Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
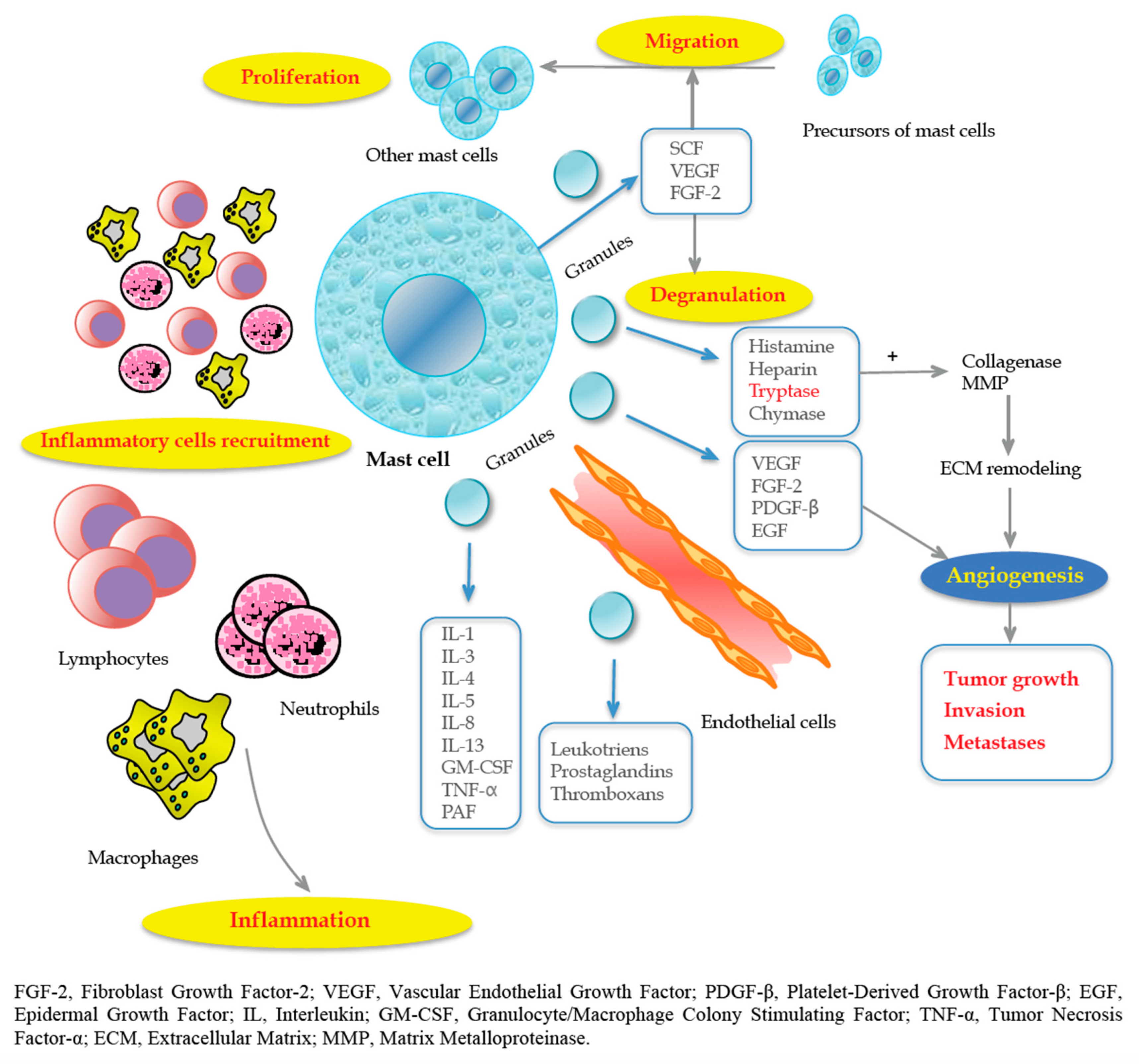
2. Mast Cells
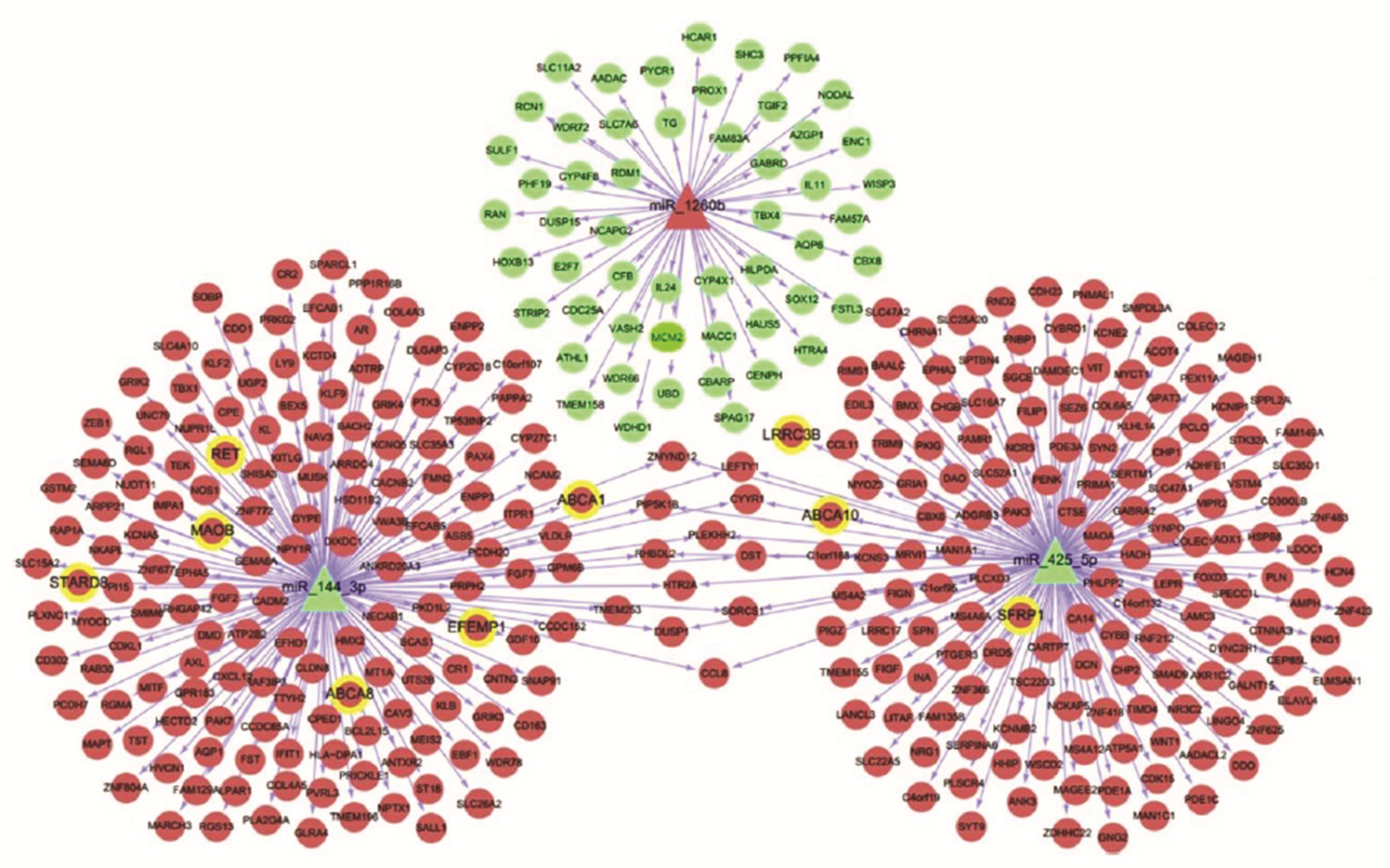
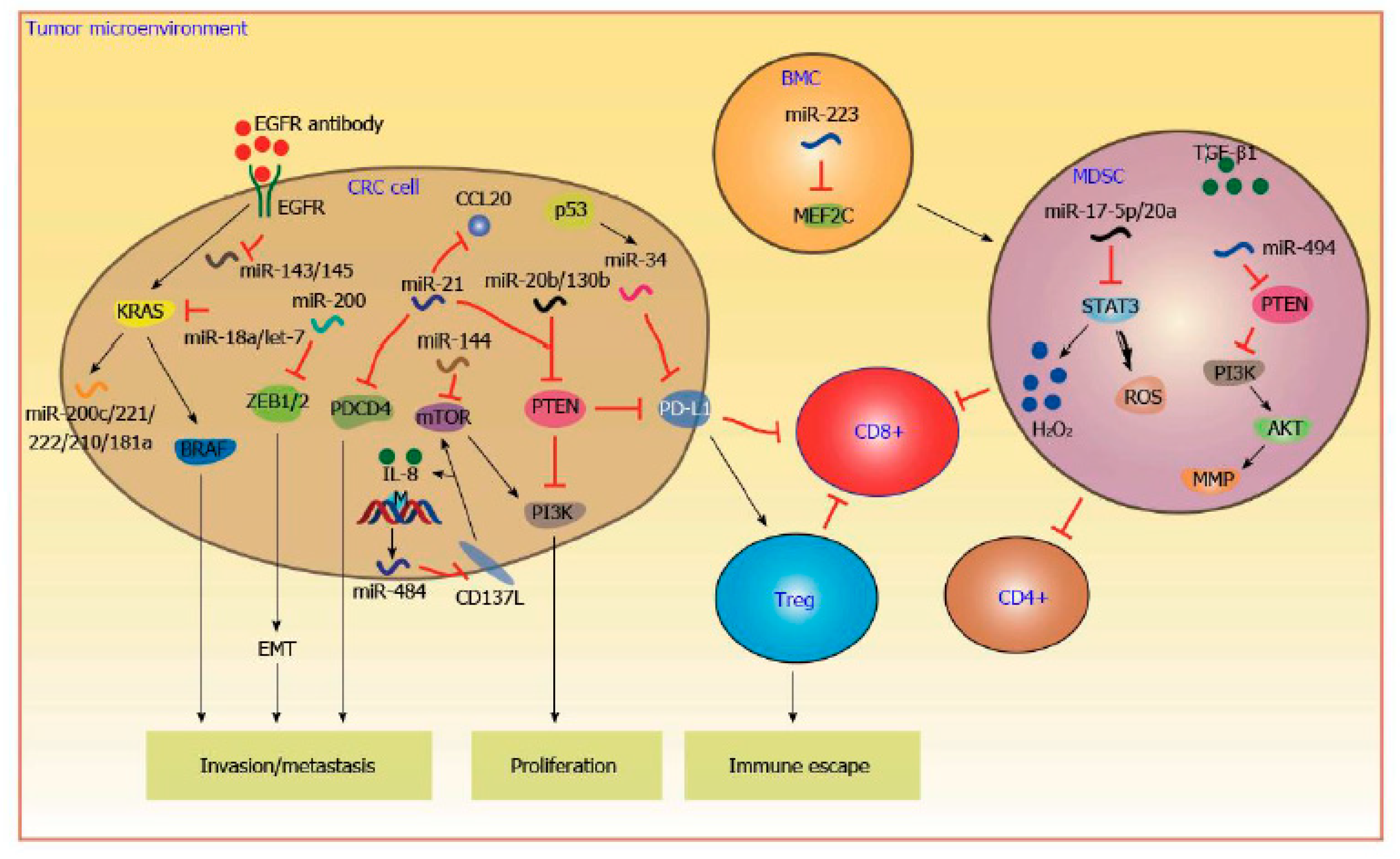
3. MicroRNA
4. KRAS
5. BRAF
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? Int. J. Mol. Sci. 2018, 19, 3733. [Google Scholar] [CrossRef]
- Alvaro, E.; Cano, J.M.; Garcia, J.L.; Brandariz, L.; Olmedillas-Lopez, S.; Arriba, M.; Rueda, D.; Rodriguez, Y.; Canete, A.; Arribas, J.; et al. Clinical and Molecular Comparative Study of Colorectal Cancer Based on Age-of-Onset and Tumor Location: Two Main Criteria for Subclassifying Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 968. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Gallo, G.; Sena, G.; Vescio, G.; Papandrea, M.; Sacco, R.; Trompetto, M.; Sammarco, G. The prognostic value of KRAS and BRAF in stage I-III colorectal cancer. A systematic review. Ann. Ital. Chir. 2019, 90, 127–137. [Google Scholar]
- Pellino, G.; Gallo, G.; Pallante, P.; Capasso, R.; De Stefano, A.; Maretto, I.; Malapelle, U.; Shengyang, Q.; Nikolaou, S.; Barina, A.; et al. Noninvasive Biomarkers of Colorectal Cancer: Role in Diagnosis and Personalised Treatment Perspectives. Gastroenterol. Res. Pract. 2018, 2018, 2397863. [Google Scholar] [CrossRef]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190. [Google Scholar] [CrossRef]
- Hui, L.; Chen, Y. Tumor Microenvironment: Sanctuary of the Devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Gallo, G.; Kotze, P.G.; Spinelli, A. Surgery in ulcerative colitis: When? How? Best Pract. Res. Clin. Gastroenterol. 2018, 32, 71–78. [Google Scholar] [CrossRef]
- Pellino, G.; Marcellinaro, R.; Candilio, G.; De Fatico, G.S.; Guadagno, E.; Campione, S.; Santangelo, G.; Reginelli, A.; Sciaudone, G.; Riegler, G.; et al. The Experience of a Referral Centre and Literature Overview of GIST and Carcinoid Tumours in Inflammatory Bowel Diseases. Int. J. Surg. 2016, 28, S133–S141. [Google Scholar] [CrossRef] [PubMed]
- Wisselink, D.D.; Braakhuis, L.L.F.; Gallo, G.; van Grevenstein, W.M.U.; van Dieren, S.; Kok, N.F.M.; de Reuver, P.R.; Tanis, P.J.; de Hingh, I.H.J.T. Systematic review of published literature on Oxaliplatin and Mitomycin C as chemotherapeutic agents for Hyperthermic Intraperitoneal Chemotherapy in patients with peritoneal metastases from colorectal cancer. Crit. Rev. Oncol. Hematol. 2019, 142, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.B.; Bapat, B. Epigenetic Biomarkers in Colorectal Cancer Diagnostics. Expert Rev. Mol. Diagn. 2012, 12, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Greegor, D.H. Diagnosis of Large-Bowel Cancer in the Asymptomatic Patient. JAMA 1967, 201, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, D.; Tokino, T.; Hamilton, S.R.; Kinzler, K.W.; Levin, B.; Frost, P.; Vogelstein, B. Identification of Ras oncogene mutations in the stool of patients with curable colorectal tumors. Science 1992, 256, 102–105. [Google Scholar] [CrossRef]
- Von Karsa, L.; Patnick, J.; Segnan, N.; Atkin, W.; Halloran, S.; Lansdorp-Vogelaar, I.; Malila, N.; Minozzi, S.; Moss, S.; Quirke, P.; et al. European Guidelines for Quality Assurance in Colorectal Cancer Screening and Diagnosis: Overview and Introduction to the Full Supplement Publication. Endoscopy 2013, 45, 51–59. [Google Scholar]
- Rosman, A.S.; Korsten, M.A. Meta-analysis comparing CT colonography, air contrast barium enema, and colonoscopy. Am. J. Med. 2007, 120, 203–210. [Google Scholar] [CrossRef]
- Rockey, D.C.; Paulson, E.; Niedzwiecki, D.; Davis, W.; Bosworth, H.B.; Sanders, L.; Yee, J.; Henderson, J.; Hatten, P.; Burdick, S.; et al. Analysis of air contrast barium enema, computed tomographic colonography, and colonoscopy: Prospective comparison. Lancet 2005, 365, 305–311. [Google Scholar] [CrossRef]
