Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence


 ,
, 

Abstract
1. Introduction
2. The PI3K/Akt/mTOR Signaling Pathway
2.1. Regulation of the PI3K/Akt/mTOR Pathway in Normal Hematopoiesis
2.2. Constitutive PI3K/Akt/mTOR Activation in AML
2.3. Targeting the PI3K/Akt/mTOR Signaling Pathway in AML
3. Crosstalk of the PI3K/Akt/mTOR Signaling Pathway with Other Signaling Pathways in AML
4. Clinical Implications of PI3K/Akt/mTOR Inhibitors in AML
4.1. Buparlisib
4.2. Gedatolisib
4.3. Idelalisib
4.4. Dactolisib
4.5. MK-2206
4.6. Afuresertib
4.7. Uprosertib
4.8. Sirolimus
4.9. Everolimus
5. Clinical Strategies to Overcome Resistance to PI3K/Akt/mTOR Inhibitors in AML
5.1. PI3K/Akt/mTOR Inhibitors in Combination with Epigenetic Targeting
5.2. PI3K/Akt/mTOR Inhibitors in Combination with Bcl-2 Inhibitors
5.3. PI3K/Akt/mTOR Inhibitors in Combination with Kinase Inhibitors
5.4. PI3K/Akt/mTOR Inhibitors in Combination with DNA Repair Inhibitors
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Saultz, J.N.; Garzon, R. Acute Myeloid Leukemia: A Concise Review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Wang, F.; Loghavi, S.; Bueso-Ramos, C.; Gumbs, C.; Little, L.; Song, X.; Zhang, J.; Kadia, T.; Borthakur, G.; et al. Late relapse in acute myeloid leukemia (AML): Clonal evolution or therapy-related leukemia? Blood Cancer J. 2019, 9, 7. [Google Scholar] [CrossRef]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Riether, C.; Schürch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M. Advances in understanding the leukaemia microenvironment. Br. J. Haematol. 2014, 164, 767–778. [Google Scholar] [CrossRef]
- Zhou, H.-S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef]
- Wang, X.; Huang, S.; Chen, J.-L. Understanding of leukemic stem cells and their clinical implications. Mol. Cancer 2017, 16, 2. [Google Scholar] [CrossRef]
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; Cappellini, A.; Ognibene, A.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim. Biophys. Acta 2010, 1803, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Guida, M.; Accordi, B.; Mediani, L.; Martelli, A.M.; Barozzi, P.; Petricoin, E.; Liotta, L.; Milani, G.; Giordan, M.; et al. Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia 2014, 28, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.-A.; Tien, H.-F. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J. Biomed. Sci. 2020, 27. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Zhou, F.; Ge, Z.; Chen, B. Quizartinib (AC220): A promising option for acute myeloid leukemia. Drug Des. Dev. Ther. 2019, 13, 1117–1125. [Google Scholar] [CrossRef]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef]
- Guzman, M.L.; Swiderski, C.F.; Howard, D.S.; Grimes, B.A.; Rossi, R.M.; Szilvassy, S.J.; Jordan, C.T. Preferential induction of apoptosis for primary human leukemic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16220–16225. [Google Scholar] [CrossRef]
- Zhou, J.; Ching, Y.Q.; Chng, W.-J. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: From molecular pathogenesis to therapeutic target. Oncotarget 2015, 6, 5490–5500. [Google Scholar] [CrossRef]
- Li, F.; Sethi, G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 2010, 1805, 167–180. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Zhou, J.; Chooi, J.-Y.; Ching, Y.Q.; Quah, J.Y.; Toh, S.H.-M.; Ng, Y.; Tan, T.Z.; Chng, W.-J. NF-κB promotes the stem-like properties of leukemia cells by activation of LIN28B. World J. Stem Cells 2018, 10, 34–42. [Google Scholar] [CrossRef]
- Birkenkamp, K.U.; Geugien, M.; Schepers, H.; Westra, J.; Lemmink, H.H.; Vellenga, E. Constitutive NF-kappaB DNA-binding activity in AML is frequently mediated by a Ras/PI3-K/PKB-dependent pathway. Leukemia 2004, 18, 103–112. [Google Scholar] [CrossRef]
- Xu, Q.; Simpson, S.-E.; Scialla, T.J.; Bagg, A.; Carroll, M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003, 102, 972–980. [Google Scholar] [CrossRef]
- Xu, Q.; Thompson, J.E.; Carroll, M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 2005, 106, 4261–4268. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Frasson, C.; Chiarini, F.; D’Avella, D.; Accordi, B.; Anselmi, L.; Barozzi, P.; Forghieri, F.; Luppi, M.; Martelli, A.M.; et al. Dual inhibition of PI3K/mTOR signaling in chemoresistant AML primary cells. Adv. Biol. Regul. 2018, 68, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Grandage, V.L.; Gale, R.E.; Linch, D.C.; Khwaja, A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia 2005, 19, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Vachhani, P.; Bose, P.; Rahmani, M.; Grant, S. Rational combination of dual PI3K/mTOR blockade and Bcl-2/-xL inhibition in AML. Physiol. Genom. 2014, 46, 448–456. [Google Scholar] [CrossRef]
- Markham, A. Copanlisib: First Global Approval. Drugs 2017, 77, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A.M. Duvelisib: A 2018 Novel FDA-Approved Small Molecule Inhibiting Phosphoinositide. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef]
- Miller, B.W.; Przepiorka, D.; de Claro, R.A.; Lee, K.; Nie, L.; Simpson, N.; Gudi, R.; Saber, H.; Shord, S.; Bullock, J.; et al. FDA approval: Idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin. Cancer Res. 2015, 21, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Nepstad, I.; Hatfield, K.J.; Gronningsaeter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Vadas, O.; Burke, J.E.; Zhang, X.; Berndt, A.; Williams, R.L. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci. Signal. 2011, 4, re2. [Google Scholar] [CrossRef] [PubMed]
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef]
- Brock, C.; Schaefer, M.; Reusch, H.P.; Czupalla, C.; Michalke, M.; Spicher, K.; Schultz, G.; Nürnberg, B. Roles of Gβγ in membrane recruitment and activation of p110γ/p101 phosphoinositide 3-kinase γ. J. Cell Biol. 2003, 160, 89–99. [Google Scholar] [CrossRef]
- Kok, K.; Nock, G.E.; Verrall, E.A.G.; Mitchell, M.P.; Hommes, D.W.; Peppelenbosch, M.P.; Vanhaesebroeck, B. Regulation of p110delta PI 3-kinase gene expression. PLoS ONE 2009, 4, e5145. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Kiyatkin, A.; Aksamitiene, E.; Markevich, N.I.; Borisov, N.M.; Hoek, J.B.; Kholodenko, B.N. Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. J. Biol. Chem. 2006, 281, 19925–19938. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef]
- Toker, A.; Marmiroli, S. Signaling Specificity in the Akt Pathway in Biology and Disease. Adv. Biol. Regul. 2014, 55, 28–38. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.M.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Turner, K.M.; Alfred Yung, W.K.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-oncology 2014, 16, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hung, M.-C. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2010, 2, 19–42. [Google Scholar]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stöckli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Hazen, A.L.; Smith, M.J.; Desponts, C.; Winter, O.; Moser, K.; Kerr, W.G. SHIP is required for a functional hematopoietic stem cell niche. Blood 2009, 113, 2924–2933. [Google Scholar] [CrossRef]
