Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers


 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Patients
2.2. NGS Sequencing
2.3. Variants’ Classification
2.4. Statistical Analysis
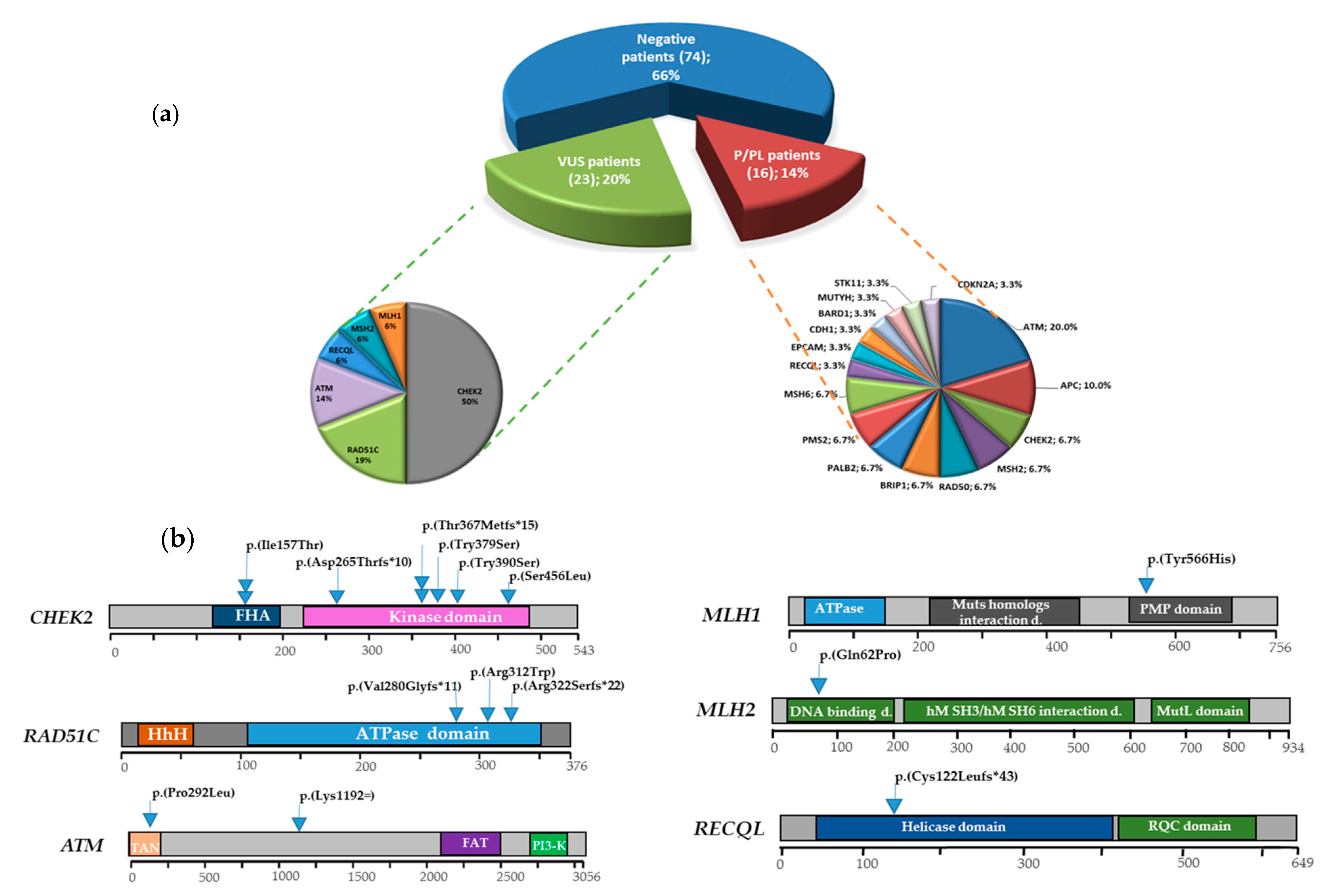
3. Results
3.1. Multigene Panel Results
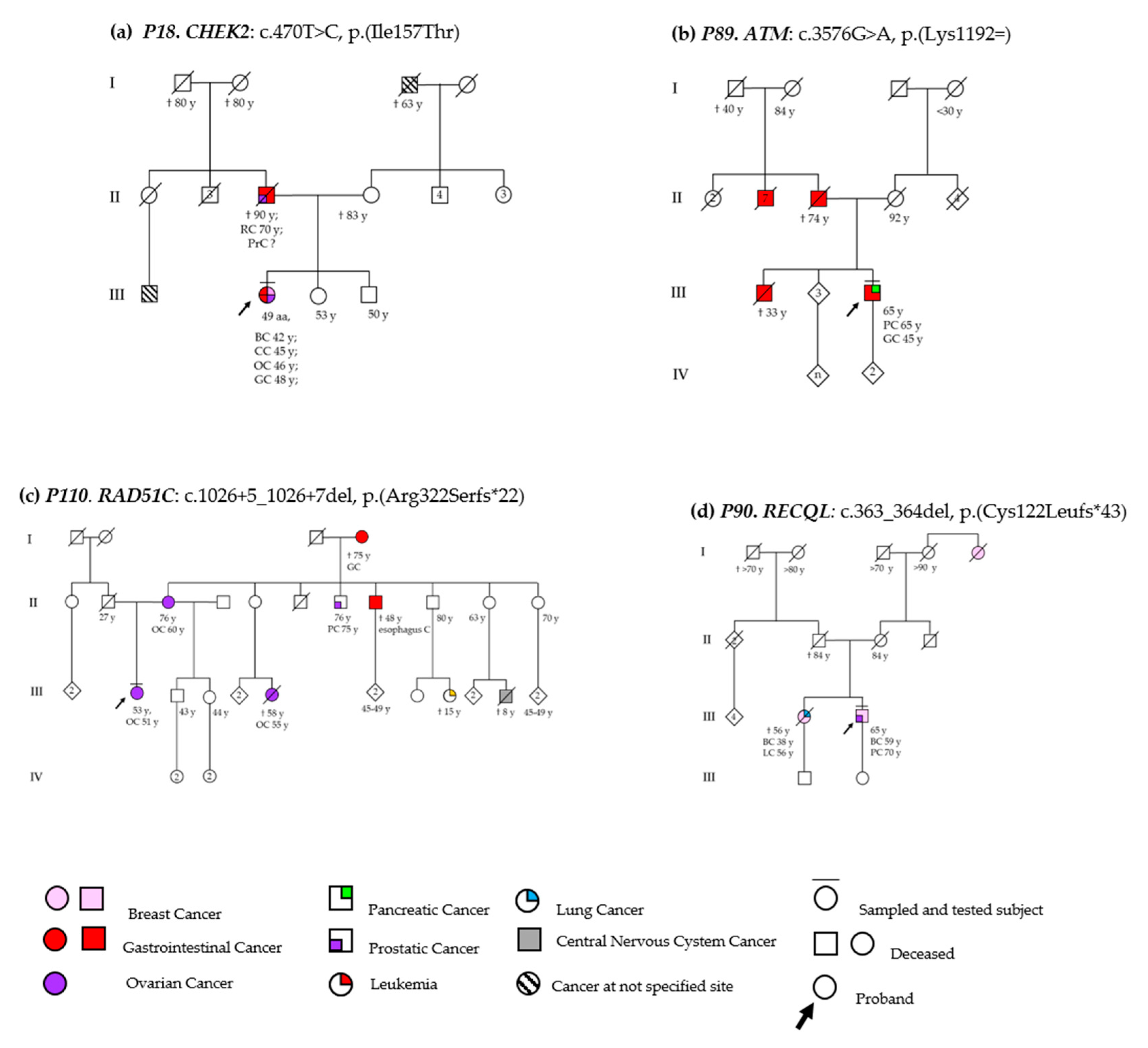
3.2. DSBs-HR Genes Variants and Related Phenotypes
3.2.1. ATM
3.2.2. RAD51C
3.3. MMR Genes Variants and Related Phenotypes
3.3.1. MLH1
3.3.2. MSH2
3.4. Cell Cycle Control Gene Variants and Related Phenotypes
CHEK2
3.5. DNA Repair Helicase Gene Variant-Related Phenotypes
RECQL
3.6. VUS Variants
3.7. Genotype-Phenotype Correlations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shih, H.A.; Couch, F.J.; Nathanson, K.L.; Blackwood, M.A.; Rebbeck, T.R.; Armstrong, K.A.; Calzone, K.; Stopfer, J.; Seal, S.; Stratton, M.R.; et al. BRCA1 and BRCA2 mutation frequency in women evaluated in a breast cancer risk evaluation clinic. J. Clin. Oncol. 2002, 20, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Stadler, Z.K.; Thom, P.; Robson, M.E.; Weitzel, J.N.; Kauff, N.D.; Hurley, K.E.; Devlin, V.; Gold, B.; Klein, R.J.; Offit, K. Genome-Wide Association Studies of Cancer. J. Clin. Oncol. 2010, 28, 4255–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-M.; Wang, H.-C.; Chen, S.-T.; Hsu, G.-C.; Shen, C.-Y.; Yu, J.-C. Breast cancer risk is associated with the genes encoding the DNA double-strand break repair Mre11/Rad50/Nbs1 complex. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2024–2032. [Google Scholar] [CrossRef] [Green Version]
- Tung, N.; Silver, D.P. Chek2 DNA damage response pathway and inherited breast cancer risk. J. Clin. Oncol. 2011, 29, 3813–3815. [Google Scholar] [CrossRef]
- Lee, C.; Banerjee, T.; Gillespie, J.; Ceravolo, A.; Parvinsmith, M.R.; Starita, L.M.; Fields, S.; Toland, A.E.; Parvin, J.D. Functional Analysis of BARD1 Missense Variants in Homology-Directed Repair of DNA Double Strand Breaks. Hum. Mutat. 2015, 36, 1205–1214. [Google Scholar] [CrossRef]
- Cantor, S.B.; Guillemette, S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011, 7, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-S.; Chen, J.; Cui, F.; Wang, H.; Wang, S.; Hang, W.; Zeng, Q.; Quan, C.-S.; Zhai, Y.-X.; Wang, J.-W.; et al. LKB1 is a DNA damage response protein that regulates cellular sensitivity to PARP inhibitors. Oncotarget 2016, 7, 73389–73401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana Dos Santos, E.; Lallemand, F.; Petitalot, A.; Caputo, S.M.; Rouleau, E. HRness in Breast and Ovarian Cancers. Int. J. Mol. Sci. 2020, 21, 3850. [Google Scholar] [CrossRef] [PubMed]
- Sarrió, D.; Moreno-Bueno, G.; Hardisson, D.; Sánchez-Estévez, C.; Guo, M.; Herman, J.G.; Gamallo, C.; Esteller, M.; Palacios, J. Epigenetic and genetic alterations of APC and CDH1 genes in lobular breast cancer: Relationships with abnormal E-cadherin and catenin expression and microsatellite instability. Int. J. Cancer 2003, 106, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Sicinska, E.; Geng, Y.; Ahnström, M.; Zagozdzon, A.; Kong, Y.; Gardner, H.; Kiyokawa, H.; Harris, L.N.; Stål, O.; et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006, 9, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Debniak, T.; Górski, B.; Huzarski, T.; Byrski, T.; Cybulski, C.; Mackiewicz, A.; Gozdecka-Grodecka, S.; Gronwald, J.; Kowalska, E.; Haus, O.; et al. A common variant of CDKN2A (p16) predisposes to breast cancer. J. Med. Genet. 2005, 42, 763–765. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.; Bonnet, F.; Sfar, S.; Lafitte, M.; Lafon, D.; Sierankowski, G.; Brouste, V.; Banneau, G.; Tunon de Lara, C.; Debled, M.; et al. Comprehensive analysis of PTEN status in breast carcinomas. Int. J. Cancer 2013, 133, 323–334. [Google Scholar] [CrossRef]