- Graser, A.; Stieber, P.; Nagel, D.; Schäfer, C.; Horst, D.; Becker, C.R.; Nikolaou, K.; Lottes, A.; Geisbüsch, S.; Kramer, H.; et al. Comparison of CT colonography, colonoscopy, sigmoidoscopy and faecal occult blood tests for the detection of advanced adenoma in an average risk population. Gut 2009, 58, 241–248. [Google Scholar] [CrossRef]
- Gallo, G. Preoperative Colorectal-Cancer Detection: Do We Need Anything Else? An Invited Brief Commentary on Is CT Scan More Accurate than Endoscopy in Identifying Distance from the Anal Verge for Left-sided Colon Cancer? A Comparative Cohort Analysis. J. Investig. Surg. 2020, 33, 281–282. [Google Scholar] [CrossRef]
- Ehrlich, P. Beiträge zur Theorie und Praxis der Histologischen Färbung. In The Collected Papers of Paul Ehrlich; Elsevier: Pergamon, Turkey, 2013; pp. 29–64. [Google Scholar]
- Crivellato, E.; Beltrami, C.A.; Mallardi, F.; Ribatti, D. Paul Ehrlich’s doctoral thesis: A milestone in the study of mast cells. Br. J. Haematol. 2003, 123, 19–21. [Google Scholar] [PubMed]
- Blank, U. The mechanism of exocytosis in Mast Cells. Adv. Exp. Med. Biol. 2011, 716, 107–122. [Google Scholar] [PubMed]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.; Silverman, A.J.; Vitkovic, L.; Lederhendler, I.I. Mast cells in the brain: Evidence and functional significance. Trends. Neurosci. 1996, 19, 25–31. [Google Scholar] [CrossRef]
- Okayama, Y.; Kawakami, T. Development, Migration, and Survival of Mast Cells. Immunol. Res. 2006, 34, 97–115. [Google Scholar] [CrossRef]
- Westphal, E. Uber Mastzellen. In Farbenanalytische Untersuchungen; Ehrlich, E., Ed.; Hirschwald: Berlin, Germany, 1891; pp. 17–41. [Google Scholar]
- Conti, P.; Castellani, M.L.; Kempuraj, D.; Salini, V.; Vecchiet, J.; Tetè, S.; Mastrangelo, F.; Perrella, A.; De Lutiis, M.A.; Tagen, M.; et al. Role of mast cells in tumor growth. Ann. Clin. Lab. Sci. 2007, 37, 315–322. [Google Scholar] [PubMed]
- Acikalin, M.F.; Oner, U.; Topcu, I.; Yasar, B.; Kiper, H.; Colak, E. Tumour angiogenesis and mast cell density in the prognostic assessment of colorectal carcinomas. Dig. Liver Dis. 2005, 37, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Melillo, R.M.; Guarino, V.; Avilla, E.; Galdiero, M.R.; Liotti, F.; Prevete, N.; Rossi, F.W.; Basolo, F.; Ugolini, C.; de Paulis, A.; et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 2010, 29, 6203–6215. [Google Scholar] [CrossRef] [PubMed]
- Ammendola, M.; Sacco, R.; Donato, G.; Zuccala, V.; Russo, E.; Luposella, M.; Vescio, G.; Rizzuto, A.; Patruno, R.; De Sarro, G.; et al. Mast cell positivity to tryptase correlates with metastatic lymph nodes in gastrointestinal cancer patients treated surgically. Oncology 2013, 85, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell. Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Ammendola, M.; Rosario, S.; Giuseppe, S.; Giuseppe, D.; Montemurro, S.; Ruggieri, E.; Patruno, R.; Marech, I.; Cariello, M.; Vacca, A.; et al. Correlation between serum tryptase, mast cells positive to tryptase and microvascular density in colo-rectal cancer patients: Possible biological-clinical significance. PLoS ONE 2014, 9, e99512. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, S.; Shen, P.; Wu, B.; Sun, T.; Sun, H.; Ji, F.; Zhou, D. SCF/c-KIT signaling promotes mucus secretion of colonic goblet cells and development of mucinous colorectal adenocarcinoma. Am. J. Cancer Res. 2018, 8, 1064–1073. [Google Scholar] [PubMed]
- Marech, I.; Ammendola, M.; Gadaleta, C.; Zizzo, N.; Oakley, C.; Gadaleta, C.D.; Ranieri, G. Possible biological and translational significance of mast cells density in colorectal cancer. World J. Gastroenterol. 2014, 20, 8910–8920. [Google Scholar] [PubMed]
