The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
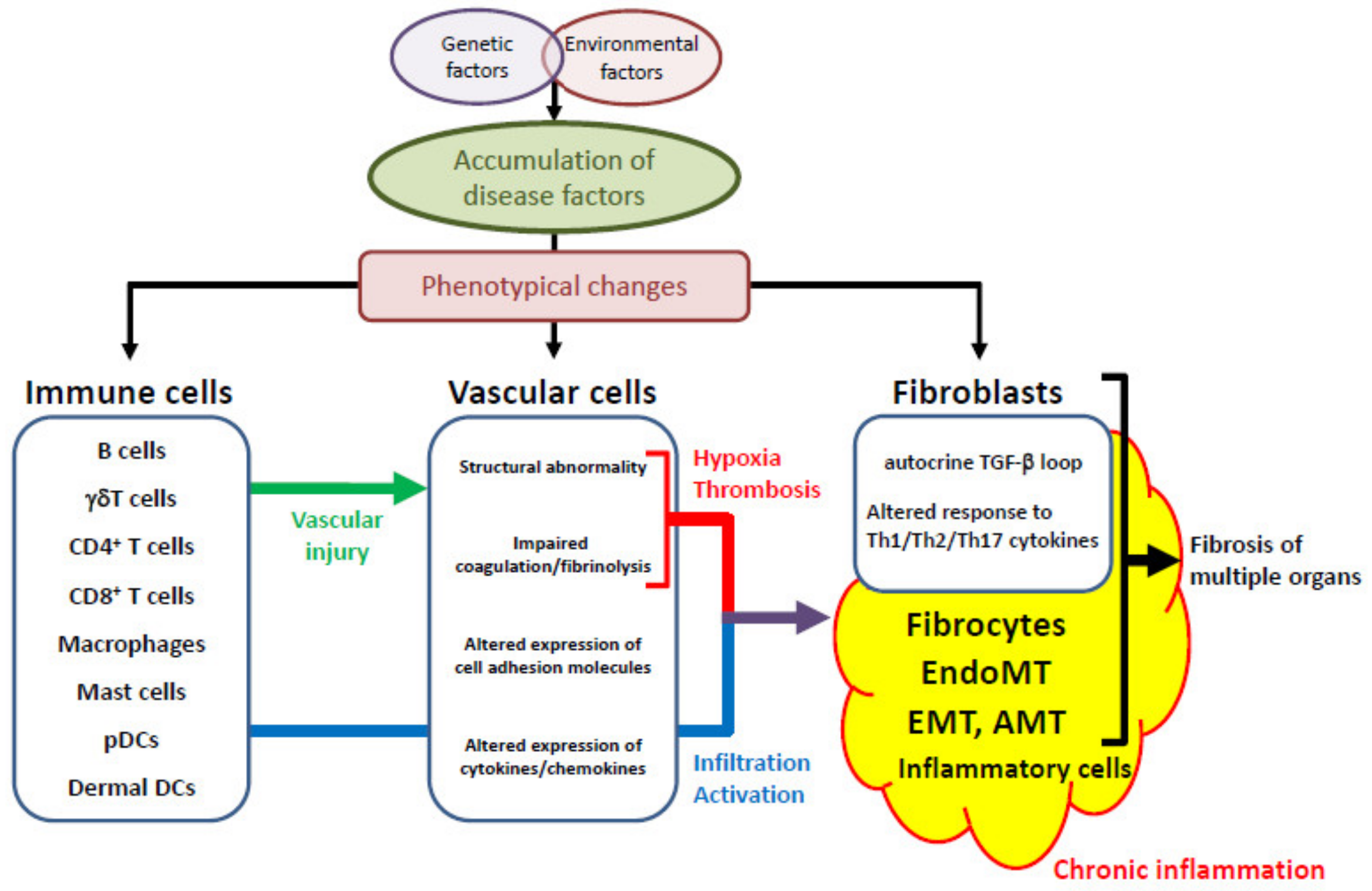
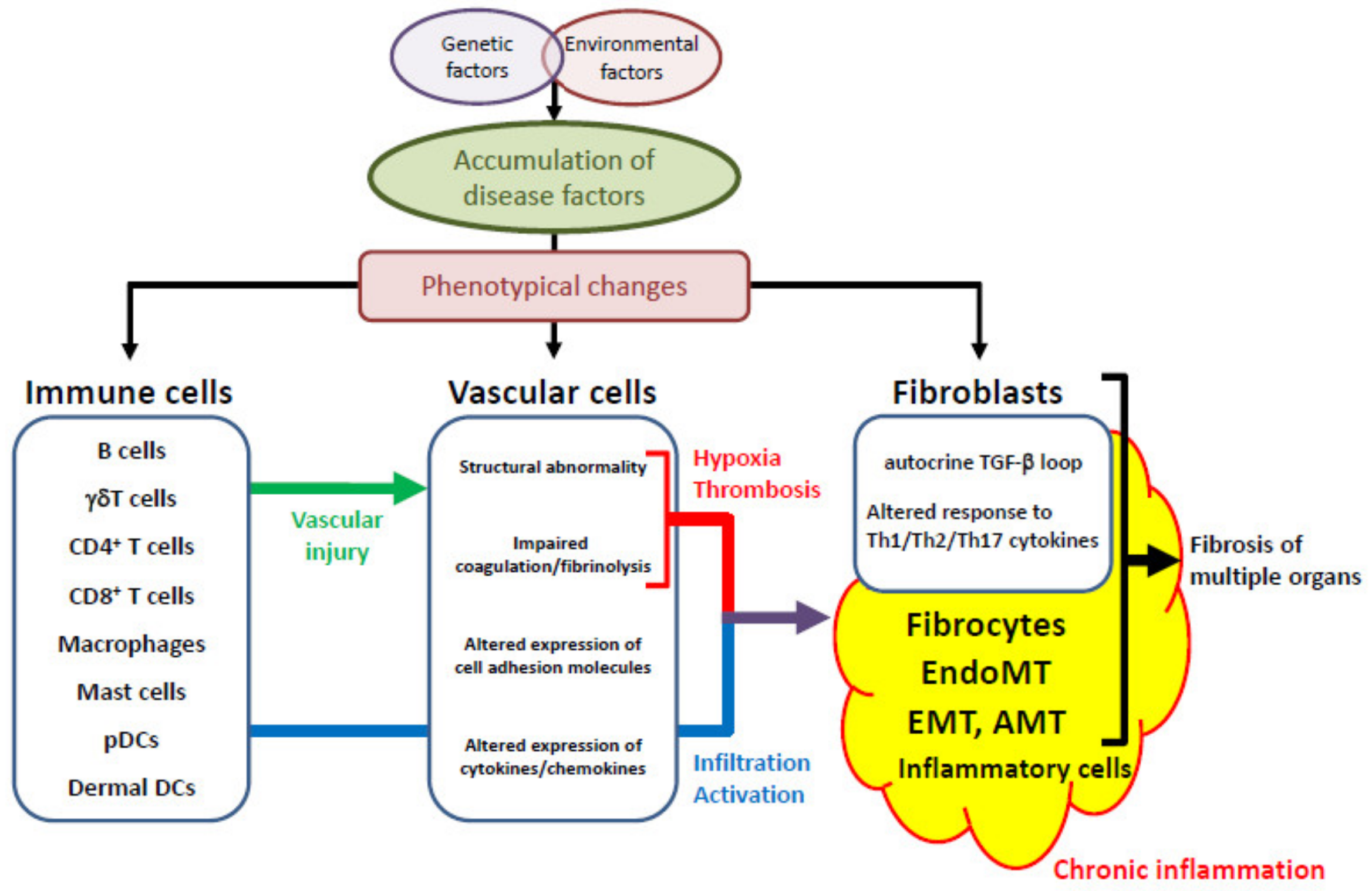
2. The Common SSc-Specific Pathologic Cascade across Multiple Organs
2.1. Genetic Factors and Environmental Influences
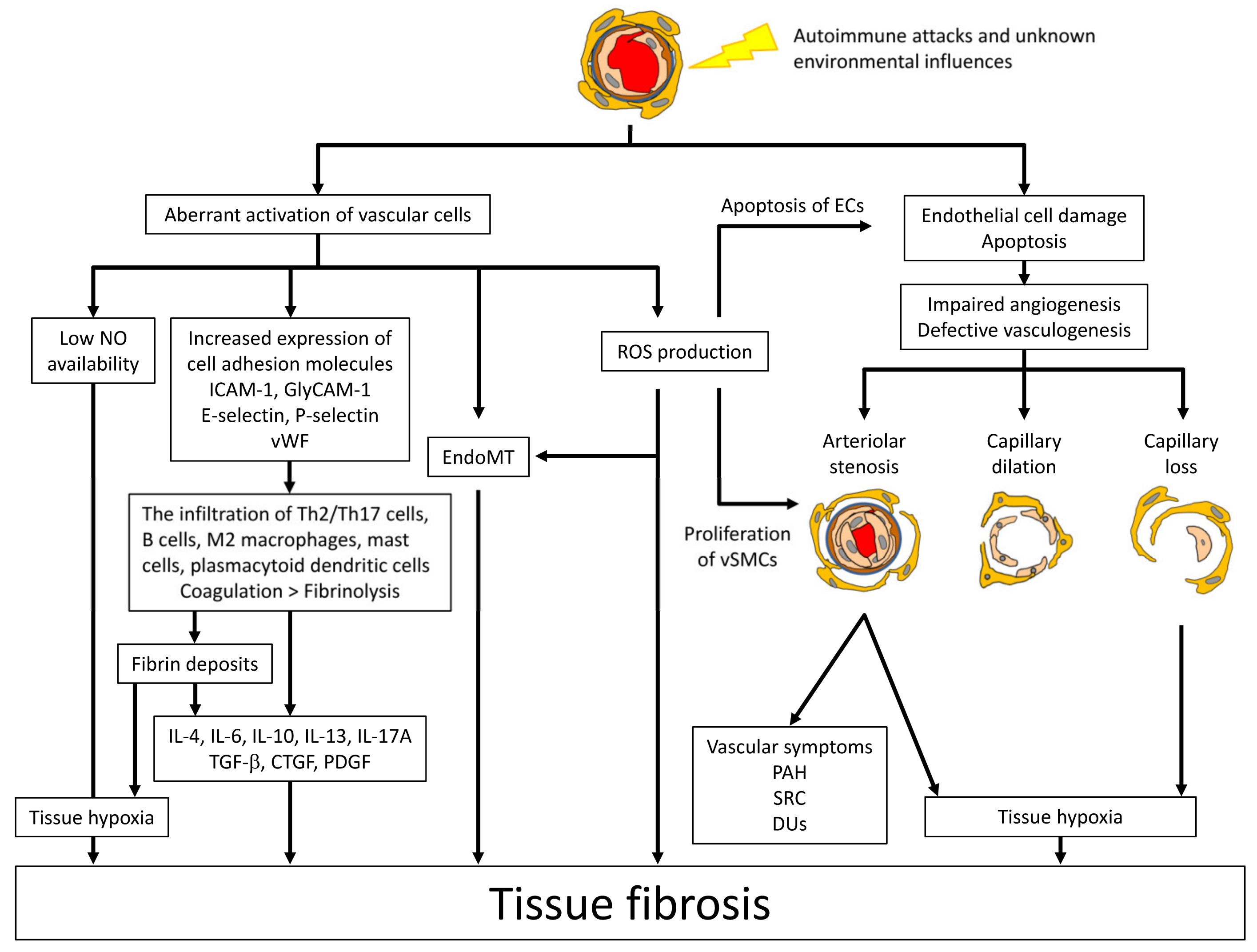
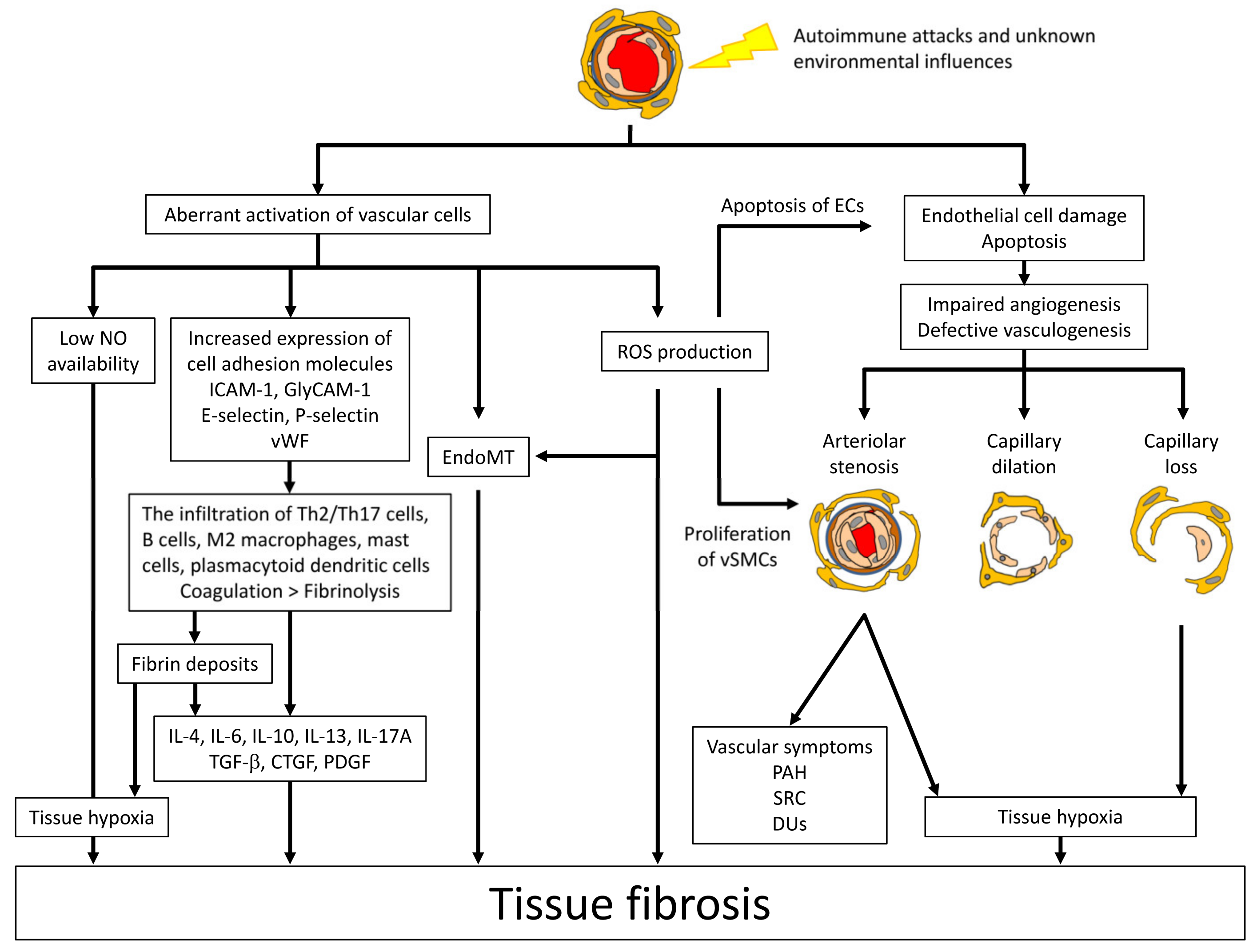
2.2. Vascular Injury, an Initial Event of SSc Development
2.3. Aberrant Vascular Reaction to Initial Vascular Injury
2.4. Inflammation
2.5. The Activation of Fibroblasts
2.6. The Role of Agonistic Autoantibodies
3. The Additional Organ-Specific Pathologies
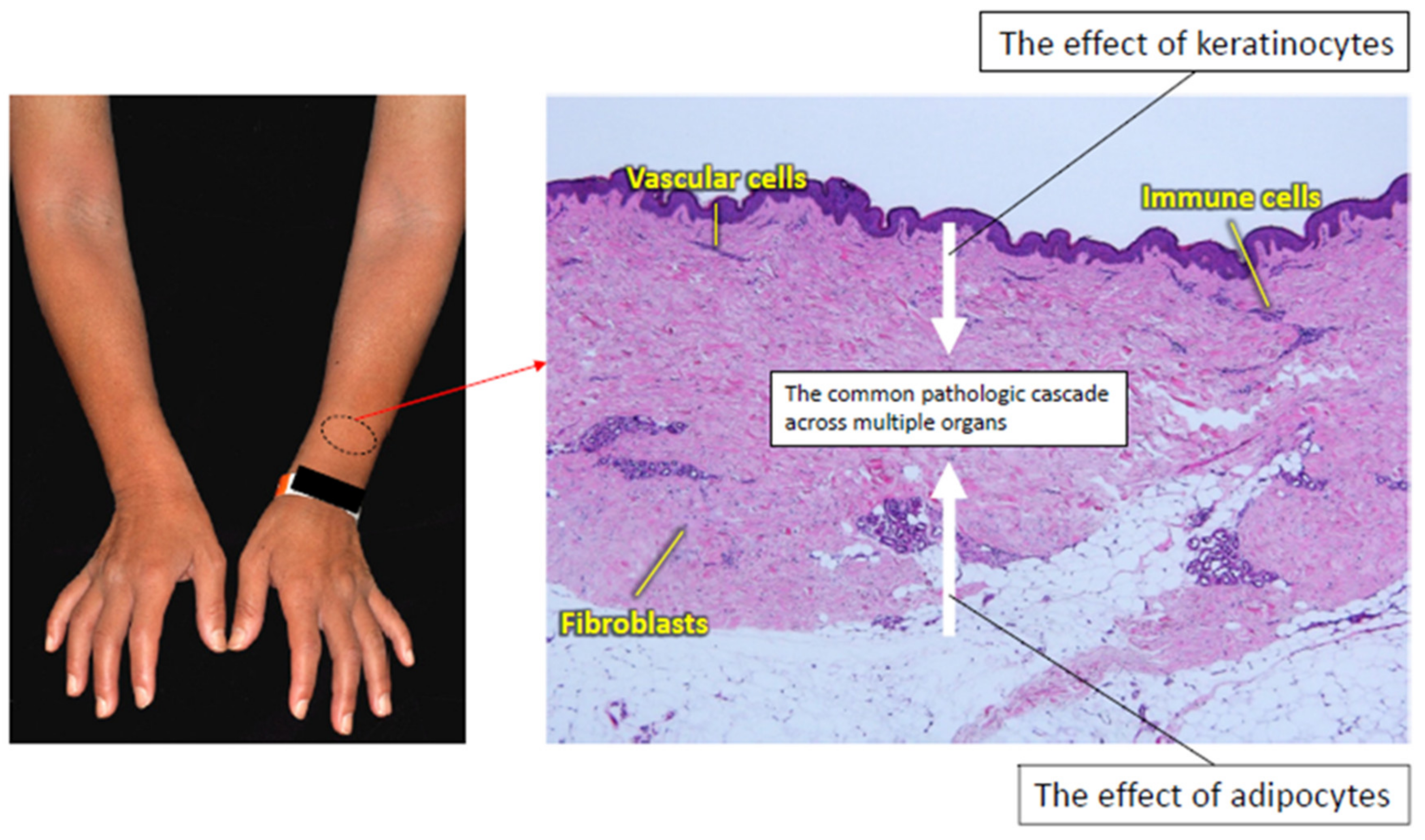
3.1. The Skin
3.1.1. Keratinocytes
3.1.2. Adipocytes
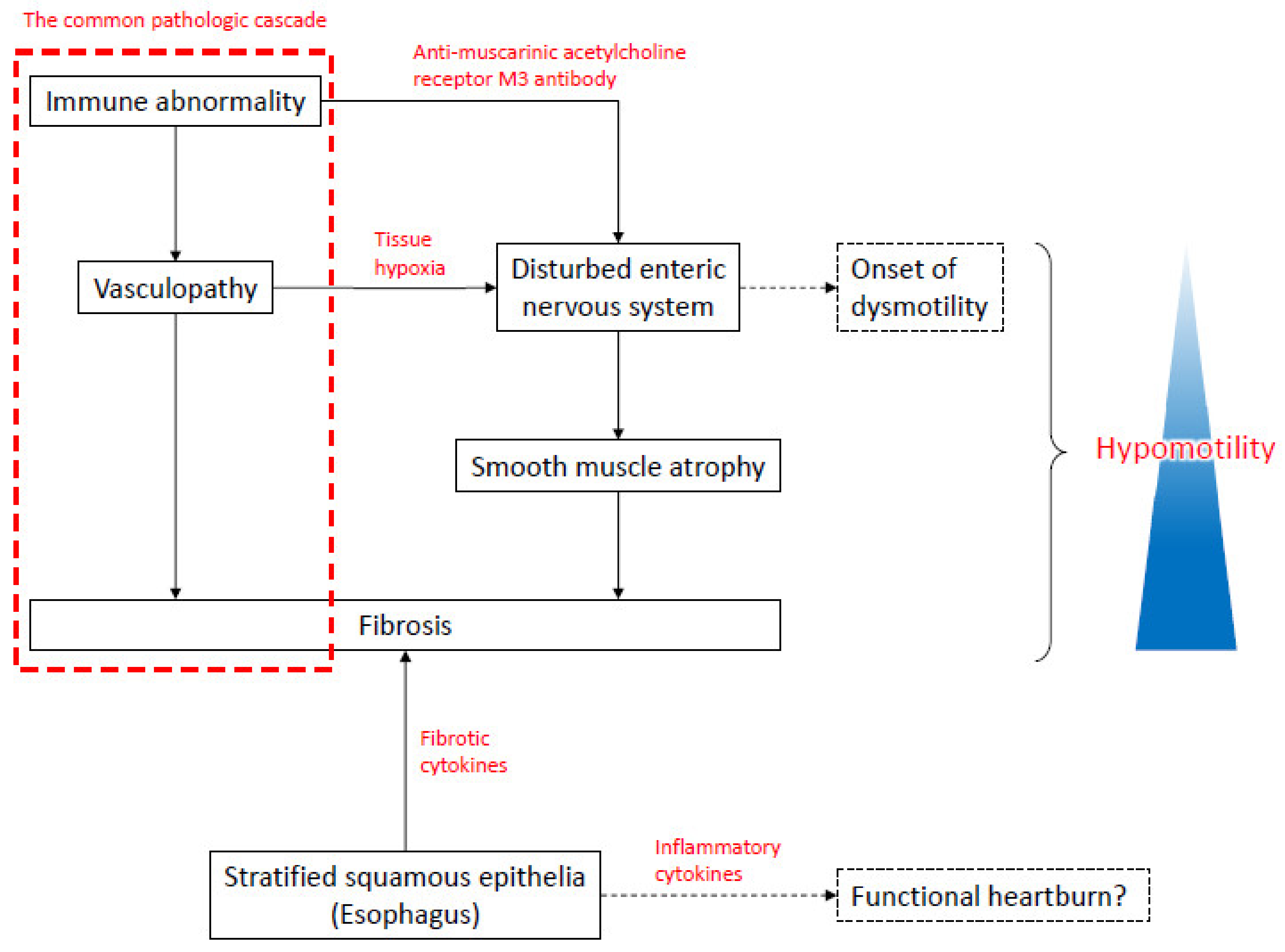
3.2. GI Involvement
4. Lung Involvement
5. Heart
6. Liver
7. SRC
8. Conclusions
Funding
Conflicts of Interest
References
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic sclerosis. Nat. Rev. Dis. Primers 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y. Systemic sclerosis. J. Dermatol. 