- Woo, J.-S.; Chung, M.S.; Paik, S.S. Clinicopathological Significance of SMAD4 Expression in Breast Cancer. J. Breast Dis. 2019, 7, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Schon, K.; Tischkowitz, M. Clinical implications of germline mutations in breast cancer: TP53. Breast Cancer Res. Treat. 2018, 167, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Brosh, R.M. RECQL: A new breast cancer susceptibility gene. Cell Cycle 2015, 14, 3540–3543. [Google Scholar] [CrossRef] [Green Version]
- Vierkoetter, K.R.; Ayabe, A.R.; VanDrunen, M.; Ahn, H.J.; Shimizu, D.M.; Terada, K.Y. Lynch Syndrome in patients with clear cell and endometrioid cancers of the ovary. Gynecol. Oncol. 2014, 135, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitin, A.G.; Chudakova, D.A.; Enikeev, R.F.; Sakaeva, D.; Druzhkov, M.; Shigapova, L.H.; Brovkina, O.I.; Shagimardanova, E.I.; Gusev, O.A.; Gordiev, M.G. Lynch Syndrome Germline Mutations in Breast Cancer: Next Generation Sequencing Case-Control Study of 1263 Participants. Front. Oncol. 2020, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Rizzolo, P.; Silvestri, V.; Bucalo, A.; Zelli, V.; Valentini, V.; Catucci, I.; Zanna, I.; Masala, G.; Bianchi, S.; Spinelli, A.M.; et al. Contribution of MUTYH Variants to Male Breast Cancer Risk: Results from a Multicenter Study in Italy. Front. Oncol. 2018, 8, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Nishimura, M.; Onoe, T.; Sakai, H.; Urakawa, Y.; Onda, T.; Yaegashi, N. PARP inhibitors for BRCA wild type ovarian cancer; gene alterations, homologous recombination deficiency and combination therapy. Jpn. J. Clin. Oncol. 2019, 49, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients with Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Hu, L.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xu, Y.; Xie, Y. Germline mutation in DNA-repair genes is associated with poor survival in BRCA1/2-negative breast cancer patients. Cancer Sci. 2019, 110, 3368–3374. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network: NCCN Clinical Pratice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Available online: https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 12 June 2020).
- Colas, C.; Golmard, L.; de Pauw, A.; Caputo, S.M.; Stoppa-Lyonnet, D. “Decoding hereditary breast cancer” benefits and questions from multigene panel testing. Breast 2019, 45, 29–35. [Google Scholar] [CrossRef]
- Ragamin, A.; Yigit, G.; Bousset, K.; Beleggia, F.; Verheijen, F.W.; de Wit, M.-C.Y.; Strom, T.M.; Dörk, T.; Wollnik, B.; Mancini, G.M.S. Human RAD50 deficiency: Confirmation of a distinctive phenotype. Am. J. Med. Genet. A 2020, 182, 1378–1386. [Google Scholar] [CrossRef] [Green Version]
- Debnath, S.; Sharma, S. RECQ1 Helicase in Genomic Stability and Cancer. Genes 2020, 11, 622. [Google Scholar] [CrossRef]
- Leachman, S.A.; Lucero, O.M.; Sampson, J.E.; Cassidy, P.; Bruno, W.; Queirolo, P.; Ghiorzo, P. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017, 36, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Stankovic, T.; Kidd, A.M.; Sutcliffe, A.; McGuire, G.M.; Robinson, P.; Weber, P.; Bedenham, T.; Bradwell, A.R.; Easton, D.F.; Lennox, G.G.; et al. ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: Expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am. J. Hum. Genet. 1998, 62, 334–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitui, M.; Nahas, S.A.; Du, L.T.; Yang, Z.; Lai, C.H.; Nakamura, K.; Arroyo, S.; Scott, S.; Purayidom, A.; Concannon, P.; et al. Functional and computational assessment of missense variants in the ataxia-telangiectasia mutated (ATM) gene: Mutations with increased cancer risk. Hum. Mutat. 2009, 30, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker-Catania, S.G.; Chen, G.; Hwang, M.J.; Wang, Z.; Sun, X.; Sanal, O.; Bernatowska-Matuszkiewicz, E.; Chessa, L.; Lee, E.Y.; Gatti, R.A. Ataxia-telangiectasia: Phenotype/genotype studies of ATM protein expression, mutations, and radiosensitivity. Mol. Genet. Metab. 2000, 70, 122–133. [Google Scholar] [CrossRef]