- Yu, Y.; Blokhuis, B.; Derks, Y.; Kumari, S.; Garssen, J.; Redegeld, F. Human mast cells promote colon cancer growth via bidirectional crosstalk: Studies in 2D and 3D coculture models. Oncoimmunology 2018, 7, e1504729. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Cimpean, A.M.; Tamma, R.; Ruggieri, S.; Nico, B.; Toma, A.; Ribatti, D. Mast cells in breast cancer angiogenesis. Crit. Rev. Oncol. Hematol. 2017, 115, 23–26. [Google Scholar] [CrossRef]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef]
- Engel, C.J.; Bennett, S.T.; Chambers, A.F.; Doig, G.S.; Kerkvliet, N.; O’Malley, F.P. Tumor angiogenesis predicts recurrence in invasive colorectal cancer when controlled for Dukes staging. Am. J. Surg. Pathol. 1996, 20, 1260–1265. [Google Scholar] [CrossRef]
- Frank, R.E.; Saclarides, T.J.; Leurgans, S.; Speziale, N.J.; Drab, E.A.; Rubin, D.B. Tumor angiogenesis as a predictor of recurrence and survival in patients with node-negative colon cancer. Ann. Surg. 1995, 222, 695–699. [Google Scholar] [CrossRef]
- Detoraki, A.; Staiano, R.I.; Granata, F.; Giannattasio, G.; Prevete, N.; de Paulis, A.; Ribatti, D.; Genovese, A.; Triggiani, M.; Marone, G. Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J. Allergy Clin. Immunol. 2009, 123, 1142–1149. [Google Scholar] [CrossRef]
- Sammarco, G.; Varricchi, G.; Ferraro, V.; Ammendola, M.; De Fazio, M.; Altomare, D.F.; Luposella, M.; Maltese, L.; Currò, G.; Marone, G.; et al. Mast Cells, Angiogenesis and Lymphangiogenesis in Human Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 2106. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhai, L.; Xue, R.; Shi, J.; Zeng, Q.; Gao, C. Mast cell tryptase contributes to pancreatic cancer growth through promoting angiogenesis via activation of angiopoietin-1. Int. J. Mol. Sci. 2016, 17, 834. [Google Scholar] [CrossRef]
- Payne, V.; Kam, P.C.A. Mast cell tryptase: A review of its physiology and clinical significance. Anaesthesia 2004, 59, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Marichal, T.; Tsai, M.; Galli, S.J. Mast cells: Potential positive and negative roles in tumor biology. Cancer Immunol. Res. 2013, 1, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Adams, W.J.; Morris, D.L. Short-course cimetidine and survival with colorectal cancer. Lancet 1994, 344, 1768–1769. [Google Scholar] [CrossRef]
- Matsumoto, S. Cimetidine and survival with colorectal cancer. Lancet 1995, 346, 115. [Google Scholar] [CrossRef]
- Gulubova, M.; Vlaykova, T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J. Gastroenterol. Hepatol. 2009, 24, 1265–1275. [Google Scholar] [CrossRef]
- Ammendola, M.; Zuccalà, V.; Patruno, R.; Russo, E.; Luposella, M.; Amorosi, A.; Vescio, G.; Sammarco, G.; Montemurro, S.; De Sarro, G.; et al. Tryptase-positive mast cells and angiogenesis in keloids: A new possible post-surgical target for prevention. Updates Surg. 2013, 65, 53–57. [Google Scholar] [CrossRef]
- Patruno, R.; Marech, I.; Zizzo, N.; Ammendola, M.; Nardulli, P.; Gadaleta, C.; Introna, M.; Capriuolo, G.; Rubini, R.A.; Ribatti, D.; et al. c-Kit expression, angiogenesis, and grading in canine mast cell tumour: A unique model to study c-Kit driven human malignancies. BioMed. Res. Int. 2014, 2014, 730246. [Google Scholar] [CrossRef]
- Ammendola, M.; Sacco, R.; Sammarco, G.; Luposella, M.; Patruno, R.; Gadaleta, C.D.; De Sarro, G.; Ranieri, G. Mast Cell-Targeted Strategies in Cancer Therapy. Transfus. Med. Hemother. 2016, 43, 109–113. [Google Scholar] [CrossRef]
- Ferrero, G.; Cordero, F.; Tarallo, S.; Arigoni, M.; Riccardo, F.; Gallo, G.; Ronco, G.; Allasia, M.; Kulkarni, N.; Matullo, G.; et al. Small non-coding RNA profiling in human biofluids and surrogate tissues from healthy individuals: Description of the diverse and most represented species. Oncotarget 2017, 9, 3097–3111. [Google Scholar] [CrossRef] [PubMed]