- Luo, J.-M.; Liu, Z.-L.; Hao, H.-L.; Wang, F.-X.; Dong, Z.-R.; Ohno, R. Mutation analysis of SHIP gene in acute leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2004, 12, 420–426. [Google Scholar] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Park, S.R.; Yoo, Y.J.; Ban, Y.-H.; Yoon, Y.J. Biosynthesis of rapamycin and its regulation: Past achievements and recent progress. J. Antibiot. 2010, 63, 434–441. [Google Scholar] [CrossRef]
- Gibbons, J.J.; Abraham, R.T.; Yu, K. Mammalian target of rapamycin: Discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin. Oncol. 2009, 36 (Suppl. S3), S3–S17. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef]
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J. Biol. Chem. 2003, 278, 10189–10194. [Google Scholar] [CrossRef] [PubMed]
- Chiang, G.G.; Abraham, R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.W.Y.; Fryer, L.G.D.; Carling, D.; Shepherd, P.R. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J. Biol. Chem. 2004, 279, 15719–15722. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Blenis, J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef]
- Hardie, D.G. New roles for the LKB1-->AMPK pathway. Curr. Opin. Cell Biol. 2005, 17, 167–173. [Google Scholar] [CrossRef]
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428. [Google Scholar] [CrossRef]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef]
- Carriere, A.; Romeo, Y.; Acosta-Jaquez, H.A.; Moreau, J.; Bonneil, E.; Thibault, P.; Fingar, D.C.; Roux, P.P. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 567–577. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Han, J.W.; Pearson, R.B.; Dennis, P.B.; Thomas, G. Rapamycin, wortmannin, and the methylxanthine SQ20006 inactivate p70s6k by inducing dephosphorylation of the same subset of sites. J. Biol. Chem. 1995, 270, 21396–21403. [Google Scholar] [CrossRef]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: From protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol. 2004, 24, 3112–3124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.J.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [CrossRef] [PubMed]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.A.; Ikenoue, T.; Nakada, D.; Lee, J.Y.; Guan, K.-L.; Morrison, S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012, 11, 415–428. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.-L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef]
- Tothova, Z.; Gilliland, D.G. FoxO transcription factors and stem cell homeostasis: Insights from the hematopoietic system. Cell Stem Cell 2007, 1, 140–152. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Fransecky, L.; Mochmann, L.H.; Baldus, C.D. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol. Cell. Ther. 2015, 3, 2. [Google Scholar] [CrossRef]
- Gao, Y.; Gao, J.; Li, M.; Zheng, Y.; Wang, Y.; Zhang, H.; Wang, W.; Chu, Y.; Wang, X.; Xu, M.; et al. Rheb1 promotes tumor progression through mTORC1 in MLL-AF9-initiated murine acute myeloid leukemia. J. Hematol. Oncol. 2016, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Kobayashi, M.; Ramdas, B.; Chatterjee, A.; Ma, P.; Mali, R.S.; Carlesso, N.; Liu, Y.; Plas, D.R.; Chan, R.J.; et al. S6K1 regulates hematopoietic stem cell self-renewal and leukemia maintenance. J. Clin. Investig. 2016, 126, 2621–2625. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Kapur, R. Role of mTORC1-S6K1 signaling pathway in regulation of hematopoietic stem cell and acute myeloid leukemia. Exp. Hematol. 2017, 50, 13–21. [Google Scholar] [CrossRef]
- Zabkiewicz, J.; Pearn, L.; Hills, R.K.; Morgan, R.G.; Tonks, A.; Burnett, A.K.; Darley, R.L. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica 2014, 99, 858–864. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760. [Google Scholar] [CrossRef]
- Zeng, Z.; Shi, Y.X.; Tsao, T.; Qiu, Y.; Kornblau, S.M.; Baggerly, K.A.; Liu, W.; Jessen, K.; Liu, Y.; Kantarjian, H.; et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012, 120, 2679–2689. [Google Scholar] [CrossRef]
- Reikvam, H.; Nepstad, I.; Bruserud, Ø.; Hatfield, K.J. Pharmacological targeting of the PI3K/mTOR pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013, 4, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Gallay, N.; Dos Santos, C.; Cuzin, L.; Bousquet, M.; Simmonet Gouy, V.; Chaussade, C.; Attal, M.; Payrastre, B.; Demur, C.; Récher, C. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 2009, 23, 1029–1038. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef]
- Min, Y.H.; Eom, J.I.; Cheong, J.W.; Maeng, H.O.; Kim, J.Y.; Jeung, H.K.; Lee, S.T.; Lee, M.H.; Hahn, J.S.; Ko, Y.W. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: Its significance as a prognostic variable. Leukemia 2003, 17, 995–997. [Google Scholar] [CrossRef]
- Martelli, A.M.; Nyåkern, M.; Tabellini, G.; Bortul, R.; Tazzari, P.L.; Evangelisti, C.; Cocco, L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006, 20, 911–928. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Aasebø, E.; Hernandez-Valladares, M.; Brenner, A.K.; Bartaula-Brevik, S.; Berven, F.; Selheim, F.; Skavland, J.; Gjertsen, B.T.; et al. Two acute myeloid leukemia patient subsets are identified based on the constitutive PI3K-Akt-mTOR signaling of their leukemic cells; a functional, proteomic, and transcriptomic comparison. Expert Opin. Ther. Targets 2018, 22, 639–653. [Google Scholar] [CrossRef]
- Chapuis, N.; Tamburini, J.; Cornillet-Lefebvre, P.; Gillot, L.; Bardet, V.; Willems, L.; Park, S.; Green, A.S.; Ifrah, N.; Dreyfus, F.; et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: Therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 2010, 95, 415–423. [Google Scholar] [CrossRef]
- Brandts, C.H.; Sargin, B.; Rode, M.; Biermann, C.; Lindtner, B.; Schwäble, J.; Buerger, H.; Müller-Tidow, C.; Choudhary, C.; McMahon, M.; et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005, 65, 9643–9650. [Google Scholar] [CrossRef]
- Tamburini, J.; Green, A.S.; Bardet, V.; Chapuis, N.; Park, S.; Willems, L.; Uzunov, M.; Ifrah, N.; Dreyfus, F.; Lacombe, C.; et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009, 114, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.; Minden, M.D.; Hedley, D.W. Constitutive phosphorylation of the S6 ribosomal protein via mTOR and ERK signaling in the peripheral blasts of acute leukemia patients. Exp. Hematol. 2006, 34, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Heiser, D.; Li, L.; Kaplan, I.; Collector, M.; Huso, D.; Sharkis, S.J.; Civin, C.; Small, D. FLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasm. Cell Stem Cell 2012, 11, 346–358. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Kikushige, Y.; Yoshimoto, G.; Miyamoto, T.; Iino, T.; Mori, Y.; Iwasaki, H.; Niiro, H.; Takenaka, K.; Nagafuji, K.; Harada, M.; et al. Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J. Immunol. 2008, 180, 7358–7367. [Google Scholar] [CrossRef]
