Measurable Residual Disease in Acute Myeloid Leukemia Using Flow Cytometry: A Review of Where We Are and Where We Are Going


Abstract

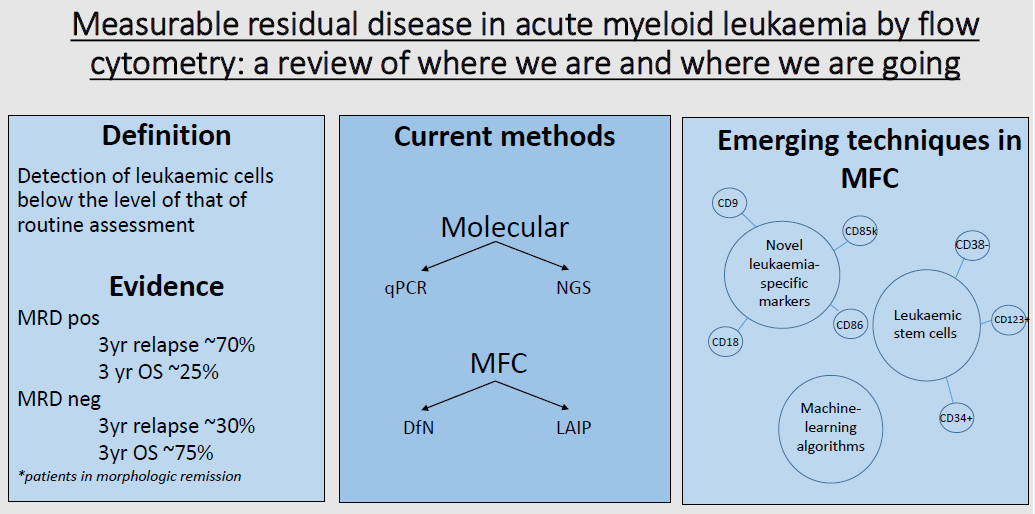
1. Introduction
2. Use of Flow Cytometry in Diagnosis
3. Current Methods of Assessing Measurable Residual Disease
3.1. Molecular Methods
3.2. Flow Cytometric Methods
4. Role of MRD Assessment in Acute Myeloid Leukaemia
5. New and Emerging Strategies in Multiparametric Flow Cytometry
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.; Fenaux, P.; Larson, R.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Rollig, C.; Bornhauser, M.; Thiede, C.; Taube, F.; Kramer, M.; Mohr, B.; Aulitzky, W.; Bodenstein, H.; Tischler, H.-J.; Stuhlmann, R.; et al. Long-term prognosis of acute myeloid leukaemia according to the new genetic risk classification of the European LeukaemiaNet recommendations: Evaluation of the proposed reporting system. J. Clin. Oncol. 2011, 29, 2758–2765. [Google Scholar] [CrossRef]
- Vora, A.; Goulden, N.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Moorman, A.; Wade, R. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2014, 15, 809–818. [Google Scholar] [CrossRef]
- Pui, C.; Pei, D.; Coustan-Smith, E.; Jeha, S.; Cheng, C.; Bowman, W.; Sandlund, J.; Ribeiro, R.; Rubnitz, J.; Inaba, H.; et al. Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: A prospective study. Lancet Oncol. 2015, 16, 465–474. [Google Scholar] [CrossRef]
- Schuurhuis, G.; Heuser, M.; Freeman, S.; Bene, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.; Hourigan, C.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukaemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Corbacioglu, A.; Paschka, P.; Weber, D.; Gaidzik, V.; Jahn, N.; Kundgen, A.; Kindler, T.; Amen Wattad, M.; Lubbert, M.; et al. Minimal residual disease monitoring in acute myeloid leukaemia (AML) with translocation t(8;21)(q22;q22): Results of the AML Study Group (AMLSG). Blood 2016, 128, 1297. [Google Scholar] [CrossRef]
- San Miguel, J.; Martinez, A.; Macedo, A.; Vidriales, M.; Lopez-Berges, C.; Gonzalez, M.; Caballero, D.; Garcia-Marcos, M.; Ramos, F.; Fernandez-Calvo, J.; et al. Immunophenotyping investigation of minimal residual disease is a useful approach for predicting relapse in acute myeloid leukaemia patients. Blood 1997, 90, 2465–2470. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Maurillo, L.; Gattei, V.; Del Poeta, G.; Del Principe, M.; Cox, M.; Panetta, P.; Irno Consalvo, M.; Mazzone, C.; Neri, B.; et al. The kinetics of reduction of minimal residual disease impacts on duration on duration of response and survival of patients with acute myeloid leukaemia. Leukaemia 2006, 20, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.; Simpson, M.; Jovanovic, J.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of minimal residual disease in standard-risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef]
- Araki, D.; Wood, B.; Othus, M.; Radich, J.; Halpern, A.; Zhou, Y.; Mielcarek, M.; Estey, E.; Appelbaum, F.; Walter, R. Allogeneic haematopoietic cell transplantation for Acute Myeloid Leukaemia: Time to move toward a minimal residual disease-based definition of complete remission? J. Clin. Oncol. 2016, 34, 329–336. [Google Scholar] [CrossRef]