- Schetter, A.J.; Leung, S.Y.; Sohn, J.J.; Zanetti, K.A.; Bowman, E.D.; Yanaihara, N.; Yuen, A.T.; Chan, T.L.; Kwong, D.L.W.; Au, G.K.H.; et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 2008, 299, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Calin, G.A. Epigenetics and miRNAs in human cancer. Adv. Genet. 2010, 70, 87–99. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene lin-4 Encodes Small RNAs with Antisense Complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional Regulation of the Heterochronic Gene lin-14 by lin-4 Mediates Temporal Pattern Formation in C. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs-microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Mashall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef]
- Tan, Y.; Lin, J.J.; Yang, X.; Gou, D.M.; Fu, L.; Li, F.R.; Yu, X.F. A panel of three plasma microRNAs for colorectal cancer diagnosis. Cancer Epidemiol. 2019, 60, 67–76. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Poursheikhani, A.; Abbaszadegan, M.R.; Nokhandani, N.; Kerachian, M.A. Integration analysis of long non-coding RNA (lncRNA) role in tumorigenesis of colon adenocarcinoma. BMC Med. Genom. 2020, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Galamb, O. Epithelial-to-mesenchymal and mesenchymal-toepithelial transitions in the colon. World J. Gastroenterol. 2012, 18, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainer, D.Y.; Srivastava, D. miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef]
- Chauhan, N.; Dasmana, A.; Jaggi, M.; Chauhan, S.C.; Yallapu, M.M. miR-205: A Potential Biomedicine for Cancer Therapy. Cells 2020, 9, 1957. [Google Scholar] [CrossRef]
- Li, X.; Nie, J.; Mei, Q.; Han, W.D. MicroRNAs: Novel immunotherapeutic targets in colorectal carcinoma. World J. Gastroenterol. 2016, 22, 5317–5331. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef]
- Hollis, M.; Nair, K.; Vyas, A.; Chaturvedi, L.S.; Gambhir, S.; Vyas, D. MicroRNAs potential utility in colon cancer: Early detection, prognosis, and chemosensitivity. World J. Gastroenterol. 2015, 21, 8284–8292. [Google Scholar] [CrossRef]
- Ghareib, A.F.; Mohamed, R.H.; Abd El-Fatah, A.R.; Saadawy, S.F. Assessment of Serum MicroRNA-21 Gene Expression for Diagnosis and Prognosis of Colorectal Cancer. J. Gastrointest. Cancer 2020, 51, 818–823. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Moreno, Z.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Nagel, R.; le Sage, C.; Diosdado, B.; van der Waal, M.; Oude Vrielink, J.A.F.; Brolijn, A.; Meijer, G.A.; Agami, R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008, 68, 5795–5802. [Google Scholar] [CrossRef]
- Fang, Z.; Tang, J.; Bai, Y.; Lin, H.; You, H.; Jin, H.; Lin, L.; You, P.; Li, J.; Dai, Z.; et al. Plasma levels of microRNA-24, microRNA-320a, and microRNA-423-5p are potential biomarkers for colorectal carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 86. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Weiner, H.L. Control of the gut microbiome by fecal microRNA. Microb. Cell 2016, 3, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.E.; Ahmed, N.C.; Vos, P.W.; Bonnerup, C.; Atkins, J.N.; Casey, M.; Nuovo, G.J.; Naziri, W.; Wiley, J.E.; Mota, H.; et al. Diagnostic microRNA markers to screen for sporadic human colon cancer in stool: I. Proof of principle. Cancer Genom. Proteom. 2013, 10, 93–113. [Google Scholar]
