.; Landtblom, A.M.; Lundberg, P. Increased concentrations of glutamate and glutamine in normal-appearing white matter of patients with multiple sclerosis and normal MR imaging brain scans. PLoS ONE 2013, 8, e61817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrez, R.; Stys, P.K.; Vivien, D.; Lipton, S.A.; Docagne, F. Mechanisms of glutamate toxicity in multiple sclerosis: Biomarker and therapeutic opportunities. Lancet Neurol. 2016, 15, 1089–1102. [Google Scholar] [CrossRef]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef]
- Thurtell, M.J.; Joshi, A.C.; Leone, A.C.; Tomsak, R.L.; Kosmorsky, G.S.; Stahl, J.S.; Leigh, R.J. Crossover trial of gabapentin and memantine as treatment for acquired nystagmus. Ann. Neurol. 2010, 67, 676–680. [Google Scholar] [CrossRef] [Green Version]
- Metzler, B.; Gfeller, P.; Guinet, E. Restricting Glutamine or Glutamine-Dependent Purine and Pyrimidine Syntheses Promotes Human T Cells with High FOXP3 Expression and Regulatory Properties. J. Immunol. 2016, 196, 3618. [Google Scholar] [CrossRef] [Green Version]
- Castell, L. Glutamine supplementation in vitro and in vivo, in exercise and in immunodepression. Sports Med. 2003, 33, 323–345. [Google Scholar] [CrossRef]
- Agostini, F.; Biolo, G. Effect of physical activity on glutamine metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, N.P.; Blannin, A.K.; Clark, A.M.; Cook, L.; Robson, P.J.; Gleeson, M. The effects of high-intensity intermittent exercise on the plasma concentrations of glutamine and organic acids. Eur. J. Appl. Physiol. Occup. Physiol. 1998, 77, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Kane, D.A. Lactate oxidation at the mitochondria: a lactate-malate-aspartate shuttle at work. Front. Neurosci. 2014, 8, 366. [Google Scholar] [CrossRef] [Green Version]
- Herbst, E.A.; Holloway, G.P. Exercise increases mitochondrial glutamate oxidation in the mouse cerebral cortex. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Et Metab. 2016, 41, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.-P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, M.; Gao, Y.; Gao, J.; Yang, W.; Liang, H.; Ji, Q.; Li, Y.; Liu, H.; Huang, J. Rheb1-mTORC1 maintains macrophage differentiation and phagocytosis in mice. Exp. Cell Res. 2016, 344, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Lexell, J.; Deierborg, T. Effects of Physical Exercise on Neuroinflammation, Neuroplasticity, Neurodegeneration, and Behavior: What We Can Learn From Animal Models in Clinical Settings. Neurorehabil. Neural Repair 2015, 29, 577–589. [Google Scholar] [CrossRef]
- Jang, B.-C.; Paik, J.-H.; Kim, S.-P.; Shin, D.-H.; Song, D.-K.; Park, J.-G.; Suh, M.-H.; Park, J.-W.; Suh, S.-I. Catalase induced expression of inflammatory mediators via activation of NF-κB, PI3K/AKT, p70S6K, and JNKs in BV2 microglia. Cell. Signal. 2005, 17, 625–633. [Google Scholar] [CrossRef]
- Heidt, S.; Roelen, D.L.; Eijsink, C.; van Kooten, C.; Claas, F.H.; Mulder, A. Effects of immunosuppressive drugs on purified human B cells: Evidence supporting the use of MMF and rapamycin. Transplantation 2008, 86, 1292–1300. [Google Scholar] [CrossRef]
- Russo, C.D.; Navarra, P.; Lisi, L. Chapter 20—mTOR in Multiple Sclerosis: The Emerging Role in the Regulation of Glial Biology. In Molecules to Medicine with mTOR; Maiese, K., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 331–343. [Google Scholar] [CrossRef]