- Gilad, S.; Chessa, L.; Khosravi, R.; Russell, P.; Galanty, Y.; Piane, M.; Gatti, R.A.; Jorgensen, T.J.; Shiloh, Y.; Bar-Shira, A. Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am. J. Hum. Genet. 1998, 62, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, S.; Funaro, A.; Porcedda, P.; Turinetto, V.; Migone, N.; Gatti, R.A.; Brusco, A. ATM mutations in Italian families with ataxia telangiectasia include two distinct large genomic deletions. Hum. Mutat. 2006, 27, 1061. [Google Scholar] [CrossRef]
- Chessa, L.; Piane, M.; Magliozzi, M.; Torrente, I.; Savio, C.; Lulli, P.; De Luca, A.; Dallapiccola, B. Founder effects for ATM gene mutations in Italian Ataxia Telangiectasia families. Ann. Hum. Genet. 2009, 73, 532–539. [Google Scholar] [CrossRef]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Choi, B.-S. Structural and functional characterization of the N-terminal domain of human Rad51D. Int. J. Biochem. Cell Biol. 2011, 43, 416–422. [Google Scholar] [CrossRef]
- Gayarre, J.; Martín-Gimeno, P.; Osorio, A.; Paumard, B.; Barroso, A.; Fernández, V.; de la Hoya, M.; Rojo, A.; Caldés, T.; Palacios, J.; et al. Characterisation of the novel deleterious RAD51C p.Arg312Trp variant and prioritisation criteria for functional analysis of RAD51C missense changes. Br. J. Cancer 2017, 117, 1048–1062. [Google Scholar] [CrossRef]
- Golmard, L.; Caux-Moncoutier, V.; Davy, G.; Al Ageeli, E.; Poirot, B.; Tirapo, C.; Michaux, D.; Barbaroux, C.; d’Enghien, C.D.; Nicolas, A.; et al. Germline mutation in the RAD51B gene confers predisposition to breast cancer. BMC Cancer 2013, 13, 484. [Google Scholar] [CrossRef]
- Janatova, M.; Soukupova, J.; Stribrna, J.; Kleiblova, P.; Vocka, M.; Boudova, P.; Kleibl, Z.; Pohlreich, P. Mutation Analysis of the RAD51C and RAD51D Genes in High-Risk Ovarian Cancer Patients and Families from the Czech Republic. PLoS ONE 2015, 10, e0127711. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Hoyer, J.; Vasileiou, G.; Wunderle, M.; Lux, M.P.; Fasching, P.A.; Krumbiegel, M.; Uebe, S.; Reuter, M.; Beckmann, M.W.; et al. Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int. J. Cancer 2017, 140, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Loizidou, M.A.; Neophytou, I.; Papamichael, D.; Kountourakis, P.; Vassiliou, V.; Marcou, Y.; Kakouri, E.; Ioannidis, G.; Philippou, C.; Spanou, E.; et al. The mutational spectrum of Lynch syndrome in cyprus. PLoS ONE 2014, 9, e105501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Xu, X.; Tsvetkov, L.M.; Stern, D.F. Chk2 activation and phosphorylation-dependent oligomerization. Mol. Cell. Biol. 2002, 22, 4419–4432. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Kim, S.H.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Jorczak, M.M.; Sgroi, D.C.; Garber, J.E.; Li, F.P.; Nichols, K.E.; et al. Destabilization of CHK2 by a missense mutation associated with Li-Fraumeni Syndrome. Cancer Res. 2001, 61, 8062–8067. [Google Scholar]
- Liu, C.; Wang, Y.; Wang, Q.-S.; Wang, Y.-J. The CHEK2 I157T variant and breast cancer susceptibility: A systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2012, 13, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, C.; Górski, B.; Huzarski, T.; Masojć, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Teodorczyk, U.; Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Starzyńska, T.; Lawniczak, M.; Domagała, P.; Ferenc, K.; Marlicz, K.; Banaszkiewicz, Z.; et al. The risk of gastric cancer in carriers of CHEK2 mutations. Fam. Cancer 2013, 12, 473–478. [Google Scholar] [CrossRef]
- Wójcicka, A.; Czetwertyńska, M.; Świerniak, M.; Długosińska, J.; Maciąg, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Płoski, R.; et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer 2014, 53, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Roeb, W.; Higgins, J.; King, M.-C. Response to DNA damage of CHEK2 missense mutations in familial breast cancer. Hum. Mol. Genet. 2012, 21, 2738–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agiannitopoulos, K.; Papadopoulou, E.; Tsaousis, G.N.; Pepe, G.; Kampouri, S.; Kocdor, M.A.; Nasioulas, G. Characterization of the c.793-1G > A splicing variant in CHEK2 gene as pathogenic: A case report. BMC Med. Genet. 2019, 20, 131. [Google Scholar] [CrossRef] [Green Version]