- Campana, D.; Pui, C. Minimal residual disease-guided therapy in childhood acute lymphoblastic leukaemia. Blood 2017, 129, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Zeijlemaker, W.; Kelder, A.; Cloos, J.; Schuurhuis, G.J. Immunophenotypic detection of measurable residual (stem cell) disease using LAIP approach in acute myeloid leukemia. Curr. Protoc. Cytom. 2019, 91, e66. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.; Stewart, C.; Dodge, R.; Leget, G.; Sule, N.; Mrozek, K.; Schiffer, C.; Powell, B.; Kolitz, J.; Moore, J.; et al. High frequency of immunophenotype changes in acute myeloid leukaemia at relapse: Implications for residual disease detection (Cancer and Leukaemia Group B Study 8361). Blood 2001, 97, 3574–3580. [Google Scholar] [CrossRef] [PubMed]
- Buldini, B.; Maurer-Granofszky, M.; Varotto, E.; Dworzak, M. Flow-cytometric monitoring of minimal residual disease in paediatric patients with acute myeloid leukaemia: Recent advances and future strategies. Front. Paediatr. 2019, 7, 412. [Google Scholar] [CrossRef] [PubMed]
- Wood, B. Flow cytometry in the diagnosis and monitoring of acute myeloid leukaemia in children. Haematopathology 2015, 8, 191–199. [Google Scholar] [CrossRef]
- Arber, D.; Orazi, A.; Hasserjian, R.; Brunning, R.; Le Beau, M.; Porwit, A.; Tefferi, A.; Levine, R.; Bloomfield, C.; Cazzola, M.; et al. Introduction and overview of the classification of myeloid neoplasms. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S., Campo, E., Harris, N., Jaffe, E., Pileri, S., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Chapter 1; pp. 17–20. [Google Scholar]
- Gajendra, S. Flow cytometry in acute leukaemia. Clin. Oncol. 2016, 1, 1166. [Google Scholar]
- Arber, D.; Brunning, R.; le Beau, M.; Falini, B.; Vardiman, J.; Porwit, A.; Thiele, J.; Foucar, K.; Dohner, H.; Bloomfield, C. Acute myeloid leukaemia and related precursor neoplasms. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S., Campo, E., Harris, N., Jaffe, E., Pileri, S., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Chapter 8; pp. 130–145. [Google Scholar]
- Bain, B.; Bene, M. Morphological and immunophenotypic clues to the WHO categories of acute myeloid leukaemia. Acta Haematol. 2019, 141, 232–244. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, H.; Ruan, G.; Qin, Y.; Shi, H.; Lai, Y.; Chang, Y.; Wang, Y.; Lu, D.; Hao, L.; et al. NPM1-mutated acute myeloid leukaemia of monocytic or myeloid origin exhibit distinct immunophenotypes. Leuk. Res. 2013, 37, 737–741. [Google Scholar] [CrossRef]
- Chang, H.; Salma, F.; Yi, Q.; Patterson, B.; Brien, B.; Minden, M. Prognostic relevance of immunophenotyping in 379 patients with acute myeloid leukaemia. Leuk. Res. 2004, 28, 43–48. [Google Scholar] [CrossRef]
- Wood, B. Principles of minimal residual disease detection for haematopoietic neoplasms by flow cytometry. Clin. Cytom. 2015, 90, 47–53. [Google Scholar] [CrossRef]
- Wood, B. Acute myeloid leukaemia minimal residual disease detection: The difference from normal approach. Curr. Protoc. Cytom. 2020, 93, e73. [Google Scholar] [PubMed]
- Hubmann, M.; Kohnke, T.; Hoster, E.; Schneider, S.; Dufour, A.; Zellmeier, E.; Fiegl, M.; Braess, J.; Bohlander, S.; Subklewe, M.; et al. Molecular response assessment by quantitative real-time polymerase chain reaction after induction therapy in NPM1-mutated patients identifies those at high risk of relapse. Haematologica 2014, 99, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; O’Brien, M.; Hills, R.; Daly, S.; Wheatley, K.; Burnett, A. Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: Results of the United Kingdom MRC AML-15 trial. Blood 2012, 120, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.; Gradowska, P.; Meijer, R.; Cloos, J.; et al. Molecular minimal residual disease in acute myeloid leukaemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.; Ayala, R.; Martinez-Lopez, J. Minimal residual disease monitoring with next-generation sequencing methodologies in haematological malignancies. Int. J. Mol. Sci. 2019, 20, 2832. [Google Scholar] [CrossRef]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C.; Hambach, L.; Stadler, M.; Koenecke, C.; Flintrop, M.; et al. Measurable residual disease monitoring by NGS before allogeneic haematopoietic cell transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef]