- Chen, W.; Drakos, E.; Grammatikakis, I.; Schlette, E.J.; Li, J.; Leventaki, V.; Staikou-Drakopoulou, E.; Patsouris, E.; Panayiotidis, P.; Medeiros, L.J.; et al. mTOR signaling is activated by FLT3 kinase and promotes survival of FLT3-mutated acute myeloid leukemia cells. Mol. Cancer 2010, 9, 292. [Google Scholar] [CrossRef]
- Zhang, S.; Broxmeyer, H.E. p85 subunit of PI3 kinase does not bind to human Flt3 receptor, but associates with SHP2, SHIP, and a tyrosine-phosphorylated 100-kDa protein in Flt3 ligand-stimulated hematopoietic cells. Biochem. Biophys. Res. Commun. 1999, 254, 440–445. [Google Scholar] [CrossRef]
- Scheijen, B.; Ngo, H.T.; Kang, H.; Griffin, J.D. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 2004, 23, 3338–3349. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Santo, E.E.; Stroeken, P.; Sluis, P.V.; Koster, J.; Versteeg, R.; Westerhout, E.M. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer Res. 2013, 73, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Sujobert, P.; Bardet, V.; Cornillet-Lefebvre, P.; Hayflick, J.S.; Prie, N.; Verdier, F.; Vanhaesebroeck, B.; Muller, O.; Pesce, F.; Ifrah, N.; et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005, 106, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.; Graf, S. Idelalisib for the treatment of non-Hodgkin lymphoma. Expert Opin. Pharmacother. 2016, 17, 265–274. [Google Scholar] [CrossRef]
- Nguyen, L.X.T.; Sesay, A.; Mitchell, B.S. Effect of CAL-101, a PI3Kδ inhibitor, on ribosomal rna synthesis and cell proliferation in acute myeloid leukemia cells. Blood Cancer J. 2014, 4, e228. [Google Scholar] [CrossRef] [PubMed]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Kaplan, J.B.; Altman, J.K.; Giles, F.J.; Platanias, L.C. Targeting mTOR signaling pathways and related negative feedback loops for the treatment of acute myeloid leukemia. Cancer Biol. Ther. 2015, 16, 648–656. [Google Scholar] [CrossRef]
- Chapuis, N.; Tamburini, J.; Green, A.S.; Vignon, C.; Bardet, V.; Neyret, A.; Pannetier, M.; Willems, L.; Park, S.; Macone, A.; et al. Dual Inhibition of PI3K and mTORC1/2 Signaling by NVP-BEZ235 as a New Therapeutic Strategy for Acute Myeloid Leukemia. Clin. Cancer Res. 2010, 16, 5424–5435. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef]
- Hayakawa, F.; Towatari, M.; Kiyoi, H.; Tanimoto, M.; Kitamura, T.; Saito, H.; Naoe, T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000, 19, 624–631. [Google Scholar] [CrossRef]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.-T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rebechi, M.T.; Pratz, K.W. Genomic instability is a principle pathologic feature of FLT3 ITD kinase activity in acute myeloid leukemia leading to clonal evolution and disease progression. Leuk. Lymphoma 2017, 58, 2040–2050. [Google Scholar] [CrossRef]
- Ng, K.L.N.; Lynn, C.; Leung, T.W.Y.; So, E.C.W.; Leung, A.Y.H. Targeting DNA Damage and Repair in Acute Myeloid Leukemia Carrying Internal Tandem Duplication of Fms-like Tyrosine Kinase 3 (FLT3-ITD)-a Mechanistic Study. Blood 2019, 134, 1261. [Google Scholar] [CrossRef]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Kodaki, T.; Woscholski, R.; Hallberg, B.; Rodriguez-Viciana, P.; Downward, J.; Parker, P.J. The activation of phosphatidylinositol 3-kinase by Ras. Curr. Biol. 1994, 4, 798–806. [Google Scholar] [CrossRef]
- She, Q.-B.; Solit, D.B.; Ye, Q.; O’Reilly, K.E.; Lobo, J.; Rosen, N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 2005, 8, 287–297. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.-Y.; Lai, C.-C.; Chang, C.-J.; Huang, W.-C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Rakus, D.; Gizak, A.; Steelman, L.S.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Yang, L.V.; Montalto, G.; Cervello, M.; et al. Effects of mutations in Wnt/β-catenin, hedgehog, Notch and PI3K pathways on GSK-3 activity-Diverse effects on cell growth, metabolism and cancer. Biochim. Biophys. Acta 2016, 1863, 2942–2976. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Birkenkamp, K.U.; Geugien, M.; Lemmink, H.H.; Kruijer, W.; Vellenga, E. Regulation of constitutive STAT5 phosphorylation in acute myeloid leukemia blasts. Leukemia 2001, 15, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Müller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. JAK2/STAT5 Inhibition Circumvents Resistance to PI3K/mTOR Blockade: A Rationale for Cotargeting These Pathways in Metastatic Breast Cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef]
- Spiekermann, K.; Bagrintseva, K.; Schwab, R.; Schmieja, K.; Hiddemann, W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin. Cancer Res. 2003, 9, 2140–2150. [Google Scholar]
- Choudhary, C.; Brandts, C.; Schwable, J.; Tickenbrock, L.; Sargin, B.; Ueker, A.; Böhmer, F.-D.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood 2007, 110, 370–374. [Google Scholar] [CrossRef]
- Green, A.S.; Maciel, T.T.; Hospital, M.-A.; Yin, C.; Mazed, F.; Townsend, E.C.; Pilorge, S.; Lambert, M.; Paubelle, E.; Jacquel, A.; et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci. Adv. 2015, 1, e1500221. [Google Scholar] [CrossRef]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015, 5, e325. [Google Scholar] [CrossRef]
- Kim, K.-T.; Baird, K.; Ahn, J.-Y.; Meltzer, P.; Lilly, M.; Levis, M.; Small, D. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood 2005, 105, 1759–1767. [Google Scholar] [CrossRef]
- Natarajan, K.; Xie, Y.; Burcu, M.; Linn, D.E.; Qiu, Y.; Baer, M.R. Pim-1 kinase phosphorylates and stabilizes 130 kDa FLT3 and promotes aberrant STAT5 signaling in acute myeloid leukemia with FLT3 internal tandem duplication. PLoS ONE 2013, 8, e74653. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer. Biomolecules 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Beharry, Z.M.; Harris, T.E.; Lilly, M.B.; Smith, C.D.; Mahajan, S.; Kraft, A.S. PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol. Ther. 2009, 8, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Thompson, C.B. The survival kinases Akt and Pim as potential pharmacological targets. J. Clin. Investig. 2005, 115, 2618–2624. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Mahajan, S.; Wang, W.; Kraft, A.S. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer Res. 2013, 73, 3402–3411. [Google Scholar] [CrossRef]
- Song, J.H.; Padi, S.K.R.; Luevano, L.A.; Minden, M.D.; DeAngelo, D.J.; Hardiman, G.; Ball, L.E.; Warfel, N.A.; Kraft, A.S. Insulin receptor substrate 1 is a substrate of the Pim protein kinases. Oncotarget 2016, 7, 20152–20165. [Google Scholar] [CrossRef]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef]