- Leedom, T.P.; LaDuca, H.; McFarland, R.; Li, S.; Dolinsky, J.S.; Chao, E.C. Breast cancer risk is similar for CHEK2 founder and non-founder mutation carriers. Cancer Genet. 2016, 209, 403–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojć, B.; Deebniak, T.; Górski, B.; Blecharz, P.; et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yu, H.; Zheng, S.L.; Na, R.; Mamawala, M.; Landis, T.; Wiley, K.; Petkewicz, J.; Shah, S.; Shi, Z.; et al. A comprehensive evaluation of CHEK2 germline mutations in men with prostate cancer. Prostate 2018, 78, 607–615. [Google Scholar] [CrossRef]
- Hale, V.; Weischer, M.; Park, J.Y. CHEK2 (∗) 1100delC Mutation and Risk of Prostate Cancer. Prostate Cancer 2014, 2014, 294575. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, T.; Chen, L.; Xie, D.; Wang, F.; Fu, L.; Cheng, C.; Li, Y.; Zhu, X.; Miao, G. A Germline CHEK2 Mutation in a Family with Papillary Thyroid Cancer. Thyroid 2020, 30, 924–930. [Google Scholar] [CrossRef]
- Näslund-Koch, C.; Nordestgaard, B.G.; Bojesen, S.E. Increased Risk for Other Cancers in Addition to Breast Cancer for CHEK2*1100delC Heterozygotes Estimated from the Copenhagen General Population Study. J. Clin. Oncol. 2016, 34, 1208–1216. [Google Scholar] [CrossRef]
- Cai, Z.; Chehab, N.H.; Pavletich, N.P. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol. Cell 2009, 35, 818–829. [Google Scholar] [CrossRef]
- Desrichard, A.; Bidet, Y.; Uhrhammer, N.; Bignon, Y.-J. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res. 2011, 13, R119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, K.N.; Wubbenhorst, B.; D’Andrea, K.; Garman, B.; Long, J.M.; Powers, J.; Rathbun, K.; Stopfer, J.E.; Zhu, J.; Bradbury, A.R.; et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet. Med. 2015, 17, 630–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers 2018, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulski, C.; Carrot-Zhang, J.; Kluźniak, W.; Rivera, B.; Kashyap, A.; Wokołorczyk, D.; Giroux, S.; Nadaf, J.; Hamel, N.; Zhang, S.; et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet. 2015, 47, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.M.; Eccles, D.M.; Romero, I.L.; Al-Mulla, F.; Balmaña, J.; Biancolella, M.; Bslok, R.; Caligo, M.A.; Calvello, M.; Capone, G.L.; et al. Genetic Testing and Clinical Management Practices for Variants in Non-BRCA1/2 Breast (and Breast/Ovarian) Cancer Susceptibility Genes: An International Survey by the Evidence-Based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) Clinical Working Group. JCO Precis. Oncol. 2018, 2. [Google Scholar] [CrossRef]
- Alvarado, M.; Tiller, G.E.; Chung, J.; Haque, R. Prevalence of mutations in a diverse cohort of 3162 women tested via the same multigene cancer panel in a managed care health plan. J. Community Genet. 2020, 11, 359–366. [Google Scholar] [CrossRef]
- Rosenthal, S.H.; Sun, W.; Zhang, K.; Liu, Y.; Nguyen, Q.; Gerasimova, A.; Nery, C.; Cheng, L.; Castonguay, C.; Hiller, E.; et al. Development and Validation of a 34-Gene Inherited Cancer Predisposition Panel Using Next-Generation Sequencing. Biomed. Res. Int. 2020, 2020, 3289023. [Google Scholar] [CrossRef]
- Suszynska, M.; Klonowska, K.; Jasinska, A.J.; Kozlowski, P. Large-scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes—Providing evidence of cancer predisposition genes. Gynecol. Oncol. 2019, 153, 452–462. [Google Scholar] [CrossRef] [Green Version]
- Angeli, D.; Salvi, S.; Tedaldi, G. Genetic Predisposition to Breast and Ovarian Cancers: How Many and Which Genes to Test? Int. J. Mol. Sci. 2020, 21, 1128. [Google Scholar] [CrossRef] [Green Version]