- Ahlquist, D.A. Molecular detection of colorectal neoplasia. Gastroenterology 2010, 138, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, S.; Ferrero, G.; Gallo, G.; Francavilla, A.; Clerico, G.; Realis Luc, A.; Manghi, P.; Thomas, A.M.; Vineis, P.; Segata, N.; et al. Altered Fecal Small RNA Profiles in Colorectal Cancer Reflect Gut Microbiome Composition in Stool Samples. Msystems 2019, 4, e00289–e00319. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Yasunaga, M.; Takahashi, A.; Kuroda, J.; Moriya, Y.; Akasu, T.; Fujita, S.; Yamamoto, S.; Baba, H.; Matsumura, Y. MicroRNA expression profiling of exfoliated colonocytes isolated from feces for colorectal cancer screening. Cancer Prev. Res. 2010, 3, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Ng, S.S.; Dong, Y.J.; Ng, S.C.; Leung, W.W.; Lee, C.W.; Wong, Y.N.; Chan, F.K.L.; Sung, J.J.Y. Detection of miR-92a and miR-21 in stool samples as potential screening biomarkers for colorectal cancer and polyps. Gut 2012, 61, 739–745. [Google Scholar] [CrossRef]
- Link, A.; Balaguer, F.; Shen, Y.; Nagasaka, T.; Lozano, J.J.; Boland, C.R.; Goel, A. Fecal MicroRNAs as novel biomarkers for colon cancer screening. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1766–1774. [Google Scholar] [CrossRef]
- Sarshar, M.; Scribano, D.; Ambrosi, C.; Palamara, A.T.; Masotti, A. Fecal microRNAs as Innovative Biomarkers of Intestinal Diseases and Effective Players in Host-Microbiome Interactions. Cancers 2020, 12, 2174. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.Z.; O’Connor, S.M.; van Holst Pellekaan, N.G.; Young, G.P.; James, R.J. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol. Cancer Res. 2003, 1, 882–891. [Google Scholar] [PubMed]
- Ng, E.K.; Tsang, W.P.; Ng, S.S.M.; Jin, H.C.; Yu, J.; Li, J.J.; Rocken, C.; Ebert, M.P.A.; Kwok, T.T.; Sung, J.J.Y. MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br. J. Cancer 2009, 101, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Bandrés, E.; Cubedo, E.; Agirre, X.; Malumbres, R.; Zarate, R.; Ramirez, N.; Abajo, A.; Navarro, A.; Moreno, I.; Monzo, M.; et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol. Cancer 2006, 5, 29. [Google Scholar] [CrossRef]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef]
- Schepeler, T.; Reinert, J.T.; Ostenfeld, M.S.; Christensen, L.L.; Silahtaroglu, A.N.; Dyrskjot, L.; Wiuf, C.; Sorensen, F.J.; Kruhoffer, M.; Laurberg, S.; et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008, 68, 6416–6424. [Google Scholar] [CrossRef]
- Milanesi, E.; Dobre, M.; Bucuroiu, A.I.; Herlea, V.; Manuc, T.E.; Salvi, E.; De Petro, G.; Manuc, M.; Becheanu, G. miRNAs-Based Molecular Signature for KRAS Mutated and Wild Type Colorectal Cancer: An Explorative Study. J. Immunol. Res. 2020, 2020, 4927120. [Google Scholar] [CrossRef]
- Gasparello, J.; Papi, C.; Allegretti, M.; Giordani, E.; Carboni, F.; Zazza, S.; Pescarmona, E.; Romania, P.; Giacomini, P.; Scapoli, C.; et al. A distinctive microRNA (miRNA) SIgature in the Blood of Colorectal Cancer (CRC) Patients at Surgery. Cancers 2020, 12, 2410. [Google Scholar] [CrossRef]
- Caramés, C.; Cristóbal, I.; Moreno, V.; del Puerto, L.; Moreno, I.; Rodriguez, M.; Marín, J.P.; Correa, A.V.; Hernández, R.; Zenzola, V.; et al. MicroRNA-21 predicts response to preoperative chemoradiotherapy in locally advanced rectal cancer. Int. J. Colorectal Dis. 2015, 30, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Kjersem, J.B.; Ikdahl, T.; Lingjaerde, O.C.; Guren, T.; Tveit, K.M.; Kure, E.H. Plasma microRNAs predicting clinical outcome in metastatic colorectal cancer patients receiving first-line oxaliplatin-based treatment. Mol. Oncol. 2014, 8, 59–67. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, S.J.; Choo, J.; Heo, J.; Yoo, J.W.; Jung, Y.; Ree, S.H.; Im, E. miR-23a-3p is a Key Regulator of IL-17C-Induced Tumor Angiogenesis in Colorectal Cancer. Cells 2020, 9, 1363. [Google Scholar] [CrossRef] [PubMed]