- Okada, K.; Nogami, A.; Ishida, S.; Akiyama, H.; Chen, C.; Umezawa, Y.; Miura, O. FLT3-ITD induces expression of Pim kinases through STAT5 to confer resistance to the PI3K/Akt pathway inhibitors on leukemic cells by enhancing the mTORC1/Mcl-1 pathway. Oncotarget 2018, 9, 8870–8886. [Google Scholar] [CrossRef]
- Nogami, A.; Oshikawa, G.; Okada, K.; Fukutake, S.; Umezawa, Y.; Nagao, T.; Kurosu, T.; Miura, O. FLT3-ITD confers resistance to the PI3K/Akt pathway inhibitors by protecting the mTOR/4EBP1/Mcl-1 pathway through STAT5 activation in acute myeloid leukemia. Oncotarget 2015, 6, 9189–9205. [Google Scholar] [CrossRef]
- Nogami, A.; Okada, K.; Ishida, S.; Akiyama, H.; Umezawa, Y.; Miura, O. Inhibition of the STAT5/Pim Kinase Axis Enhances Cytotoxic Effects of Proteasome Inhibitors on FLT3-ITD-Positive AML Cells by Cooperatively Inhibiting the mTORC1/4EBP1/S6K/Mcl-1 Pathway. Transl. Oncol. 2019, 12, 336–349. [Google Scholar] [CrossRef]
- Watanabe, D.; Nogami, A.; Okada, K.; Akiyama, H.; Umezawa, Y.; Miura, O. FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers 2019, 11, 1827. [Google Scholar] [CrossRef] [PubMed]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Chuang, C.-C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Sillar, J.R.; Germon, Z.P.; DeIuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.-D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef]
- Stanicka, J.; Russell, E.G.; Woolley, J.F.; Cotter, T.G. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J. Biol. Chem. 2015, 290, 9348–9361. [Google Scholar] [CrossRef]
- Mi, T.; Wang, Z.; Bunting, K.D. The Cooperative Relationship between STAT5 and Reactive Oxygen Species in Leukemia: Mechanism and Therapeutic Potential. Cancers 2018, 10, 359. [Google Scholar] [CrossRef]
- Bourgeais, J.; Gouilleux-Gruart, V.; Gouilleux, F. Oxidative metabolism in cancer: A STAT affair? Jak-Stat 2013, 2, e25764. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Jayavelu, A.K.; Stanicka, J.; Roche, S.L.; O’Brien, R.L.; Scholl, S.; Böhmer, F.-D.; Cotter, T.G. Nuclear membrane-localised NOX4D generates pro-survival ROS in FLT3-ITD-expressing AML. Oncotarget 2017, 8, 105440–105457. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Wada, Y.; Kitagishi, Y.; Matsuda, S. Link between PI3K/AKT/PTEN Pathway and NOX Proteinin Diseases. Aging Dis. 2014, 5, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Nakaso, K.; Yano, H.; Fukuhara, Y.; Takeshima, T.; Wada-Isoe, K.; Nakashima, K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003, 546, 181–184. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential Roles of the PI3 Kinase/Akt Pathway in Regulating Nrf2-Dependent Antioxidant Functions in the RPE. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef]
- Vazquez, F.; Grossman, S.R.; Takahashi, Y.; Rokas, M.V.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J. Biol. Chem. 2001, 276, 48627–48630. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Hasskarl, J. Everolimus. Recent Results Cancer Res. 2018, 211, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Maira, S.-M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef]
- Burger, M.T.; Pecchi, S.; Wagman, A.; Ni, Z.-J.; Knapp, M.; Hendrickson, T.; Atallah, G.; Pfister, K.; Zhang, Y.; Bartulis, S.; et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011, 2, 774–779. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Guan, W.; Qian, M.; Yao, Z.; Yin, X.; Zhao, H. NVP-BKM120 inhibits colon cancer growth via FoxO3a-dependent PUMA induction. Oncotarget 2017, 8, 83052–83062. [Google Scholar] [CrossRef]
- Jin, L.; Jin, M.-H.; Nam, A.-R.; Park, J.-E.; Bang, J.-H.; Oh, D.-Y.; Bang, Y.-J. Anti-tumor effects of NVP-BKM120 alone or in combination with MEK162 in biliary tract cancer. Cancer Lett. 2017, 411, 162–170. [Google Scholar] [CrossRef]
- Yang, L.; Yang, G.; Ding, Y.; Huang, Y.; Liu, S.; Zhou, L.; Wei, W.; Wang, J.; Hu, G. Combined treatment with PI3K inhibitor BKM120 and PARP inhibitor olaparib is effective in inhibiting the gastric cancer cells with ARID1A deficiency. Oncol. Rep. 2018, 40, 479–487. [Google Scholar] [CrossRef]
- Allegretti, M.; Ricciardi, M.R.; Licchetta, R.; Mirabilii, S.; Orecchioni, S.; Reggiani, F.; Talarico, G.; Foà, R.; Bertolini, F.; Amadori, S.; et al. The pan-class I phosphatidyl-inositol-3 kinase inhibitor NVP-BKM120 demonstrates anti-leukemic activity in acute myeloid leukemia. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Sadeghi, S.; Esmaeili, S.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Bashash, D. PI3K abrogation using pan PI3K inhibitor BKM120 give rise to a weighty anti-cancer effect on AML-derived KG-1 cells by inducing apoptosis and G2/M arrest. Turk. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ragon, B.K.; Kantarjian, H.; Jabbour, E.; Ravandi, F.; Cortes, J.; Borthakur, G.; DeBose, L.; Zeng, Z.; Schneider, H.; Pemmaraju, N.; et al. Buparlisib, a PI3K inhibitor, demonstrates acceptable tolerability and preliminary activity in a phase I trial of patients with advanced leukemias. Am. J. Hematol. 2017, 92, 7–11. [Google Scholar] [CrossRef]
- Venkatesan, A.M.; Dehnhardt, C.M.; Delos Santos, E.; Chen, Z.; Dos Santos, O.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis(morpholino-1,3,5-triazine) derivatives: Potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: Discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J. Med. Chem. 2010, 53, 2636–2645. [Google Scholar] [CrossRef] [PubMed]
- Mallon, R.; Feldberg, L.R.; Lucas, J.; Chaudhary, I.; Dehnhardt, C.; Santos, E.D.; Chen, Z.; dos Santos, O.; Ayral-Kaloustian, S.; Venkatesan, A.; et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin. Cancer Res. 2011, 17, 3193–3203. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Alsina, M.; Soares, H.P.; Braña, I.; Britten, C.D.; Del Conte, G.; Ezeh, P.; Houk, B.; Kern, K.A.; Leong, S.; et al. A Multi-Arm Phase I Study of the PI3K/mTOR Inhibitors PF-04691502 and Gedatolisib (PF-05212384) plus Irinotecan or the MEK Inhibitor PD-0325901 in Advanced Cancer. Target Oncol. 2017, 12, 775–785. [Google Scholar] [CrossRef]
- Langdon, S.P.; Kay, C.; Um, I.H.; Dodds, M.; Muir, M.; Sellar, G.; Kan, J.; Gourley, C.; Harrison, D.J. Evaluation of the dual mTOR/PI3K inhibitors Gedatolisib (PF-05212384) and PF-04691502 against ovarian cancer xenograft models. Sci. Rep. 2019, 9, 18742. [Google Scholar] [CrossRef]
- Iezzi, A.; Caiola, E.; Broggini, M. Activity of Pan-Class I Isoform PI3K/mTOR Inhibitor PF-05212384 in Combination with Crizotinib in Ovarian Cancer Xenografts and PDX. Transl. Oncol. 2016, 9, 458–465. [Google Scholar] [CrossRef][Green Version]
- Del Campo, J.M.; Birrer, M.; Davis, C.; Fujiwara, K.; Gollerkeri, A.; Gore, M.; Houk, B.; Lau, S.; Poveda, A.; González-Martín, A.; et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol. Oncol. 2016, 142, 62–69. [Google Scholar] [CrossRef]
- Soares, H.P.; Ming, M.; Mellon, M.; Young, S.H.; Han, L.; Sinnet-Smith, J.; Rozengurt, E. Dual PI3K/mTOR inhibitors induce rapid over-activation of the MEK/ERK pathway in human pancreatic cancer cells through suppression of mTORC2. Mol. Cancer Ther. 2015, 14, 1014–1023. [Google Scholar] [CrossRef]