- Weischer, M.; Heerfordt, I.M.; Bojesen, S.E.; Eigentler, T.; Garbe, C.; Röcken, M.; Hölmich, L.R.; Schmidt, H.; Klyver, H.; Bastholt, L.; et al. CHEK2*1100delC and risk of malignant melanoma: Danish and German studies and meta-analysis. J. Investig. Dermatol. 2012, 132, 299–303. [Google Scholar] [CrossRef]
- Fulk, K.; Milam, M.R.; Li, S.; Yussuf, A.; Black, M.H.; Chao, E.C.; LaDuca, H.; Stany, M.P. Women with breast and uterine cancer are more likely to harbor germline mutations than women with breast or uterine cancer alone: A case for expanded gene testing. Gynecol. Oncol. 2019, 152, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Rizzolo, P.; Zelli, V.; Silvestri, V.; Valentini, V.; Zanna, I.; Bianchi, S.; Masala, G.; Spinelli, A.M.; Tibiletti, M.G.; Russo, A.; et al. Insight into genetic susceptibility to male breast cancer by multigene panel testing: Results from a multicenter study in Italy. Int. J. Cancer 2019, 145, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Siołek, M.; Cybulski, C.; Gąsior-Perczak, D.; Kowalik, A.; Kozak-Klonowska, B.; Kowalska, A.; Chłopek, M.; Kluźniak, W.; Wokołorczyk, D.; Pałyga, I.; et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int. J. Cancer 2015, 137, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Isinger, A.; Bhat, M.; Borg, A.; Nilbert, M. CHEK2 1100delC in patients with metachronous cancers of the breast and the colorectum. BMC Cancer 2006, 6, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvai, K.J.; Roberts, M.E.; Torene, R.I.; Susswein, L.R.; Marshall, M.L.; Zhang, Z.; Carter, N.J.; Yackowski, L.; Rinella, E.S.; Klein, R.T.; et al. Age-adjusted association of homologous recombination genes with ovarian cancer using clinical exomes as controls. Hered. Cancer Clin. Pract. 2019, 17, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbari, M.R.; Tonin, P.; Foulkes, W.D.; Ghadirian, P.; Tischkowitz, M.; Narod, S.A. RAD51C germline mutations in breast and ovarian cancer patients. Breast Cancer Res. 2010, 12, 404. [Google Scholar] [CrossRef]
- Hoyer, J.; Vasileiou, G.; Uebe, S.; Wunderle, M.; Kraus, C.; Fasching, P.A.; Thiel, C.T.; Hartmann, A.; Beckmann, M.W.; Lux, M.P.; et al. Addition of triple negativity of breast cancer as an indicator for germline mutations in predisposing genes increases sensitivity of clinical selection criteria. BMC Cancer 2018, 18, 926. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.-S.; Tao, H.-Q.; He, X.-J.; Long, M.; Yu, S.; Xia, Y.-J.; Wei, Z.; Xiong, Z.; Jones, S.; He, Y.; et al. Prevalence of deleterious ATM germline mutations in gastric cancer patients. Oncotarget 2015, 6, 40953–40958. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhao, Z.; Zhang, Y.; Bao, C.; Cui, L.; Cai, S.; Bai, Y.; Shen, L.; Zhang, X. Pathogenic Germline Mutations in Chinese Patients with Gastric Cancer Identified by Next-Generation Sequencing. Oncology 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Fein, D.; Ramirez, C.B.; Do, K.; Saif, M.W. PARP Inhibitors in Pancreatic Cancer: From Phase I to Plenary Session. Pancreas (Fairfax) 2019, 3, e5–e8. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Dumont, T.; Steen, J.A.; Winship, I.; Park, D.J.; Pope, B.J.; Hammet, F.; Mahmoodi, M.; Tsimiklis, H.; Theys, D.; Clendenning, M.; et al. Mismatch repair gene pathogenic germline variants in a population-based cohort of breast cancer. Fam. Cancer 2020, 19, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, A. Mutations in conserved functional domains of human RecQ helicases are associated with diseases and cancer: A review. Biophys. Chem. 2020, 265, 106433. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Y.; Xia, Y.; Xu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Mutations in RECQL Gene Are Associated with Predisposition to Breast Cancer. PLoS Genet. 2015, 11, e1005228. [Google Scholar] [CrossRef]
- Bogdanova, N.; Pfeifer, K.; Schürmann, P.; Antonenkova, N.; Siggelkow, W.; Christiansen, H.; Hillemanns, P.; Park-Simon, T.-W.; Dörk, T. Analysis of a RECQL splicing mutation, c.1667_1667+3delAGTA, in breast cancer patients and controls from Central Europe. Fam. Cancer 2017, 16, 181–186. [Google Scholar] [CrossRef]