- Amerizadeh, F.; Khazaei, M.; Maftouh, M.; Mardani, R.; Bahrami, A. miRNA Targeting Angiogenesis as a Potential Therapeutic Approach in the Treatment of Colorectal Cancers. Curr. Pharm. Des. 2018, 24, 4668–4674. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, A.; Graziano, F.; Vincenzi, B.; Canestrari, E.; Perrone, G.; Galluccio, N.; Catalano, V.; Loupakis, F.; Rabitti, C.; Santini, D.; et al. High let-7° microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapyrefractory metastatic disease. Oncologist 2012, 17, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. Croce CM. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef]
- Wagenaar, T.R.; Zabludoff, S.; Ahn, S.M.; Allerson, C.; Arlt, H.; Baffa, R.; Cao, H.; Davis, S.; Garcia-Echeverria, C.; Gaur, R.; et al. Anti-miR-21 Suppresses Hepatocellular Carcinoma Growth via Broad Transcriptional Network Deregulation. Mol. Cancer Res. 2015, 13, 1009–1021. [Google Scholar] [CrossRef]
- Nishida, N.; Yamashita, S.; Mimori, K.; Sudo, T.; Tanaka, F.; Shibata, K.; Yamamoto, H.; Ishii, H.; Doki, Y.; Mori, M. MicroRNA-10b is a prognostic indicator in colorectal cancer and confers resistance to the chemotherapeutic agent 5-fluorouracil in colorectal cancer cells. Ann. Surg. Oncol. 2012, 19, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Poorebrahim, M.; Sadeghi, S.; Ghanbarian, M.; Kalhor, H.; Mehrtash, A.; Teimoori-Toolabi, L. Identification of candidate genes and miRNAs for sensitizing resistant colorectal cancer cells to oxaliplatin and irinotecan. Cancer Chemother. Pharmacol. 2020, 85, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Bekaii-Saab, T. A Comprehensive Review of Sequencing and Combination Strategies of Targeted Agents in Metastatic Colorectal Cancer. Oncologist 2018, 23, 25–34. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug. Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Irahara, N.; Baba, Y.; Nosho, K.; Shima, K.; Yan, L.; Dias-Santagata, D.; Iafrate, A.J.; Fuchs, C.S.; Haigis, K.M.; Ogino, S. NRAS mutations are rare in colorectal cancer. Diagn. Mol. Pathol. 2010, 19, 157–163. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Thorrez, L.; Siegfried, G.; Meulemans, S.; Evrard, S.; Tejpar, S.; Khatib, A.-M.; Creemers, J.W.M. The proprotein convertase furin is a pro-oncogenic driver in KRAS and BRAF driven colorectal cancer. Oncogene 2020, 39, 3571–3587. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Shibata, N.; Ura, T.; Takahari, D.; Shitara, K.; Muro, K.; Yatabe, Y. Cycleave polymerase chain reaction method is practically applicable for V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS)/V-raf murine sarcoma viral oncogene homolog B1 (BRAF) genotyping in colorectal cancer. Transl. Res. 2010, 156, 98–105. [Google Scholar] [PubMed]
- Smakman, N.; Borel Rinkes, I.H.M.; Voest, E.E.; Kranenburg, O. Control of colorectal metastasis formation by K-Ras. Biochim. Biophys. Acta 2005, 1756, 103–114. [Google Scholar] [CrossRef]
- Allgayer, H.; Wang, H.; Shirasawa, S.; Sasazuki, T.; Boyd, D. Targeted disruption of the K-ras oncogene in an invasive colon cancer cell line down-regulates urokinase receptor expression and plasminogen-dependent proteolysis. Br. J. Cancer 1999, 80, 1884–1891. [Google Scholar] [CrossRef][Green Version]
- Di Mauro, C.; Pesapane, A.; Formisano, L.; Rosa, R.; D’amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen acrivator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef]
- Yamamoto, H.; Itoh, F.; Senota, A.; Adachi, Y.; Yoshimoto, M.; Endoh, T.; Hinoda, Y.; Yachi, A.; Imai, K. Expression of matrix metalloproteinase matrilysin (MMP-7) was induced by activated Ki-ras via AP-1 activation in SW1417 colon cancer cells. J. Clin. Lab. Anal. 1995, 9, 297–301. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Curtin, K.; Schaffer, D.; Robertson, M.; Leppert, M.; Slattery, M.L. Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: A population-based study. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1193–1197. [Google Scholar]