- Shah, K.; Moharram, S.A.; Kazi, J.U. Acute leukemia cells resistant to PI3K/mTOR inhibition display upregulation of P2RY14 expression. Clin. Epigenetics 2018, 10, 83. [Google Scholar] [CrossRef]
- Vargaftig, J.; Farhat, H.; Ades, L.; Briaux, A.; Benoist, C.; Turbiez, I.; Vey, N.; Glaisner, S.; Callens, C. Phase 2 Trial of Single Agent Gedatolisib (PF-05212384), a Dual PI3K/mTOR Inhibitor, for Adverse Prognosis and Relapse/Refractory AML: Clinical and Transcriptomic Results. Blood 2018, 132, 5233. [Google Scholar] [CrossRef]
- Furman, R.R.; Byrd, J.C.; Brown, J.R.; Coutre, S.E.; Benson, D.M.; Wagner-Johnston, N.D.; Flinn, I.W.; Kahl, B.S.; Spurgeon, S.E.; Lannutti, B.; et al. CAL-101, An Isoform-Selective Inhibitor of Phosphatidylinositol 3-Kinase P110δ, Demonstrates Clinical Activity and Pharmacodynamic Effects In Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia. Blood 2010, 116, 55. [Google Scholar] [CrossRef]
- Kahl, B.S.; Spurgeon, S.E.; Furman, R.R.; Flinn, I.W.; Coutre, S.E.; Brown, J.R.; Benson, D.M.; Byrd, J.C.; Peterman, S.; Cho, Y.; et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014, 123, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Kahl, B.S.; Leonard, J.P.; Furman, R.R.; Brown, J.R.; Byrd, J.C.; Wagner-Johnston, N.D.; Coutre, S.E.; Benson, D.M.; Peterman, S.; et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-δ, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood 2014, 123, 3406–3413. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Byrd, J.C.; Coutre, S.E.; Benson, D.M.; Flinn, I.W.; Wagner-Johnston, N.D.; Spurgeon, S.E.; Kahl, B.S.; Bello, C.; Webb, H.K.; et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014, 123, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Byrd, J.C.; Furman, R.R.; Brown, J.R.; Benson, D.M.; Coutre, S.E.; Kahl, B.S.; Smith, B.D.; Wagner-Johnston, N.D.; Spurgeon, S.E.; et al. Evidence of Clinical Activity in a Phase 1 Study of CAL-101, An Oral P110Δ Isoform-Selective Inhibitor of Phosphatidylinositol 3-Kinase, in Patients with Relapsed or Refractory B-Cell Malignancies. Blood 2009, 114, 922. [Google Scholar] [CrossRef]
- Pongas, G.; Fojo, T. BEZ235: When Promising Science Meets Clinical Reality. Oncologist 2016, 21, 1033–1034. [Google Scholar] [CrossRef]
- Carlo, M.I.; Molina, A.M.; Lakhman, Y.; Patil, S.; Woo, K.; DeLuca, J.; Lee, C.-H.; Hsieh, J.J.; Feldman, D.R.; Motzer, R.J.; et al. A Phase Ib Study of BEZ235, a Dual Inhibitor of Phosphatidylinositol 3-Kinase (PI3K) and Mammalian Target of Rapamycin (mTOR), in Patients with Advanced Renal Cell Carcinoma. Oncologist 2016, 21, 787–788. [Google Scholar] [CrossRef]
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klümpen, H.-J.; Capdevila, J.; et al. Phase II Study of BEZ235 versus Everolimus in Patients with Mammalian Target of Rapamycin Inhibitor-Naïve Advanced Pancreatic Neuroendocrine Tumors. Oncologist 2018, 23, 766-e90. [Google Scholar] [CrossRef]
- Huang, J.-C.; Cui, Z.-F.; Chen, S.-M.; Yang, L.-J.; Lian, H.-K.; Liu, B.; Su, Z.-H.; Liu, J.-S.; Wang, M.; Hu, Z.-B.; et al. NVP-BEZ235 synergizes cisplatin sensitivity in osteosarcoma. Oncotarget 2018, 9, 10483–10496. [Google Scholar] [CrossRef]
- Ma, Y.; Jin, Z.; Yu, K.; Liu, Q. NVP-BEZ235-induced autophagy as a potential therapeutic approach for multiple myeloma. Am. J. Transl. Res. 2019, 11, 87–105. [Google Scholar] [PubMed]
- Mitchell, R.; Hopcroft, L.E.M.; Baquero, P.; Allan, E.K.; Hewit, K.; James, D.; Hamilton, G.; Mukhopadhyay, A.; O’Prey, J.; Hair, A.; et al. Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by mTOR and Autophagy Inhibition. J. Natl. Cancer Inst. 2018, 110, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-Y.; Hu, L.-S.; Han, H.-J.; Song, C.-Y.; Huang, Y.-X.; Guo, K.-Y. [NVP-BEZ235 inhibits proliferation and colony-forming capability of CD34(+)CD38(-) human acute myeloid leukemia stem cells]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2013, 21, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Jiang, L.; Ln, X.-H.; Tseng, K.-F.; Liu, Y.; Zhang, X.; Dong, R.-H.; Lu, Z.-G.; Wang, X.-J. The PI3K/mTOR dual inhibitor BEZ235 suppresses proliferation and migration and reverses multidrug resistance in acute myeloid leukemia. Acta Pharmacol. Sin. 2017, 38, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Wunderle, L.; Badura, S.; Lang, F.; Wolf, A.; Schleyer, E.; Serve, H.; Goekbuget, N.; Pfeifer, H.; Bug, G.; Ottmann, O.G. Safety and Efficacy of BEZ235, a Dual PI3-Kinase /mTOR Inhibitor, In Adult Patients with Relapsed or Refractory Acute Leukemia: Results of a Phase I Study. Blood 2013, 122, 2675. [Google Scholar] [CrossRef]
- Lu, J.-W.; Lin, Y.-M.; Lai, Y.-L.; Chen, C.-Y.; Hu, C.-Y.; Tien, H.-F.; Ou, D.-L.; Lin, L.-I. MK-2206 induces apoptosis of AML cells and enhances the cytotoxicity of cytarabine. Med. Oncol. 2015, 32, 206. [Google Scholar] [CrossRef]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III PIK3CA-Mutant ER-Positive and HER2-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 6823–6832. [Google Scholar] [CrossRef]
- Akcakanat, A.; Meric-Bernstam, F. MK-2206 window of opportunity study in breast cancer. Ann. Transl. Med. 2018, 6. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Walter, R.B.; Faderl, S.H.; Jabbour, E.J.; Zeng, Z.; Borthakur, G.; Huang, X.; Kadia, T.M.; Ruvolo, P.P.; Feliu, J.B.; et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin. Cancer Res. 2014, 20, 2226–2235. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef]
- Blagden, S.P.; Hamilton, A.L.; Mileshkin, L.; Wong, S.; Michael, A.; Hall, M.; Goh, J.C.; Lisyanskaya, A.S.; DeSilvio, M.; Frangou, E.; et al. Phase IB Dose Escalation and Expansion Study of AKT Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum-resistant Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Naymagon, L.; Abdul-Hay, M. Novel agents in the treatment of multiple myeloma: A review about the future. J. Hematol. Oncol. 2016, 9, 52. [Google Scholar] [CrossRef]
- Spencer, A.; Yoon, S.-S.; Harrison, S.J.; Morris, S.R.; Smith, D.A.; Brigandi, R.A.; Gauvin, J.; Kumar, R.; Opalinska, J.B.; Chen, C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 2014, 124, 2190–2195. [Google Scholar] [CrossRef] [PubMed]
- Algazi, A.P.; Esteve-Puig, R.; Nosrati, A.; Hinds, B.; Hobbs-Muthukumar, A.; Nandoskar, P.; Ortiz-Urda, S.; Chapman, P.B.; Daud, A. Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma. Pigment Cell Melanoma Res. 2018, 31, 110–114. [Google Scholar] [CrossRef]