2018, 45, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C.; Cho, M.; Chatterjee, S.; Aguilar, M.B.; Reveille, J.D.; Mayes, M.D. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. 2001, 44, 1359–1362. [Google Scholar] [CrossRef]
- Feghali-Bostwick, C.; Medsger, T.A.; Wright, T.M. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum 2003, 48, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Broen, J.C.; Radstake, T.R.; Rossato, M. The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 671–681. [Google Scholar] [CrossRef]
- Yamashita, K.; Kawasaki, A.; Matsushita, T.; Furukawa, H.; Kondo, Y.; Okiyama, N.; Nagaoka, S.; Shimada, K.; Sugii, S.; Katayama, M.; et al. Association of functional (GA)n microsatellite polymorphism in the FLI1 gene with susceptibility to human systemic sclerosis. Rheumatology 2020. [Google Scholar] [CrossRef]
- Dieudé, P.; Guedj, M.; Wipff, J.; Avouac, J.; Fajardy, I.; Diot, E.; Granel, B.; Sibilia, J.; Cabane, J.; Mouthon, L.; et al. Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: A new perspective for pulmonary fibrosis. Arthritis Rheum. 2009, 60, 225–233. [Google Scholar] [CrossRef]
- Ito, I.; Kawaguchi, Y.; Kawasaki, A.; Hasegawa, M.; Ohashi, J.; Hikami, K.; Kawamoto, M.; Fujimoto, M.; Takehara, K.; Sato, S.; et al. Association of a functional polymorphism in the IRF5 region with systemic sclerosis in a Japanese population. Arthritis Rheum. 2009, 60, 1845–1850. [Google Scholar] [CrossRef]
- Dieude, P.; Dawidowicz, K.; Guedj, M.; Legrain, Y.; Wipff, J.; Hachulla, E.; Diot, E.; Sibilia, J.; Mouthon, L.; Cabane, J.; et al. Phenotype-haplotype correlation of IRF5 in systemic sclerosis: Role of 2 haplotypes in disease severity. J. Rheumatol. 2010, 37, 987–992. [Google Scholar] [CrossRef] [Green Version]
- Radstake, T.R.; Gorlova, O.; Rueda, B.; Martin, J.E.; Alizadeh, B.Z.; Palomino-Morales, R.; Coenen, M.J.; Vonk, M.C.; Voskuyl, A.E.; Schuerwegh, A.J.; et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat. Genet. 2010, 42, 426–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Chen, B.; Ma, B.; Nie, S. Association between IRF5 polymorphisms and autoimmune diseases: A meta-analysis. Genet. Mol. Res. 2014, 13, 4473–4485. [Google Scholar] [CrossRef]