- Weichhart, T.; Säemann, M.D. The multiple facets of mTOR in immunity. Trends Immunol. 2009, 30, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.; Mangano, K.; Amoroso, A.; Mazzarino, M.C.; Imbesi, R.; Castrogiovanni, P.; Coco, M.; Meroni, P.; Nicoletti, F. Treatment with rapamycin ameliorates clinical and histological signs of protracted relapsing experimental allergic encephalomyelitis in Dark Agouti rats and induces expansion of peripheral CD4+CD25+Foxp3+ regulatory T cells. J. Autoimmun. 2009, 33, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Pearce, E.J. MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells. Immunity 2017, 46, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Chaube, B.; Bhat, M.K. AMPK, a key regulator of metabolic/energy homeostasis and mitochondrial biogenesis in cancer cells. Cell Death Dis. 2016, 7, e2044. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Khan, M.; Rattan, R.; Mangalam, A.; Makkar, R.S.; Meester, C.d.; Bertrand, L.; Singh, I.; Chen, Y.; Viollet, B.; et al. Loss of AMPK exacerbates experimental autoimmune encephalomyelitis disease severity. Biochem. Biophys. Res. Commun. 2009, 386, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.-X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARδ Promotes Running Endurance by Preserving Glucose. Cell Metab. 2017, 25, 1186–1193.e1184. [Google Scholar] [CrossRef]
- Agostini, D.; Natalucci, V.; Baldelli, G.; De Santi, M.; Donati Zeppa, S.; Vallorani, L.; Annibalini, G.; Lucertini, F.; Federici, A.; Izzo, R.; et al. New Insights into the Role of Exercise in Inhibiting mTOR Signaling in Triple-Negative Breast Cancer. Oxid Med. Cell Longev. 2018, 2018, 5896786. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Freitag, J.; Berod, L.; Kamradt, T.; Sparwasser, T. Immunometabolism and autoimmunity. Immunol. Cell Biol. 2016, 94, 925–934. [Google Scholar] [CrossRef] [PubMed]
- De Gasperis-Brigante, C.D.; Parker, J.L.; O’Connor, P.W.; Bruno, T.R. Reducing clinical trial risk in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 5, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanain, H.H.; Chon, S.Y.; Gupta, S.L. Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and -alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. In J. Biol. Chem.; 1993; Volume 268, pp. 5077–5084. [Google Scholar]
- Schröcksnadel, K.; Wirleitner, B.; Winkler, C.; Fuchs, D. Monitoring tryptophan metabolism in chronic immune activation. Clin. Chim. Acta 2006, 364, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Orabona, C.; Puccetti, P.; Vacca, C.; Bicciato, S.; Luchini, A.; Fallarino, F.; Bianchi, R.; Velardi, E.; Perruccio, K.; Velardi, A.; et al. Toward the identification of a tolerogenic signature in IDO-competent dendritic cells. Blood 2006, 107, 2846–2854. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Zhang, G.-X.; Gran, B.; Fallarino, F.; Yu, S.; Li, H.; Cullimore, M.L.; Rostami, A.; Xu, H. IDO Upregulates Regulatory T Cells via Tryptophan Catabolite and Suppresses Encephalitogenic T Cell Responses in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2010, 185, 5953. [Google Scholar] [CrossRef]
- Xiao, B.G.; Liu, X.; Link, H. Antigen-specific T cell functions are suppressed over the estrogen-dendritic cell-indoleamine 2,3-dioxygenase axis. Steroids 2004, 69, 653–659. [Google Scholar] [CrossRef]
- Negrotto, L.; Correale, J. Amino Acid Catabolism in Multiple Sclerosis Affects Immune Homeostasis. J. Immunol. 2017, 198, 1900–1909. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [Green Version]
- Amirkhani, A.; Rajda, C.; Arvidsson, B.; Bencsik, K.; Boda, K.; Seres, E.; Markides, K.E.; Vécsei, L.; Bergquist, J. Interferon-β affects the tryptophan metabolism in multiple sclerosis patients. Eur. J. Neurol. 2005, 12, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Rajda, C.; Galla, Z.; Polyák, H.; Maróti, Z.; Babarczy, K.; Pukoli, D.; Vécsei, L. Cerebrospinal Fluid Neurofilament Light Chain Is Associated with Kynurenine Pathway Metabolite Changes in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Bansi, J.; Koliamitra, C.; Bloch, W.; Joisten, N.; Schenk, A.; Watson, M.; Kool, J.; Langdon, D.; Dalgas, U.; Kesselring, J. Persons with secondary progressive and relapsing remitting multiple sclerosis reveal different responses of tryptophan metabolism to acute endurance exercise and training. J. Neuroimmunol. 