- Westin, S.N.; Sill, M.W.; Coleman, R.L.; Waggoner, S.; Moore, K.N.; Mathews, C.A.; Martin, L.P.; Modesitt, S.C.; Lee, S.; Ju, Z.; et al. Safety lead-in of the MEK inhibitor trametinib in combination with GSK2141795, an AKT inhibitor, in patients with recurrent endometrial cancer: An NRG Oncology/GOG study. Gynecol. Oncol. 2019, 155, 420–428. [Google Scholar] [CrossRef]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 Inhibitor Trametinib in Combination with AKT Inhibitor GSK2141795 in Patients With Acute Myeloid Leukemia With RAS Mutations: A Phase II Study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440.e13. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, R.; Tseng, K.; Li, K.; Lu, Z.; Liu, Y.; Han, K.; Gan, Z.; Lin, S.; Hu, H.; et al. Sirolimus induces apoptosis and reverses multidrug resistance in human osteosarcoma cells in vitro via increasing microRNA-34b expression. Acta Pharmacol. Sin. 2016, 37, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Imrali, A.; Mao, X.; Yeste-Velasco, M.; Shamash, J.; Lu, Y. Rapamycin inhibits prostate cancer cell growth through cyclin D1 and enhances the cytotoxic efficacy of cisplatin. Am. J. Cancer Res. 2016, 6, 1772–1784. [Google Scholar]
- Yao, C.; Liu, J.; Shao, L. Rapamycin inhibits the proliferation and apoptosis of gastric cancer cells by down regulating the expression of survivin. Hepatogastroenterology 2011, 58, 1075–1080. [Google Scholar]
- Wagner, F.; Henningsen, B.; Lederer, C.; Eichenmüller, M.; Gödeke, J.; Müller-Höcker, J.; von Schweinitz, D.; Kappler, R. Rapamycin blocks hepatoblastoma growth in vitro and in vivo implicating new treatment options in high-risk patients. Eur. J. Cancer 2012, 48, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands-Where We Are and Where to Go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Colon-Otero, G.; Weroha, S.J.; Foster, N.R.; Haluska, P.; Hou, X.; Wahner-Hendrickson, A.E.; Jatoi, A.; Block, M.S.; Dinh, T.A.; Robertson, M.W.; et al. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol. Oncol. 2017, 146, 64–68. [Google Scholar] [CrossRef]
- Zhu, A.X.; Abrams, T.A.; Miksad, R.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Zheng, H.; Muzikansky, A.; Clark, J.W.; Kwak, E.L.; Schrag, D.; et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer 2011, 117, 5094–5102. [Google Scholar] [CrossRef]
- Perl, A.E.; Kasner, M.T.; Shank, D.; Luger, S.M.; Carroll, M. Single-cell pharmacodynamic monitoring of S6 ribosomal protein phosphorylation in AML blasts during a clinical trial combining the mTOR inhibitor sirolimus and intensive chemotherapy. Clin. Cancer Res. 2012, 18, 1716–1725. [Google Scholar] [CrossRef]
- Kasner, M.; Luger, S.M.; Jeschke, G.R.; Mick, R.; Carroll, M.; Perl, A.E. Single-Cell Pharmacodynamic Monitoring of S6 Ribosomal Protein in AML Blasts During Trials Combining Sirolimus and Intensive Chemotherapy: Target Inhibition Enhances Response. Blood 2011, 118, 230. [Google Scholar] [CrossRef]
- Kasner, M.T.; Mick, R.; Jeschke, G.R.; Carabasi, M.; Filicko-O’Hara, J.; Flomenberg, N.; Frey, N.V.; Hexner, E.O.; Luger, S.M.; Loren, A.W.; et al. Sirolimus enhances remission induction in patients with high risk acute myeloid leukemia and mTORC1 target inhibition. Investig. New Drugs 2018, 36, 657–666. [Google Scholar] [CrossRef]
- Perl, A.E.; Kasner, M.T.; Tsai, D.E.; Vogl, D.T.; Loren, A.W.; Schuster, S.J.; Porter, D.L.; Stadtmauer, E.A.; Goldstein, S.C.; Frey, N.V.; et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin. Cancer Res. 2009, 15, 6732–6739. [Google Scholar] [CrossRef]
- Raimondo, L.; D’Amato, V.; Servetto, A.; Rosa, R.; Marciano, R.; Formisano, L.; Mauro, C.D.; Orsini, R.C.; Cascetta, P.; Ciciola, P.; et al. Everolimus induces Met inactivation by disrupting the FKBP12/Met complex. Oncotarget 2016, 7, 40073–40084. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, J.; Chapuis, N.; Bardet, V.; Park, S.; Sujobert, P.; Willems, L.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: Rationale for therapeutic inhibition of both pathways. Blood 2008, 111, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chapuis, N.; Saint Marcoux, F.; Recher, C.; Prebet, T.; Chevallier, P.; Cahn, J.-Y.; Leguay, T.; Bories, P.; Witz, F.; et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia 2013, 27, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Tiong, I.S.; Tan, P.; McManus, J.; Cummings, N.; Sadawarte, S.; Catalano, J.; Hills, R.; Wei, A. Phase Ib study of the mTOR inhibitor everolimus with low dose cytarabine in elderly acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 493–496. [Google Scholar] [CrossRef]
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2018, 32, 235–248. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef]
- Martens, J.H.A.; Stunnenberg, H.G. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett. 2010, 584, 2662–2669. [Google Scholar] [CrossRef]
- San José-Enériz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef]
- Gardin, C.; Dombret, H. Hypomethylating Agents as a Therapy for AML. Curr. Hematol. Malig. Rep. 2017, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Shallis, R.; Stahl, M.; Zeidan, A.M. Epigenetic therapy combinations in acute myeloid leukemia: What are the options? Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jiang, W.; Hou, P. Emerging role of PI3K/AKT in tumor-related epigenetic regulation. Semin. Cancer Biol. 2019, 59, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Roberts, T.M.; Zhao, J.J. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 123–131. [Google Scholar] [CrossRef]
- Long, J.; Jia, M.-Y.; Fang, W.-Y.; Chen, X.-J.; Mu, L.-L.; Wang, Z.-Y.; Shen, Y.; Xiang, R.-F.; Wang, L.-N.; Wang, L.; et al. FLT3 inhibition upregulates HDAC8 via FOXO to inactivate p53 and promote maintenance of FLT3-ITD+ acute myeloid leukemia. Blood 2020, 135, 1472–1483. [Google Scholar] [CrossRef]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Koeffler, H.P.; Yokoyama, A. Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia 2008, 22, 2159–2168. [Google Scholar] [CrossRef]
- Thépot, S.; Lainey, E.; Cluzeau, T.; Sébert, M.; Leroy, C.; Adès, L.; Tailler, M.; Galluzzi, L.; Baran-Marszak, F.; Roudot, H.; et al. Hypomethylating agents reactivate FOXO3A in acute myeloid leukemia. Cell Cycle 2011, 10, 2323–2330. [Google Scholar] [CrossRef]
- Rahmani, M.; Aust, M.M.; Benson, E.C.; Wallace, L.; Friedberg, J.; Grant, S. PI3K/mTOR inhibition markedly potentiates HDAC inhibitor activity in NHL cells through BIM- and MCL-1-dependent mechanisms in vitro and in vivo. Clin. Cancer Res. 2014, 20, 4849–4860. [Google Scholar] [CrossRef]
- Li, Y.; Chen, K.; Zhou, Y.; Xiao, Y.; Deng, M.; Jiang, Z.; Ye, W.; Wang, X.; Wei, X.; Li, J.; et al. A New Strategy to Target Acute Myeloid Leukemia Stem and Progenitor Cells Using Chidamide, a Histone Deacetylase Inhibitor. Curr. Cancer Drug Targets 2015, 15, 493–503. [Google Scholar] [CrossRef]
- Qian, X.; Li, Y.; Yu, Y.; Yang, F.; Deng, R.; Ji, J.; Jiao, L.; Li, X.; Wu, R.-Y.; Chen, W.-D.; et al. Inhibition of DNA methyltransferase as a novel therapeutic strategy to overcome acquired resistance to dual PI3K/mTOR inhibitors. Oncotarget 2015, 6, 5134–5146. [Google Scholar] [CrossRef]