- Allanore, Y.; Saad, M.; Dieudé, P.; Avouac, J.; Distler, J.H.; Amouyel, P.; Matucci-Cerinic, M.; Riemekasten, G.; Airo, P.; Melchers, I.; et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011, 7, e1002091. [Google Scholar] [CrossRef] [PubMed]
- Sharif, R.; Mayes, M.D.; Tan, F.K.; Gorlova, O.Y.; Hummers, L.K.; Shah, A.A.; Furst, D.E.; Khanna, D.; Martin, J.; Bossini-Castillo, L.; et al. IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1197–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Martinis, M.; Ciccarelli, F.; Sirufo, M.M.; Ginaldi, L. An overview of environmental risk factors in systemic sclerosis. Expert Rev. Clin. Immunol. 2016, 12, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Yang, Y.; Guo, X.; Hei, N.; Lai, S.; Assassi, S.; Liu, M.; Tan, F.; Zhou, X. Identification of an Association of TNFAIP3 Polymorphisms With Matrix Metalloproteinase Expression in Fibroblasts in an Integrative Study of Systemic Sclerosis-Associated Genetic and Environmental Factors. Arthritis Rheum. 2016, 68, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altorok, N.; Kahaleh, B. Epigenetics and systemic sclerosis. Semin Immunopathol. 2015, 37, 453–462. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Gordon, S.M.; Waldman, M.; Edison, J.D.; Little, D.J.; Stitt, R.S.; Bailey, W.T.; Hughes, J.B.; Olson, S.W. Autoantibodies are present before the clinical diagnosis of systemic sclerosis. PLoS ONE 2019, 14, e0214202. [Google Scholar] [CrossRef] [Green Version]
- Avouac, J.; Fransen, J.; Walker, U.A.; Riccieri, V.; Smith, V.; Muller, C.; Miniati, I.; Tarner, I.H.; Randone, S.B.; Cutolo, M.; et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: Results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann. Rheum. Dis. 2011, 70, 476–481. [Google Scholar] [CrossRef]
- Kahaleh, M.B.; Fan, P.S.; Otsuka, T. Gammadelta receptor bearing T cells in scleroderma: Enhanced interaction with vascular endothelial cells in vitro. Clin. Immunol. 1999, 91, 188–195. [Google Scholar] [CrossRef]