- Bazan, V.; Migliavacca, M.; Zanna, I.; Tubiolo, C.; Grassi, N.; Latteri, M.A.; La Farina, M.; Albanese, I.; Dardanoni, G.; Salerno, S.; et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann. Oncol. 2002, 13, 1438–1446. [Google Scholar] [CrossRef]
- Jiang, Y.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Cheng, H.; Ajani, J.A. Assessment of K-ras mutation: A step toward personalized medicine for patients with colorectal cancer. Cancer 2009, 115, 3609–3617. [Google Scholar] [CrossRef]
- Binefa, G.; Rodriguez-Moranta, F.; Teule, A.; Medina-Hayas, M. Colorectal cancer: From prevention to personalized medicine. World J. Gastroenterol. 2014, 20, 6786–6808. [Google Scholar] [CrossRef]
- Rose, J.S.; Serna, D.S.; Martin, L.K.; Li, X.; Weatherby, L.M.; Abdel-Misih, S.; Zhao, W.; Bekaii-Saab, T. Influence of KRAS mutation status in metachronous and synchronous metastatic colorectal adenocarcinoma. Cancer 2012, 118, 6243–6252. [Google Scholar] [CrossRef]
- Charlton, M.E.; Kahl, A.R.; Greenbaum, A.A.; Karlitz, J.J.; Lin, C.; Lynch, C.F.; Chen, V.W. KRAS Testing, Tumor Location, and Survival in Patients with Stage IV Colorectal Cancer: SEER 2010–2013. J. Natl. Compr. Cancer Netw. 2017, 15, 1484–1493. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Ntanasis-Stathopoulos, I.; Bagante, F.; Moris, D.; Cloyd, J.; Spartalis, E.; Pawlik, T.M. Clinical significance and prognostic relevance of KRAS, BRAF, PI3K and TP53 genetic mutation analysis for resectable and unresectable colorectal liver metastases: A systematic review of the current evidence. Surg. Oncol. 2018, 27, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Denbo, J.W.; Yamashita, S.; Passot, G.; Egger, M.; Chun, Y.S.; Kopetz, S.E.; Maru, D.; Brudvik, K.W.; Wei, S.H.; Conrad, C.; et al. RAS Mutation Is Associated with Decreased Survival in Patients Undergoing Repeat Hepatectomy for Colorectal Liver Metastases. J. Gastrointest. Surg. 2017, 21, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Ricci, V.; Vivenza, D.; Granetto, C.; Fabozzi, T.; Miraglio, E.; Merlano, M.C. Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World J. Gastroenterol. 2016, 22, 6944–6954. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Heitzer, E.; Geigl, J.B.; Speicher, M.R. Patient monitoring through liquid biopsies using circulating tumor DNA. Int. J. Cancer 2017, 141, 887–896. [Google Scholar] [CrossRef]
- Oellerich, M.; Schutz, E.; Beck, J.; Walson, P.D. Circulating Cell-Free DNA-Diagnostic and Prognostic Applications in Personalized Cancer Therapy. Ther. Drug. Monit. 2019, 41, 115–120. [Google Scholar] [CrossRef]
- Furuki, H.; Yamada, T.; Takahashi, G.; Iwai, T.; Koizumi, M.; Shinji, S.; Yokoyama, Y.; Takeda, K.; Taniai, N.; Uchida, E. Evaluation of liquid biopsies for detection of emerging mutated genes in metastatic colorectal cancer. Eur. J. Surg. Oncol. 2018, 44, 975–982. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline Summary From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. J. Oncol. Pract. 2017, 13, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, E.; Argiles, G.; Elez, E.; Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- Barras, D. BRAF Mutation in Colorectal Cancer: An Update. Biomark. Cancer 2015, 7, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Cohen, R.; Pudlarz, T.; Delattre, J.F.; Colle, R.; André, T. Molecular Targets for the Treatement of Metastatic Colorectal Cancer. Cancers 2020, 19, 2350. [Google Scholar] [CrossRef]
- Chen, D.; Huang, J.F.; Liu, K.; Zhang, L.Q.; Yang, Z.; Chuai, Z.R.; Wang, Y.X.; Shi, D.C.; Huang, Q.; Fu, W.L. BRAFV600E mutation and its association with clinicopathological features of colorectal cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e90607. [Google Scholar] [CrossRef]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef]