- Gupta, M.; Ansell, S.M.; Novak, A.J.; Kumar, S.; Kaufmann, S.H.; Witzig, T.E. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood 2009, 114, 2926–2935. [Google Scholar] [CrossRef] [PubMed]
- Liesveld, J.L.; O’Dwyer, K.; Walker, A.; Becker, M.W.; Ifthikharuddin, J.J.; Mulford, D.; Chen, R.; Bechelli, J.; Rosell, K.; Minhajuddin, M.; et al. A phase I study of decitabine and rapamycin in relapsed/refractory AML. Leuk. Res. 2013, 37, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Cashen, A.F. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res. 2014, 6, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Tan, P.; Tiong, I.S.; Fleming, S.; Pomilio, G.; Cummings, N.; Droogleever, M.; McManus, J.; Schwarer, A.; Catalano, J.; Patil, S.; et al. The mTOR inhibitor everolimus in combination with azacitidine in patients with relapsed/refractory acute myeloid leukemia: A phase Ib/II study. Oncotarget 2017, 8, 52269–52280. [Google Scholar] [CrossRef]
- Huang, D.C.; Strasser, A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell 2000, 103, 839–842. [Google Scholar] [CrossRef]
- Kuwana, T.; Newmeyer, D.D. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL-2 family: Key mediators of the apoptotic response to targeted anti-cancer therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C.S. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef]
- Zhou, J.-D.; Zhang, T.-J.; Xu, Z.-J.; Gu, Y.; Ma, J.-C.; Li, X.-X.; Guo, H.; Wen, X.-M.; Zhang, W.; Yang, L.; et al. BCL2 overexpression: Clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 2019, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Jiang, S.; Zhang, X.; Konopleva, M.; Estrov, Z.; Snell, V.E.; Xie, Z.; Okcu, M.F.; Sanchez-Williams, G.; Dong, J.; et al. Expression of Bcl-2-related genes in normal and AML progenitors: Changes induced by chemotherapy and retinoic acid. Leukemia 1999, 13, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Gandhi, V.; Konopleva, M. Pathways and mechanisms of venetoclax resistance. Leuk. Lymphoma 2017, 58. [Google Scholar] [CrossRef]
- Xiang, Z.; Luo, H.; Payton, J.E.; Cain, J.; Ley, T.J.; Opferman, J.T.; Tomasson, M.H. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J. Clin. Investig. 2010, 120, 2109–2118. [Google Scholar] [CrossRef]
- Lin, K.H.; Winter, P.S.; Xie, A.; Roth, C.; Martz, C.A.; Stein, E.M.; Anderson, G.R.; Tingley, J.P.; Wood, K.C. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci. Rep. 2016, 6, 27696. [Google Scholar] [CrossRef]
- Teh, T.-C.; Nguyen, N.-Y.; Moujalled, D.M.; Segal, D.; Pomilio, G.; Rijal, S.; Jabbour, A.; Cummins, K.; Lackovic, K.; Blombery, P.; et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia 2018, 32, 303–312. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 inhibition in AML: An unexpected bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef]
- Tsao, T.; Shi, Y.; Kornblau, S.; Lu, H.; Konoplev, S.; Antony, A.; Ruvolo, V.; Qiu, Y.H.; Zhang, N.; Coombes, K.R.; et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann. Hematol. 2012, 91, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A.; Hou, J.-Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined with Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Chao, J.R.; Chen, W.; Kuo, M.L.; Yen, J.J.; Yang-Yen, H.F. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol. Cell. Biol. 1999, 19, 6195–6206. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Nesterova, A.; Sable, C.; Heidenreich, K.A.; Boxer, L.M.; Heasley, L.E.; Reusch, J.E. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J. Biol. Chem. 2000, 275, 10761–10766. [Google Scholar] [CrossRef]
- Su, Y.; Li, X.; Ma, J.; Zhao, J.; Liu, S.; Wang, G.; Edwards, H.; Taub, J.W.; Lin, H.; Ge, Y. Targeting PI3K, mTOR, ERK, and Bcl-2 signaling network shows superior antileukemic activity against AML ex vivo. Biochem. Pharmacol. 2018, 148, 13–26. [Google Scholar] [CrossRef]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef]
- Bose, P.; Rahmani, M.; Grant, S. Coordinate PI3K pathway and Bcl-2 family disruption in AML. Oncotarget 2012, 3, 1499–1500. [Google Scholar] [CrossRef]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C.; Ferreira-Gonzalez, A.; Grant, S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013, 73, 1340–1351. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Singh, N.; Luevano, L.A.; Padi, S.K.R.; Okumura, K.; Olive, V.; Black, S.M.; Warfel, N.A.; Goodrich, D.W.; Kraft, A.S. Mechanisms Behind Resistance to PI3K Inhibitor Treatment Induced by the PIM Kinase. Mol. Cancer Ther. 2018, 17, 2710–2721. [Google Scholar] [CrossRef] [PubMed]
- Meja, K.; Stengel, C.; Sellar, R.; Huszar, D.; Davies, B.R.; Gale, R.E.; Linch, D.C.; Khwaja, A. PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br. J. Haematol. 2014, 167, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Koblish, H.; Li, Y.-L.; Shin, N.; Hall, L.; Wang, Q.; Wang, K.; Covington, M.; Marando, C.; Bowman, K.; Boer, J.; et al. Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies. PLoS ONE 2018, 13, e0199108. [Google Scholar] [CrossRef]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II Study of the Oral Mek Inhibitor Selumetinib in Advanced Acute Myeloid Leukemia (AML): A University of Chicago Phase II Consortium Trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef]
- Sandhöfer, N.; Metzeler, K.H.; Rothenberg, M.; Herold, T.; Tiedt, S.; Groiß, V.; Carlet, M.; Walter, G.; Hinrichsen, T.; Wachter, O.; et al. Dual PI3K/mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia 2015, 29, 828–838. [Google Scholar] [CrossRef]
- Liu, H.; Diaz-Flores, E.; Poiré, X.; Odenike, O.; Koval, G.; Malnassy, G.; Ihonor, P.; Zhang, Y.; Le Beau, M.M.; Shannon, K.; et al. Combination of a MEK Inhibitor, AZD6244, and Dual PI3K/mTOR Inhibitor, NVP-BEZ235: An Effective Therapeutic Strategy for Acute Myeloid Leukemia. Blood 2010, 116, 3978. [Google Scholar] [CrossRef]
- Juric, D.; Soria, J.-C.; Sharma, S.; Banerji, U.; Azaro, A.; Desai, J.; Ringeisen, F.P.; Kaag, A.; Radhakrishnan, R.; Hourcade-Potelleret, F.; et al. A phase 1b dose-escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. JCO 2014, 32, 9051. [Google Scholar] [CrossRef]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Lam, S.S.Y.; Leung, A.Y.H. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. [Google Scholar] [CrossRef]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 inhibitors in acute myeloid leukemia: Current status and future directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Weber, D.; Fiedler, W.; Salih, H.R.; Wulf, G.; Salwender, H.; Schroeder, T.; Kindler, T.; Lübbert, M.; Wolf, D.; et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 2019, 133, 840–851. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef] [PubMed]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.-T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Seedhouse, C.H.; Hunter, H.M.; Lloyd-Lewis, B.; Massip, A.-M.; Pallis, M.; Carter, G.I.; Grundy, M.; Shang, S.; Russell, N.H. DNA repair contributes to the drug-resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia 2006, 20, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases—The lessons from the mouse models: Inhibition ≠ deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, L.; Small, D.; Rassool, F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood 2010, 116, 5298–5305. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double strand break repair pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- Lord, C.J.; Tutt, A.N.J.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Zhao, L.; So, C.W.E. PARP-inhibitor-induced synthetic lethality for acute myeloid leukemia treatment. Exp. Hematol. 2016, 44, 902–907. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Shall, S.; MacPherson, L.J.; Twine, N.A.; Lea, N.C.; Farzaneh, F.; Mufti, G.J. Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: Potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica 2009, 94, 638–646. [Google Scholar] [CrossRef][Green Version]