- Getta, B.; Devlin, S.; Levine, R.; Arcila, M.; Mohanty, A.; Zehir, A.; Tallman, M.; Giralt, S.; Roshal, M. Multicolor flow cytometry and multigene next-generation sequencing are complementary and highly predictive for relapse in acute myeloid leukaemia after allogeneic transplantation. Biol. Blood Bone Marrow Transpl. 2017, 23, 1064–1071. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Schuurhuis, G. Minimal residual disease and leukaemic stem cells in acute myeloid leukaemia. In Leukaemia; Guenova, M., Ed.; IntechOpen: London, UK, 2013; Chapter 6; pp. 195–226. [Google Scholar]
- Heath, E.; Chan, S.; Minden, M.; Murphy, T.; Shlush, L.; Schimmer, A. Biological and clinical consequences of NPM1 mutations in AML. Leukaemia 2017, 31, 798–807. [Google Scholar] [CrossRef]
- Scholl, C.; Schlenk, R.; Eiwen, K.; Dohner, H.; Frohling, S.; Dohner, K.; Frohling, S.; Dohner, K.; AML Study Group. The prognostic value of MLL-AF9 detection in patients with t(9;11)(p22;q23)-positive acute myeloid leukaemia. Haematologica 2005, 90, 1626–1634. [Google Scholar]
- Ley, T.; Mardis, E.; Ding, L.; Fulton, B.; McLellan, M.; Chen, K.; Dooling, D.; Dunford-Shore, B.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef]
- Kottaridis, P.; Gale, R.; Frew, M.; Harrison, G.; Langabeer, S.; Belton, A.; Walker, H.; Wheatley, K.; Bowen, D.; Burnett, A.; et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukaemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: Analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 2001, 98, 1752–1759. [Google Scholar] [PubMed]
- Dillon, L.; Hayati, S.; Roloff, G.; Tunc, I.; Pirooznia, M.; Mitrofanova, A.; Hourigan, C. Targeted RNA-sequencing for the quantification of measurable residual disease in acute myeloid leukaemia. Haematologica 2019, 104, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Voso, M.; Ottone, T.; Lavorgna, S.; Venditti, A.; Maurillo, L.; Lo-Coco, F.; Buccisano, F. MRD in AML: The role of new techniques. Front. Oncol. 2019, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Hoklund, P.; Ommen, H.; Nyvold, C.; Roug, A. Sensitivity of minimal residual disease in acute myeloid leukaemia in first remission—Methodologies in relation to their clinical situation. Br. J. Haematol. 2012, 158, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Grigoriadis, G.; Westerman, D. The role of multiparametric flow cytometry in the detection of minimal residual disease in acute leukaemia. Pathology 2015, 47, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Loken, M.; Alonzo, T.; Pardo, L.; Gerbing, R.; Raimondi, S.; Hirsch, B.; Ho, P.; Franklin, J.; Cooper, T.; Gamis, A.; et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukaemia: A report from Children’s Oncology Group. Blood 2012, 120, 1581–1588. [Google Scholar] [CrossRef]
- Sievers, E.; Lange, B.; Alonzo, T.; Gerbing, R.; Bernstein, I.; Smith, F.; Arceci, R.; Woods, W.; Loken, M. Immunophenotypic evidence of leukaemia after induction therapy predicts relapse: Results from prospective children’s cancer group study of 252 patients with acute myeloid leukaemia. Blood 2003, 101, 3398–3406. [Google Scholar] [CrossRef]