- Palmer, J.R.; Polley, E.C.; Hu, C.; John, E.M.; Haiman, C.; Hart, S.N.; Gaudet, M.; Pal, T.; Anton-Culver, H.; Trentham-Dietz, A.; et al. Contribution of Germline Predisposition Gene Mutations to Breast Cancer Risk in African American Women. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef]
- Ahmed, H.; Lerner-Ellis, J.; Cybulski, C.; Metcalfe, K.; Lubiński, J.; Narod, S.A.; Akbari, M.R. Reply to “Mutations in RECQL are not associated with breast cancer risk in an Australian population”. Nat. Genet. 2018, 50, 1348–1349. [Google Scholar] [CrossRef]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.-H.; Kuo, W.-H.; Huang, A.-C.; Lu, Y.-S.; Lin, C.-H.; Kuo, S.-H.; Wang, M.-Y.; Liu, C.-Y.; Cheng, F.T.-F.; Yeh, M.-H.; et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016, 7, 8310–8320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef]
- Zhen, J.T.; Syed, J.; Nguyen, K.A.; Leapman, M.S.; Agarwal, N.; Brierley, K.; Llor, X.; Hofstatter, E.; Shuch, B. Genetic testing for hereditary prostate cancer: Current status and limitations. Cancer 2018, 124, 3105–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Panel Gene | Syndrome | Main Pathway | Cancer Related | Reference |
|---|---|---|---|---|
| ATM | Ataxia Telangiectasia (AR) | DOUBLE STRAND BREAKS REPAIR (Homologous Recombination) | Breast, ovarian, pancreatic | [2,24,26] |
| PALB2 | Fanconi Anemia (AR) | [24,26,27] | ||
| MRE11A | Ataxia Telangiectasia-like disorder (AR) | Breast | [4,27] | |
| RAD50 | Nijmegen breakage syndrome-like disorder (AR) | [4,28] | ||
| BARD1 | [6] | |||
| NBN | Nijmegen Breakage Syndrome (AR) | Breast, ovarian | [26] | |
| BRIP1 | Fanconi Anemia (AR) | [7] | ||
| RAD51C | [27] | |||
| RAD51D | [27] | |||
| STK11 | Peutz-Jeghers Syndrome (AD) | Colorectal, breast, pancreatic, gastric, small intestine, cervical, ovarian | [2,11] | |
| MSH2 | Lynch Syndrome (AD) | MISMATCH REPAIR | Colorectal, endometrial, ovarian, gastric, urothelial, pancreaticobiliary, cutaneous sebaceous neo-plasms, brain | |
| MLH1 | ||||
| MSH6 | [2,26] | |||
| PMS2 | ||||
| EPCAM | ||||
| MUTYH | MYH-Associated polyposis (AR) | BASE EXCISION REPAIR | Colorectal, duodenal, breast | [22] |
| RECQL | DNA REPAIR (helicase) | breast | [29] | |
| TP53 | Li-Fraumeni Syndrome (AD) | cell cycle control | Breast, sarcoma, brain, adrenocortical, leukemia, gastric | [2,26,27] |
| PTEN | Cowden Syndrome (AD) | Colorectal, breast, endometrial, thyroid, renal | [2,16,26,27] | |
| CHEK2 | Li-Fraumeni variant (AD) | Breast ovarian | [2,5,26] | |
| CDH1 | Hereditary diffuse gastric cancer (AD) | Gastric, breast | [2,26,27] | |
| CDK4 | Familial melanoma (AD) | Melanoma | [30] | |
| CDKN2A | Melanoma, pancreatic | [2] | ||
| SMAD4 | Juvenile polyposis Syndrome (AD) | Colorectal, Gastric | [17] | |
| APC | Familial adenomatous polyposis (AD) | Colorectal, small intestine, ampullary, gastric, desmoid, thyroid | [13] |
| n pts (F/M) | Mean Age Tot ± SD (Min–Max) | Mean Age at Onset Tot ± SD (Min–Max) | n Pts Onset ≤ 40 Years | Family History 1st, 2nd Degree (BC, OC, PrC, PC other C, multiple C) | |
|---|---|---|---|---|---|
| BC | 87 (87/0) | 54.39 ± 10.28 (36–83) | 49.25 ± 10.35 (30–82) | 16 | 82 (68, 18, 22, 9, 59, 14) |
| Male BC | 7 (0/7) | 63.1 ± 11.8 (49–78) | 60.7 ± 13.2 (43–76) | 0 | 6 (4, 2, 1, 0, 6, 3) |
| OC | 9 (9/0) | 59.56 ± 8.75 (46–71) | 52.33 ± 14.04 (28–68) | 2 | 9 (6, 3, 1, 0, 7, 0) |
| PC | 3 (1/2) | 67.7 ± 2.1 (65–69) | 65.3 ± 0.6 (65–66) | 0 | 3 (1, 0, 1, 0, 3, 0) |
| Fam | 7 (7/0) | 54.14 ± 8.63 (45–67) | - | - | 7 (5, 2, 3, 3, 6, 3) |
| Total | 113 (104/9) | 55.65 ± 10.37 (36–83) | 50.24 ± 11.19 (27–82) | 19 | 107 (84, 25, 28, 12, 81, 20) |
| Gene | Locus | Transcript | dbsnp | cDNA (HGVS) | Protein | Type of Variants | Gnomad | ACMG Classification | Count |
|---|---|---|---|---|---|---|---|---|---|