- Gafencu, G.A.; Tomuleasa, C.I.; Ghiaur, G. PARP inhibitors in acute myeloid leukaemia therapy: How a synthetic lethality approach can be a valid therapeutic alternative. Med. Hypotheses 2017, 104, 30–34. [Google Scholar] [CrossRef]
- Huang, T.-T.; Lampert, E.J.; Coots, C.; Lee, J.-M. Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat. Rev. 2020, 86, 102021. [Google Scholar] [CrossRef]
- Kumar, A.; Fernandez-Capetillo, O.; Fernadez-Capetillo, O.; Carrera, A.C. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef] [PubMed]
- González-Billalabeitia, E.; Seitzer, N.; Song, S.J.; Song, M.S.; Patnaik, A.; Liu, X.-S.; Epping, M.T.; Papa, A.; Hobbs, R.M.; Chen, M.; et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 2014, 4, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, C.; Zhang, Y.; Wang, M.; Jiang, N.; Xiang, L.; Li, T.; Roberts, T.M.; Zhao, J.J.; Cheng, H.; et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol. Oncol. 2016, 142, 548–556. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
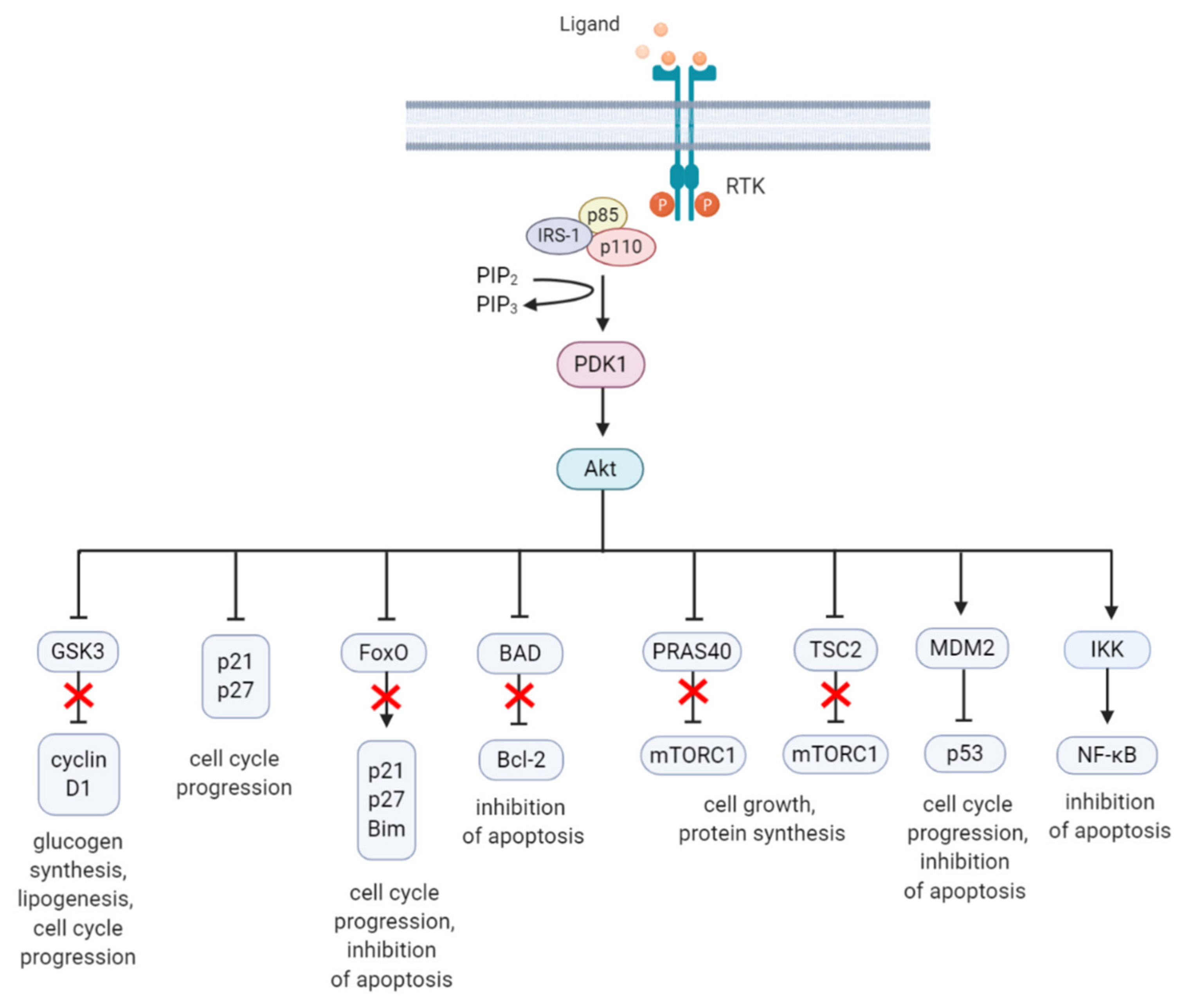{kind=link}
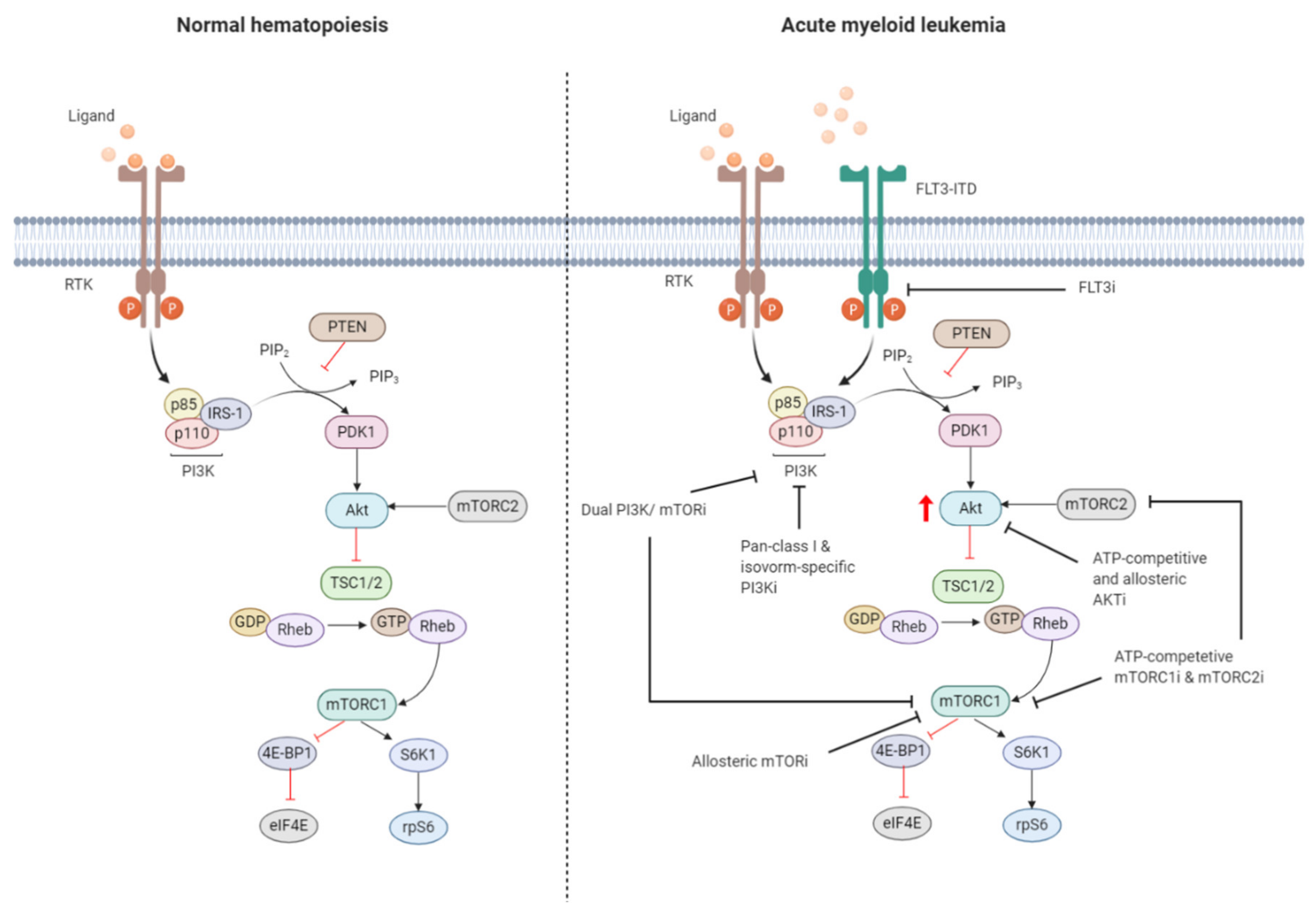{kind=link}
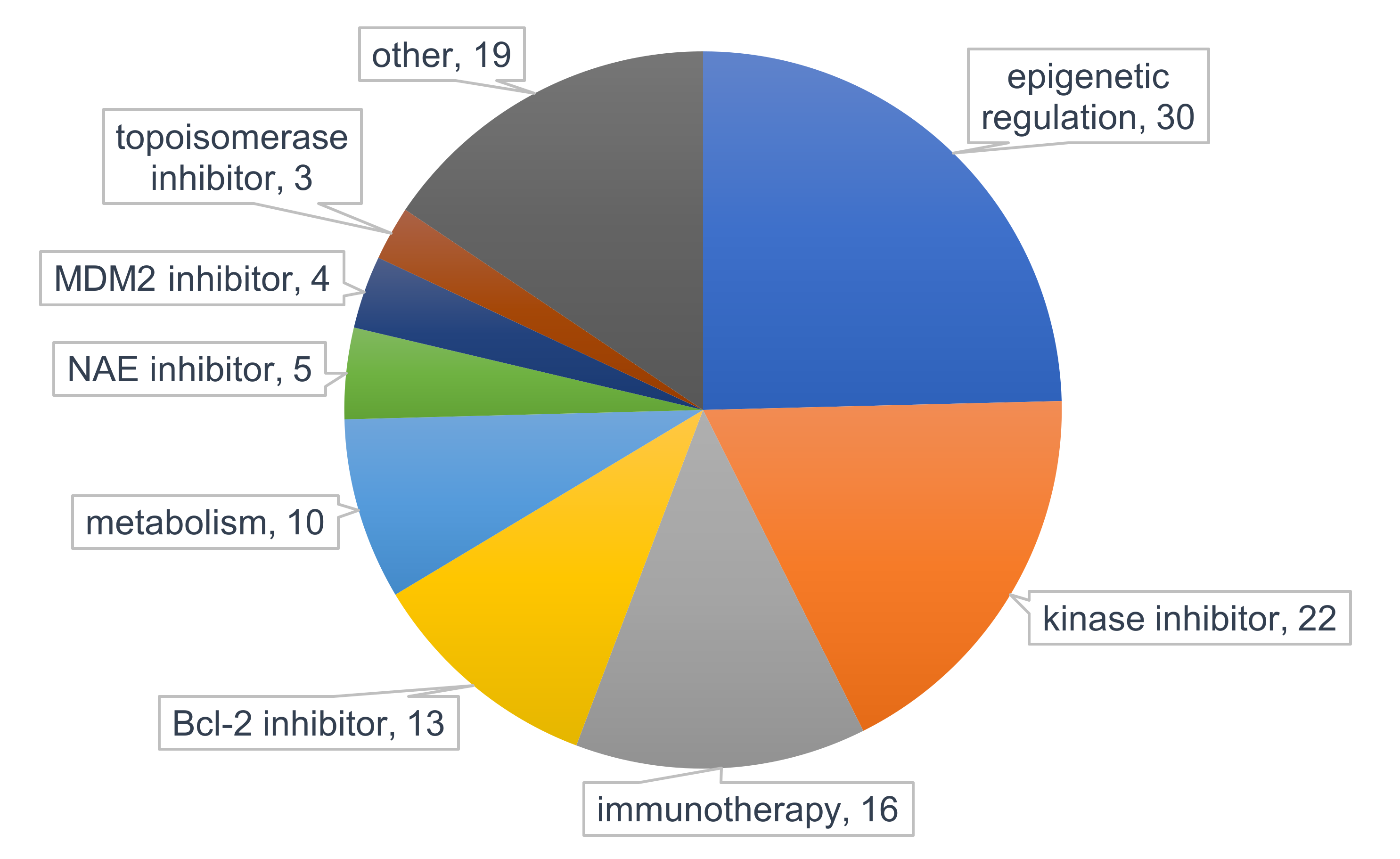{kind=link}
| Compound | Manufacturer | Generic Name | Year | Target | Disease/Condition | Reference |
|---|---|---|---|---|---|---|
| Copiktra® | Verastem Oncology, Needham, MA, USA | duvelisib | 2018 | PI3K-δ PI3K-γ | Relapsed or refractory CLL, SLL, FL | [30] |
| Aliqopa® | Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ, USA | copanlisib | 2017 | PI3K-α PI3K-δ | Relapsed FL | [29] |
| Zydelig® | Gilead Sciences, Foster City, CA, USA | idelalisib | 2014 | PI3K-δ | Relapsed CLL in combination with rituximab, relapsed FL and SLL | [31] |
| Piqray® | Novartis, Basel, Switzerland | alpelisib | 2019 | PI3K-α | In combination with fulvestrant; hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer | [181] |
| Afinitor® | Novartis, Basel, Switzerland | everolimus | 2009 | mTOR | Progressive, well-differentiated non-functional, NET of GI or lung origin with unresectable, locally advanced or metastatic disease | [182] |
| Torisel® | Pfizer Inc., New York City, NY, USA | temsirolimus | 2007 | mTOR | Advanced renal cell carcinoma | [183] |
| Rapamune® | Pfizer Inc., New York City, NY, USA | sirolimus | 1999 | mTOR | LAM, kidney transplant | [184] |
| Investigational Agent | Condition | Phase | Status | Subjects | Study Period | Trial Number |
|---|---|---|---|---|---|---|
| gedatolisib | Therapy-related AML and MDS | 2 | Terminated | 10 | 2015–2018 | NCT02438761 |
| buparlisib | Advanced Leukemias | 1 | Completed | 16 | 2012–2016 | NCT01396499 |
| idelalisib | CLL, NHL, AML, MM | 1 | Completed | 192 | 2008–2012 | NCT00710528 |
| Personalized kinase inhibitor therapy (dasatinib, idelalisib, ruxolitinib, ponatinib, sorafenib, sunitinib) + chemotherapy +/− monoclonal antibody treatment: (cyclophosphamide, cytarabine, doxorubicin, idarubicin, methotrexate, vincristine, rituximab) | ALL, AML | 1 | Recruiting | 24 | 2016–2019 | NCT02779283 |
| dactolisib | ALL, AML, CLL with crisis of blast cells | 1 | Unknown | 23 | 2017–2017 | NCT01756118 |
| MK-2206 | Relapsed or refractory AML | 2 | Completed | 19 | 2010–2018 | NCT01253447 |
| afuresertib | Hematological malignancies | 1/2 | Completed | 73 | 2009–2012 | NCT00881946 |
| uprosertib + trametinib | Recurrent or untreated adult AML | 2 | Terminated | 24 | 2013–2018 | NCT01907815 |
| everolimus + midostaurin | AML, MDS | 1 | Active, not recruiting | 29 | 2009– | NCT00819546 |
| everolimus + nilotinib | AML | 1/2 | Completed | 40 | 2008–2012 | NCT00762632 |
| everolimus + cytarabine/daunorubicin | AML | 1 | Completed | 31 | 2010–2012 | NCT01074086 |
| everolimus + cytarabine | AML | 1 | Unknown | 40 | 2008– | NCT00636922 |
| everolimus + cytarabine/daunorubicin | Relapsed AML | 1 | Unknown | 21 | 2007– | NCT00544999 |
| sirolimus + MEC | AML, myeloid leukemias, leukemia, CML | 1 | Completed | 16 | 2007–2010 | NCT00780104 |
| sirolimus + MEC | AML | NA | Completed | 36 | 2010–2016 | NCT01184898 |
| sirolimus + idaurubicin/cytarabine | AML, PEL | 1 | Completed | 55 | 2013–2019 | NCT01822015 |
| sirolimus + azacitidine | (Recurrent) AML, de novo MDS, MDS with isolated del(5q) previously treated MDS | 2 | Active, not recruiting | 57 | 2013– | NCT01869114 |
| sirolimus + decitabine | Relapsed or refractory AML | 1 | Completed | 13 | 2009–2012 | NCT00861874 |
| Response | Criteria |
|---|---|
| Complete remission (CR) | Bone marrow blasts <5%; absence of circulating blasts and blasts with Auer rods; absence of extramedullary disease; ANC ≥1.0 × 109/L (1000/µL); platelet count ≥100 × 109/L (100.000/µL) |
| CR with incomplete hematological recovery (CRi) | All CR criteria except for residual neutropenia (<1.0 × 109/L [1000/µL]) or thrombocytopenia (<100 × 109/L [100.000/µL]) |
| Partial remission (PR) | All hematologic criteria of CR; decrease of bone marrow blast percentage to 5 to 25%; and decrease of pre-treatment bone marrow blast percentage by ≥50% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darici, S.; Alkhaldi, H.; Horne, G.; Jørgensen, H.G.; Marmiroli, S.; Huang, X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. https://doi.org/10.3390/jcm9092934
Darici S, Alkhaldi H, Horne G, Jørgensen HG, Marmiroli S, Huang X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. Journal of Clinical Medicine. 2020; 9(9):2934. https://doi.org/10.3390/jcm9092934
Chicago/Turabian StyleDarici, Salihanur, Hazem Alkhaldi, Gillian Horne, Heather G. Jørgensen, Sandra Marmiroli, and Xu Huang. 2020. "Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence" Journal of Clinical Medicine 9, no. 9: 2934. https://doi.org/10.3390/jcm9092934
APA StyleDarici, S., Alkhaldi, H., Horne, G., Jørgensen, H. G., Marmiroli, S., & Huang, X. (2020). Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. Journal of Clinical Medicine, 9(9), 2934. https://doi.org/10.3390/jcm9092934