2018, 314, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Boscaro, F.; Pieraccini, G.; Calabresi, P.; Romani, L.; Di Filippo, M.; Zelante, T. Host and Microbial Tryptophan Metabolic Profiling in Multiple Sclerosis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, S.; Åkerfeldt, T.; Spjuth, O.; Burman, J.; Kultima, K. Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing⁻Remitting Multiple Sclerosis. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Strasser, B.; Geiger, D.; Schauer, M.; Gatterer, H.; Burtscher, M.; Fuchs, D. Effects of Exhaustive Aerobic Exercise on Tryptophan-Kynurenine Metabolism in Trained Athletes. PLoS ONE 2016, 11, e0153617. [Google Scholar] [CrossRef]
- Schlittler, M.; Goiny, M.; Agudelo, L.Z.; Venckunas, T.; Brazaitis, M.; Skurvydas, A.; Kamandulis, S.; Ruas, J.L.; Erhardt, S.; Westerblad, H.; et al. Endurance exercise increases skeletal muscle kynurenine aminotransferases and plasma kynurenic acid in humans. Am. J. Physiol. Cell Physiol. 2016, 310, C836–C840. [Google Scholar] [CrossRef] [Green Version]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.T.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab. 2018, 27, 378–392.e375. [Google Scholar] [CrossRef] [Green Version]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal Muscle PGC-1α1 Modulates Kynurenine Metabolism and Mediates Resilience to Stress-Induced Depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Beckerman, H.; de Groot, V.; Scholten, M.A.; Kempen, J.C.E.; Lankhorst, G.J. Physical Activity Behavior of People with Multiple Sclerosis: Understanding How They Can Become More Physically Active. Phys. Ther. 2010, 90, 1001–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.M.; Stennett, A.M.; Peacock, S.; Baker, G.; Norris, M. Associations between activity and participation in adults with multiple sclerosis: A cross sectional study. Physiotherapy 2019, 105, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpatkin, H.I. Multiple Sclerosis and Exercise: A Review of the Evidence. Int. J. Ms. Care 2005, 7, 36–41. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Pharmacological Drugs | Exercise Therapy | |||
|---|---|---|---|---|---|
| Drug | Effect | REF(S) | Effect | REF(S) | |
| Glycolysis | DMF | ↓ Glycolysis Enzyme GAPDH In Myeloid and Lymphoid Cells | [69] | ↓ Glycolysis ↑ Glucose Tolerance | [70,72,73,174] |
| Fatty Acid Metabolism | Natalizumab | ↓ Inflammatory Oxysterol 24S-OH In CSF | [80] | ↑ FAO ↓ Cholesterol LDL Synthesis | [92,95,97,98] |
| Mitochondrial Oxidative Stress | DMF | ↓ Ros ↑ Nrf2 Antioxidant Response Pathway | [175] | ↓ Ros ↑ Pgc1α ↑ Antioxidant Response | [22,113,121,122] |
| Glutamate | Memantine | ↓ Glutamate Excitotoxicity | [130] | ↓ Glutamate by Increasing Glutamate Uptake and Metabolism | [136,176] |
| mTOR/AMPK | Temsirolimus | ↓ mTOR Activity | [154] | ↓ mTOR ↑ AMPK | [95,151] |
| Kynurenine | IFN-β | ↑ Kyn/Trp Ratio | [165] | ↑ KynA (due to ↑ PGC-1α) | [155,171] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afzal, R.; Dowling, J.K.; McCoy, C.E. Impact of Exercise on Immunometabolism in Multiple Sclerosis. J. Clin. Med. 2020, 9, 3038. https://doi.org/10.3390/jcm9093038
Afzal R, Dowling JK, McCoy CE. Impact of Exercise on Immunometabolism in Multiple Sclerosis. Journal of Clinical Medicine. 2020; 9(9):3038. https://doi.org/10.3390/jcm9093038
Chicago/Turabian StyleAfzal, Remsha, Jennifer K Dowling, and Claire E McCoy. 2020. "Impact of Exercise on Immunometabolism in Multiple Sclerosis" Journal of Clinical Medicine 9, no. 9: 3038. https://doi.org/10.3390/jcm9093038
APA StyleAfzal, R., Dowling, J. K., & McCoy, C. E. (2020). Impact of Exercise on Immunometabolism in Multiple Sclerosis. Journal of Clinical Medicine, 9(9), 3038. https://doi.org/10.3390/jcm9093038