- Rubnitz, J.; Inaba, H.; Dahl, G.; Ribeiro, R.; Bowman, W.; Taub, J.; Pounds, S.; Razzouk, B.; Lacayo, N.; Cao, X.; et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: Results of the AML02 multicentre trial. Lancet Oncol. 2010, 11, 543–552. [Google Scholar] [CrossRef]
- Terwijn, M.; van Putten, W.; Kelder, A.; van der velden, V.; Brooimans, R.; Pabst, T.; Maertens, J.; Boeckx, N.; de Greef, G.; Valk, P.; et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukaemia: Prospective data from the HOVON/SAKK 42a Study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef]
- Short, N.; Ravandi, F. How close are we to incorporating measurable residual disease into clinical practice for acute myeloid leukaemia? Haematologica 2019, 104, 1532–1541. [Google Scholar] [CrossRef]
- Paietta, E. Consensus on MRD in AML? Blood 2018, 131, 1265–1266. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.; Bloomfield, C. Acute Myeloid Leukaemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.; Janssen, J.; van de Loosdrecht, A. Risk factors for relapse after allogeneic transplantation in acute myeloid leukaemia. Haematologica 2016, 101, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Lo Coco, F.; Diverio, D.; Avvisati, G.; Petti, M.; Meloni, G.; Pogliani, E.; Biondi, A.; Rossi, G.; Carlo-Stella, C.; Seller, C.; et al. Therapy of molecular relapse in acute promyelocytic leukaemia. Blood 1999, 94, 2225–2229. [Google Scholar] [CrossRef]
- Esteve, J.; Escoda, L.; Rubio, M.; Diaz-Mediavilla, J.; Gonzalez, M.; Rivas, C.; Alvarez, C.; Gonzales San Miguel, J.; Bruent, S.; Tomas, J.; et al. Outcome of patients with acute promyelocytic leukaemia failing to meet front-line treatment with all-trans retinoic acid and anthracycline-based chemotherapy (PETHEMA protocols LPA96 and LPA99): Benefit of an early intervention. Leukaemia 2007, 21, 446–452. [Google Scholar] [CrossRef]
- Kohnke, T.; Sauter, D.; Ringel, K.; Hoster, E.; Laubender, R.; Hubmann, M.; Bohlander, S.; Kakadia, P.; Schneider, S.; Dufour, A.; et al. Early assessment of minimal residual disease in AML by flow cytometry during aplasia identifies patients at increased risk of relapse. Leukaemia 2015, 29, 377–386. [Google Scholar] [CrossRef]
- Venditti, A.; Piciocchi, A.; Candoni, A.; Melillo, L.; Calafiore, V.; Cairoli, R.; de Fabritiis, P.; Storti, G.; Salutari, P.; Lanza, F.; et al. GIMEMA AML1310 trial of risk-adapted, MRD-directed therapy for young adults with newly diagnosed acute myeloid leukaemia. Blood 2019, 134, 935–945. [Google Scholar] [CrossRef]
- Zhou, Y.; Othus, M.; Araki, D.; Wood, B.; Radich, J.; Halpern, A.; Mielcarek, M.; Estey, E.; Appelbaum, R.; Walter, R. Pre- and post-transplant quantification of measurable (“minimal”) residual disease via multiparameter flow cytometry in adult acute myeloid leukaemia. Leukaemia 2016, 30, 1456–1464. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.; Oussoren-Brockhoff, Y.; Snel, A.; Veldhuizen, S.; Scholten, W.; et al. CD34+CD38- leukaemic stem cell frequency to predict outcome in acute myeloid leukaemia. Leukaemia 2018, 33, 1102–1112. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Song, G.; Shurtleff, S.; Yeoh, A.; Chng, W.; Chen, S.; Rubnitz, J.; Pui, C.; Downing, J.; Campana, D. Universal monitoring of minimal residual disease in acute myeloid leukaemia. JCI Insight 2018, 3, e98561. [Google Scholar] [CrossRef]