| APC | chr5:112176574 | NM_000038.5 | rs933729249 | c.5283C>G | p.(Asn1761Lys) | missense | / | VUS | 1 |
| chr5:112176656 | rs1554086666 | c.5365G>C | p.(Val1789Leu) | missense | / | VUS | 1 | ||
| chr5:112178958 | rs761133356 | c.7667C>T | p.(Ser2556Leu) | missense | 0.000012 | VUS | 1 | ||
| ATM | chr11:108114838 | NM_000051.3 | rs771685059 | c.655T>C | p.(Cys219Arg) | missense | 0.00000796 | VUS | 1 |
| chr11:108115727 | rs747727055 | c.875C>T | p.(Pro292Leu) | missense | 0.00000806 | LP | 1 | ||
| chr11:108151895 | rs587776551 | c.3576G>A | p.(Lys1192=) | synonymous | 0.0000159 | P | 1 | ||
| chr11:108163473 | rs1064795495 | c.4564G>A | p.(Gly1522Ser) | missense | 0.00000398 | VUS | 1 | ||
| chr11:108164131 | rs368830730 | c.4703A>G | p.(His1568Arg) | missense | 0.0000398 | VUS | 1 | ||
| chr11:108181006 | rs56399311 | c.5882A>G | p.(Tyr1961Cys) | missense | 0.0000398 | VUS | 1 | ||
| chr11:108198368 | / | c.6976-4A>G | p.? | intronic | / | VUS | 1 | ||
| chr11:108205751 | rs759779781 | c.8066A>G | p.(Glu2689Gly) | missense | 0.00000398 | VUS | 1 | ||
| BARD1 | chr2:215661789 | NM_000465.3 | rs1060501308 | c.211T>A | p.(Cys71Ser) | missense | / | VUS | 1 |
| BRIP1 | chr17:59937216 | NM_032043.2 | / | c.146G>A | p.(Gly49Glu) | missense | / | VUS | 1 |
| chr17:59934442 | rs889877039 | c.356A>G | p.(Asn119Ser) | missense | 0.000137 | VUS | 1 | ||
| CDH1 | chr16:68842738 | NM_004360.4 | rs786203207 | c.674T>C | p.(Ile225Thr) | missense | / | VUS | 1 |
| CDKN2A | chr9:21970943 | NM_001195132.1 | rs587781733 | c.415G>A | p.(Gly139Ser) | missense | / | VUS | 1 |
| CHEK2 | chr22:29121228 | NM_007194.3 | rs587781279 | c.444+3A>G | p.? | intronic | / | VUS | 1 |
| chr22:29121087 | rs17879961 | c.470T>C | p.(Ile157Thr) | missense | 0.001 | LP | 2 | ||
| chr22:29107974 | rs121908702 | c.715G>A | p.(Glu239Lys) | missense | / | VUS | 1 | ||
| chr22:29106048 | rs730881687 | c.793-1G>A | p.(Asp265Thrfs*10) | splicing | / | P | 1 | ||
| chr22:29091857 | rs555607708 | c.1100delC | p.(Thr367Metfs*15) | frameshift | 0.002 | P | 2 | ||
| chr22:29091788 | rs200928781 | c.1169A>C | p.(Tyr390Ser) | missense | / | LP | 1 | ||
| chr22:29091821 | rs267606211 | c.1136C>G | p.(Ser379Cys) | missense | / | LP | 1 | ||
| chr22:29091123 | rs876659827 | c.1367C>T | p.(Ser456Leu) | missense | / | LP | 1 | ||
| EPCAM | chr2:47602397 | NM_002354.2 | rs864622724 | c.450C>G | p.(His150Gln) | missense | / | VUS | 1 |
| MLH1 | chr3:37083787 | NM_000249.3 | rs730881743 | c.1696T>C | p.(Tyr566His) | missense | / | LP | 1 |
| MSH2 | chr2:47630512 | NM_000251.2 | rs587779113 | c.182A>C | p.(Gln61Pro) | missense | / | LP | 1 |
| chr2:47702191 | rs41295288 | c.1787A>G | p.(Asn596Ser) | missense | 0.000318 | VUS | 1 | ||
| chr2:47702251 | rs587779965 | c.1847C>G | p.(Pro616Arg) | missense | / | VUS | 1 | ||
| MSH6 | chr2:48027323 | NM_000179.2 | rs1060502883 | c.2201T>A | p.(Val734Glu) | missense | / | VUS | 1 |
| MUTYH | chr1:45798518 | NM_001128425.1 | rs890418965 | c.505-12T>G | p.? | intronic | / | VUS | 1 |
| PALB2 | chr16:23614905 | NM_024675.3 | rs879254033 | c.3436C>A | p.(Gln1146Lys) | missense | / | VUS | 2 |
| PMS2 | chr7:6045541 | NM_000535.6 | rs1583418527 | c.145G>A | p.(Ala49Thr) | missense | / | VUS | 1 |
| chr7:6027143 | rs587782640 | c.1253C>T | p.(Ser418Phe) | missense | / | VUS | 1 | ||
| chr7:6022480 | rs201671325 | c.2149G>A | p.(Val717Met) | missense | 0 | VUS | 1 | ||
| RAD50 | chr5:131925536 | NM_005732.3 | / | c.1452+7T>G | p.? | intronic | / | VUS | 1 |
| chr5:131939174 | / | c.2390G>A | p.(Arg797Lys) | missense | / | VUS | 1 | ||
| RAD51C | chr17:56798178 | NM_058216.2 | rs587782702 | c.904+5G>T | p.(Val280Glyfs*11) | intronic | / | LP | 1 |
| chr17:56801430 | rs730881932 | c.934C>T | p.(Arg312Trp) | missense | 0.00001 | LP | 1 | ||
| chr17:56809908 | rs587781410 | c.1026+5_1026+7del | p.(Arg322Serfs*22) | intronic | / | LP | 1 | ||
| RECQL | chr12:21643163 | NM_032941.2 | New | c.363_364del | p.(Cys122Leufs*43) | frameshift | / | P | 1 |