- Tie, J.; Desai, J. Targeting BRAF mutant metastatic colorectal cancer: Clinical implications and emerging therapeutic strategies. Target Oncol. 2015, 10, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Clancy, C.; Burke, J.P.; Kalady, M.F.; Coffey, J.C. BRAF mutation is associated with distinct clinicopathological characteristics in colorectal cancer: A systematic review and meta-analysis. Colorectal Dis. 2013, 15, e711–e718. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Gibbs, P.; Lipton, L.; Christie, M.; Jorissen, R.N.; Burgess, A.W.; Croxford, M.; Jones, I.; Langland, R.; Kosmider, S.; et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int. J. Cancer 2011, 128, 2075–2084. [Google Scholar] [CrossRef]
- Lochhead, P.; Kuchiba, A.; Imamura, Y.; Liao, X.; Yamauchi, M.; Nishihara, R.; Qian, Z.R.; Morikawa, T.; Shen, J.; Meyerhardt, J.A.; et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J. Natl. Cancer Inst. 2013, 105, 1151–1156. [Google Scholar] [CrossRef]
- Landau, M.S.; Kuan, S.F.; Chiosea, S.; Pai, R.K. BRAF-mutated microsatellite stable colorectal carcinoma: An aggressive adenocarcinoma with reduced CDX2 and increased cytokeratin 7 immunohistochemical expression. Hum. Pathol. 2014, 45, 1704–1712. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Dal Pozzo, C.A.; Depetris, I.; Schirripa, M.; Brignola, S.; Biason, P.; Balistreri, M.; Dal Santo, L.; Lonardi, S.; Munari, G.; et al. The heterogeneous clinical and pathological landscapes of metastatic Braf-mutated colorectal cancer. Cancer Cell Int. 2020, 20, 30. [Google Scholar] [CrossRef]
- Ogino, S.; Shima, K.; Meyerhardt, J.A.; McCleary, J.; Ng, K.; Hollis, D.; Saltz, L.B.; Mayer, R.J.; Schaefer, P.; Whittom, R.; et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: Results from intergroup trial CALGB 89803. Clin. Cancer Res. 2012, 18, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Intini, R.; Cremolini, C.; Orlandi, A.; Sartore-Bianchi, A.; Pietrantonio, F.; Pella, N.; Spallanzani, A.; Dell’Aquila, E.; Scartozzi, M.; et al. A validated prognostic classifier for (V600E)BRAF-mutated metastatic colorectal cancer: The ‘BRAF BeCool’ study. Eur. J. Cancer 2019, 118, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Martens, U.M.; Riera-Knorrenschild, J.; Greeve, J.; Florschutz, A.; Wessendorf, S.; Ettrich, T.; Kanzler, S.; Norenberg, D.; Ricke, J. FOLFOXIRI Plus Panitumumab As First-Line Treatment of RAS Wild-Type Metastatic Colorectal Cancer: The Randomized, Open-Label, Phase II VOLFI Study (AIO KRK0109). J. Clin. Oncol. 2019, 37, 3401–3411. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwye, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Sang Hong, Y.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sammarco, G.; Gallo, G.; Vescio, G.; Picciariello, A.; De Paola, G.; Trompetto, M.; Currò, G.; Ammendola, M. Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine. J. Clin. Med. 2020, 9, 2852. https://doi.org/10.3390/jcm9092852
Sammarco G, Gallo G, Vescio G, Picciariello A, De Paola G, Trompetto M, Currò G, Ammendola M. Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine. Journal of Clinical Medicine. 2020; 9(9):2852. https://doi.org/10.3390/jcm9092852
Chicago/Turabian StyleSammarco, Giuseppe, Gaetano Gallo, Giuseppina Vescio, Arcangelo Picciariello, Gilda De Paola, Mario Trompetto, Giuseppe Currò, and Michele Ammendola. 2020. "Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine" Journal of Clinical Medicine 9, no. 9: 2852. https://doi.org/10.3390/jcm9092852
APA StyleSammarco, G., Gallo, G., Vescio, G., Picciariello, A., De Paola, G., Trompetto, M., Currò, G., & Ammendola, M. (2020). Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine. Journal of Clinical Medicine, 9(9), 2852. https://doi.org/10.3390/jcm9092852