- Gao, M.; Ruan, G.; Chang, Y.; Liu, Y.; Qin, Y.; Jiang, Q.; Jiang, H.; Huang, X.; Zhao, X. The predictive value of minimal residual disease when facing the inconsistent results detected by real-time quantitative PCR and flow cytometry in NPM1-mutated acute myeloid leukaemia. Ann. Haematol. 2019, 99, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.; Hills, R.; Freeman, S.; Potter, N.; Jovanovic, J.; Ivey, A.; Kanda, A.; Runglall, M.; Foot, N.; Valganon, M.; et al. Molecular MRD status and outcome after transplantation in NPM1-mutated AML. Blood 2020, 135, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Oran, B.; Giralt, S.; Couriel, D.; Hosing, C.; Shpall, E.; de Meis, E.; Khouri, I.; Qazilbash, M.; Anderlini, P.; Kebriaei, P.; et al. Treatment of AML and MDS relapsing after reduced intensity conditioning and allogeneic haematopoietic stem cell transplantation. Leukaemia 2007, 21, 2540–2544. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Liu, D.; Liu, K.; Xu, L.; Liu, Y.; Chen, H.; Han, H.; Wang, Y.; Qin, Y.; Huang, X. Risk stratification-directed donor lymphocyte infusion could reduce relapse of standard-risk acute leukaemia patients after allogeneic haematopoietic stem cell transplantation. Blood 2012, 119, 3256–3262. [Google Scholar] [CrossRef]
- Platzbecker, U.; Wemke, M.; Radke, J.; Oelschlaegel, U.; Seltmann, F.; Kiani, A.; Klut, I.; Knoth, H.; Rollig, C.; Schetelig, J.; et al. Azacitadine for treatment of immiment relapse in MDS or AML patients after allogeneic HSCT: Results of the RELAZA trial. Leukaemia 2012, 26, 381–389. [Google Scholar] [CrossRef]
- Sockel, K.; Wemke, M.; Radke, J.; Kiani, A.; Schaich, M.; Bornhauser, M.; Ehninger, G.; Thiede, C.; Platzbecker, U. Minimal residual disease-directed preemptive treatment with azacitadine in patients with NPM1-mutant acute myeloid leukaemia and molecular relapse. Haematologica 2011, 96, 1568–1570. [Google Scholar] [CrossRef]
- Ossenkoppele, G.; Schuurhuis, G.; van de Loosdrecht, A.; Cloos, J. Can we incorporate MRD assessment into clinical practice in AML? Best Pract. Res. Clin. Haematol. 2019, 32, 186–191. [Google Scholar] [CrossRef]
- Selim, A.; Moore, A. Molecular minimal residual disease monitoring in acute myeloid leukaemia: Challenges and future directions. J. Mol. Diagn. 2018, 20, 389–397. [Google Scholar] [CrossRef]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukaemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef]
- Plesa, A.; Dumontet, C.; Mattei, E.; Tagoug, I.; Havette, S.; Sujobert, P.; Tigaud, I.; Pages, M.; Chelghoum, Y.; Baracco, F.; et al. High frequency of CD34+CD38-/low immature leukaemia stem cells is correlated with unfavourable prognosis in acute myeloid leukaemia. World J. Stem Cells 2017, 9, 227. [Google Scholar] [CrossRef]
- Al-Mawali, A.; Pinto, A.; Al-Zadjali, S. CD34+CD38-CD123+ cells are present in virtually all acute myeloid leukaemia blasts: A promising single unique phenotype for minimal residual disease detection. Acta Haematol. 2017, 138, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Daga, S.; Rosenberger, A.; Quehenberger, F.; Krisper, N.; Prietl, B.; Reinisch, A.; Zebisch, A.; Sill, H.; Wolfler, A. High GPR56 surface expression correlates with a leukaemic stem cell gene signature in CD34-positive AML. Cancer Med. 2019, 8, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Menezes, D.; Pinheiro, H.; Sandes, A.; Nunes, M.; Lyra, D.; Schimieguel, D. Role of new immunophenotypic markers on prognostic and overall survival of acute myeloid leukaemia: A systematic review and meta-analysis. Nat. Sci. Rep. 2017, 7, 4138. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Berman, S.; Soni, R.