| chr12:21643150 | rs1267616869 | c.377C>T | p.(Pro126Leu) | missense | / | VUS | 1 | ||
| STK11 | chr19:1219382 | NM_000455.4 | rs369764220 | c.434A>G | p.(Glu145Gly) | missense | 0.0000134 | VUS | 1 |
| ID Sample | Sex | Gene | P/LP Variants | Other Variants (VUS ) | Age (Years) | Age at Onset (Years) | Tumor Site | Histological Diagnosis | Grading | Other Personal History of Cancer (Onset in Years) | Family History of Cancer (n of 1st and 2nd Degree Affected Relatives) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P29 | F | ATM | c.875C>T p.(Pro292Leu) | RECQL c.377C>T p.(Pro126Leu) | 44 | 43 | breast (left) | DCIS | G1 | M(42) | M (1); CNSC (1) |
| P89 | M | ATM | c.3576G>A p.(Lys1192=) | - | 66 | 66 | pancreas | Adenocarcinoma | na | GC(45) | GC (8) |
| P113 | F | RAD51C | c.904+5G>T p.(Val280Glyfs*11) | - | 46 | 43 | ovary | HSGC | na | - | NHL(1); OC (2), TC (1) |
| P33 | F | RAD51C | c.934C>T p.(Arg312Trp) | c.1847C>G p.(Pro616Arg) | 47 | 35 | breast (left); breast (right) | CLI; IDC | G3 | - | BC (1 + 1); CC (1); BlC (1), CNSC(1) |
| P110 | F | RAD51C | c.1026+5_1026+7del p.(Arg322Serfs*22) | - | 53 | 51 | ovary | HSGC | na | - | OC (1), GC (1), EC (1); PrC (1); LC (1) |
| P67 | F | MLH1 | c.1696T>C p.(Tyr566His) | - | 39 | 39 | breast (right) | IDC | G3 | - | BC (1); PrC (2) |
| P53 | F | MSH2 | c.182A>C p.(Gln61Pro) | - | 58 | 56 | breast (left) | IDC | G3 | - | PrC (3 + 1), UC (1); unk C (1) |
| P90 | M | RECQL | c.363_364del p.(Cys122Leufs*43) | - | 72 | 59 | breast (right) | IDC | G2 | PrC | BC (1); LC (1) |
| P18 | F | CHEK2 | c.470T>C p.(Ile157Thr) | - | 47 | 42 | breast (left) | CDI | G2 | CC, OC, GC | PrC (1); RC (1) |
| P46 | F | CHEK2 | c.470T>C p.(Ile157Thr) | PMS2 c.145G>A p.(Ala49Thr) | 51 | 50 | breast (right) | DCIS | G1 | - | OC (1); UC (1); RC (1) |
| P68 | F | CHEK2 | c.793-1G>A p.(Asp265Thrfs*10) | - | 47 | 27 | tyroid | papillary | na | - | BC (1), PrC(1 + 3); GC (1); LC (1); S(1) |
| P111 | F | CHEK2 | c.1100del p.(Thr367Metfs*15) | - | 56 | 50 | breast (bil) | IDC | G2 (right) G1 (left) | - | BC (1) |
| P112 | F | CHEK2 | c.1100del p.(Thr367Metfs*15) | - | 66 | 64 | breast (bil) | IDC | G3 (right) G2 (left) | dML | BC (2 + 1), unk C (1) |
| P12 | F | CHEK2 | c.1136C>G p.(Ser379Cys) | RAD50 c.1452+7T>G | 49 | 49 | skin/foot | melanoma | na | - | BC (2 + 1); GC (1); PC (1); LC (2), head-neck C (1) |
| P60 | M | CHEK2 | c.1367C>T p.(Ser456Leu) | - | 71 | 71 | breast | DCIS | G2 | - | BC (2 + 1), OC (1 + 1); LC (1) |
| P04 | F | CHEK2 | c.1169A>C p.(Tyr390Ser) | - | 53 | 52 | uterus | Mullerian sarcoma | na | - | BC (2), CC (3 + 1) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Germani, A.; Petrucci, S.; De Marchis, L.; Libi, F.; Savio, C.; Amanti, C.; Bonifacino, A.; Campanella, B.; Capalbo, C.; Lombardi, A.; et al. Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers. J. Clin. Med. 2020, 9, 3003. https://doi.org/10.3390/jcm9093003
Germani A, Petrucci S, De Marchis L, Libi F, Savio C, Amanti C, Bonifacino A, Campanella B, Capalbo C, Lombardi A, et al. Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers. Journal of Clinical Medicine. 2020; 9(9):3003. https://doi.org/10.3390/jcm9093003
Chicago/Turabian StyleGermani, Aldo, Simona Petrucci, Laura De Marchis, Fabio Libi, Camilla Savio, Claudio Amanti, Adriana Bonifacino, Barbara Campanella, Carlo Capalbo, Augusto Lombardi, and et al. 2020. "Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers" Journal of Clinical Medicine 9, no. 9: 3003. https://doi.org/10.3390/jcm9093003
APA StyleGermani, A., Petrucci, S., De Marchis, L., Libi, F., Savio, C., Amanti, C., Bonifacino, A., Campanella, B., Capalbo, C., Lombardi, A., Maggi, S., Mattei, M., Osti, M. F., Pellegrini, P., Speranza, A., Stanzani, G., Vitale, V., Pizzuti, A., Torrisi, M. R., & Piane, M. (2020). Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers. Journal of Clinical Medicine, 9(9), 3003. https://doi.org/10.3390/jcm9093003