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.; Brennan, C.; Sadelain, M. Integrating proteomics and transcriptomics for systemic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell 2017, 32, 506–519. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualising data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Ko, B.; Wang, Y.; Li, J.; Li, C.; Weng, P.; Hsu, S.; Hou, H.; Huang, H.; Yao, M.; Lin, C.; et al. Clinically validated machine learning algorithm for detecting residual diseases with multicolor flow cytometry analysis in acute myeloid leukaemia and myelodysplastic syndrome. EBioMedicine 2018, 37, 91–100. [Google Scholar] [CrossRef]
- Gjertsen, B.; Tislevoll, B.; Eagerholt, O.; Hellesoy, M. Response evaluation in AML using mass cytometry. HemaSphere 2019, 3, S2. [Google Scholar] [CrossRef]

{kind=link}
| MRD Methodology | Sensitivity | Advantages | Disadvantages | References |
|---|---|---|---|---|
| MFC-LAIP | 1 in 103–105 |
|
| [5,13,14,15,22] |
| MFC-DfN | 1 in 103–105 |
|
| [5,14,15,22,23] |
| qPCR | 1 in 104–106 |
|
| [5,9,24,25] |
| NGS | Variable |
|
| [26,27,28,29] |
| Reference | MFC Method | Methodology/Surface Markers Used | Time Point of Assessment | Evidence-Outcomes |
|---|---|---|---|---|
| [42] | MFC-LAIP | Limit of detection 0.1% | After induction therapy |
|
| [50] | MFC-LAIP combined with qPCR | 8 color MFC assay | After consolidation |
|
| [26] | MFC-LAIP combined with NGS | Limit of detection 0.1% | After induction therapy |
|
| [49] | LAIP at time of bone marrow aplasia (day 16–18) | 31 surface markers, multiple LAIPs identified for each patient Limit of detection 0.15% | Day 16–18 of induction therapy |
|
| [10] | MFC-DfN | 10 color assay 3 tubes, 1 million events per tube. Limit of detection 0.1% | MRD assessment pre-alloSCT and outcomes post-transplant |
|
| [51] | MFC-DfN | 10 color MFC assay Surface markers: CD4, CD5, CD7, CD13, CD14, CD15, CD16, CD19, CD33, CD34, CD38, CD45, CD56, CD64, CD71, CD117, CD123, HLA-DR. Any measurable MRD considered positive | Pre-alloSCT and post-alloSCT (day +28) |
|
| [29] | MFC-DfN separately and combined with NGS | 10 color MFC assay Limit of detection 0.1% | Pre-alloSCT |
|
| [52] | Combined MFC-LAIP and LSC | Limit of detection 0.1% LSCs defined as CD34+/CD38− | After second induction |
|
| [53] | Novel leukemia-specific markers | CD9, CD18, CD25, CD32, CD44, CD47, CD52, CD54, CD59, CD64, CD68, CD86, CD93, CD96, CD97, CD99, CD123, CD200, CD300a/c, CD366, CD371, CX3CR1 | At diagnosis (to identify novel markers), compared to healthy donors and relapsed AML patients. |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dix, C.; Lo, T.-H.; Clark, G.; Abadir, E. Measurable Residual Disease in Acute Myeloid Leukemia Using Flow Cytometry: A Review of Where We Are and Where We Are Going. J. Clin. Med. 2020, 9, 1714. https://doi.org/10.3390/jcm9061714
Dix C, Lo T-H, Clark G, Abadir E. Measurable Residual Disease in Acute Myeloid Leukemia Using Flow Cytometry: A Review of Where We Are and Where We Are Going. Journal of Clinical Medicine. 2020; 9(6):1714. https://doi.org/10.3390/jcm9061714
Chicago/Turabian StyleDix, Caroline, Tsun-Ho Lo, Georgina Clark, and Edward Abadir. 2020. "Measurable Residual Disease in Acute Myeloid Leukemia Using Flow Cytometry: A Review of Where We Are and Where We Are Going" Journal of Clinical Medicine 9, no. 6: 1714. https://doi.org/10.3390/jcm9061714
APA StyleDix, C., Lo, T.-H., Clark, G., & Abadir, E. (2020). Measurable Residual Disease in Acute Myeloid Leukemia Using Flow Cytometry: A Review of Where We Are and Where We Are Going. Journal of Clinical Medicine, 9(6), 1714. https://doi.org/10.3390/jcm9061714