- Hill, M.B.; Phipps, J.L.; Cartwright, R.J.; Milford Ward, A.; Greaves, M.; Hughes, P. Antibodies to membranes of endothelial cells and fibroblasts in scleroderma. Clin. Exp. Immunol. 1996, 106, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.; Pottinger, B.E.; Woo, P.; Black, C.M.; Loizou, S.; Byron, M.A.; Pearson, J.D. Measurement and characterisation of circulating anti-endothelial cell IgG in connective tissue diseases. Clin. Exp. Immunol. 1988, 72, 450–456. [Google Scholar] [PubMed]
- Salojin, K.V.; Le Tonquèze, M.; Saraux, A.; Nassonov, E.L.; Dueymes, M.; Piette, J.C.; Youinou, P.Y. Antiendothelial cell antibodies: Useful markers of systemic sclerosis. Am. J. Med. 1997, 102, 178–185. [Google Scholar] [CrossRef]
- Sgonc, R.; Gruschwitz, M.S.; Boeck, G.; Sepp, N.; Gruber, J.; Wick, G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000, 43, 2550–2562. [Google Scholar] [CrossRef]
- Akamata, K.; Asano, Y.; Taniguchi, T.; Yamashita, T.; Saigusa, R.; Nakamura, K.; Noda, S.; Aozasa, N.; Toyama, T.; Takahashi, T.; et al. Increased expression of chemerin in endothelial cells due to Fli1 deficiency may contribute to the development of digital ulcers in systemic sclerosis. Rheumatology 2015, 54, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; Fleming, J.; Pritchard, D.K.; Amon, L.M.; Xue, J.; Arnett, H.A.; Chen, G.; Breen, P.; Buckner, J.H.; Molitor, J.A.; et al. Combined analysis of monocyte and lymphocyte messenger RNA expression with serum protein profiles in patients with scleroderma. Arthritis Rheum. 2008, 58, 1465–1474. [Google Scholar] [CrossRef]
- Takahashi, T.; Asano, Y.; Nakamura, K.; Yamashita, T.; Saigusa, R.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Yoshizaki, A.; Tamaki, Z.; et al. A potential contribution of antimicrobial peptide LL-37 to tissue fibrosis and vasculopathy in systemic sclerosis. Br. J. Dermatol 2016, 175, 1195–1203. [Google Scholar] [CrossRef]
- Toyama, T.; Asano, Y.; Miyagawa, T.; Nakamura, K.; Hirabayashi, M.; Yamashita, T.; Saigusa, R.; Miura, S.; Ichimura, Y.; Takahashi, T.; et al. The impact of transcriptional factor Fli1 deficiency on the regulation of angiogenesis. Exp. Dermatol. 2017. [Google Scholar] [CrossRef]
- Asano, Y.; Stawski, L.; Hant, F.; Highland, K.; Silver, R.; Szalai, G.; Watson, D.K.; Trojanowska, M. Endothelial Fli1 deficiency impairs vascular homeostasis: A role in scleroderma vasculopathy. Am. J. Pathol. 2010, 176, 1983–1998. [Google Scholar] [CrossRef]
- Kuwana, M.; Okazaki, Y. Impaired in vivo neovascularization capacity of endothelial progenitor cells in patients with systemic sclerosis. Arthritis Rheum. 2013. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Okazaki, Y.; Seta, N.; Satoh, T.; Takahashi, K.; Ikezawa, Z.; Kuwana, M. Enhanced angiogenic potency of monocytic endothelial progenitor cells in patients with systemic sclerosis. Arthritis Res. Ther. 2010, 12, R205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwana, M.; Okazaki, Y.; Yasuoka, H.; Kawakami, Y.; Ikeda, Y. Defective vasculogenesis in systemic sclerosis. Lancet 2004, 364, 603–610. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Yanaba, K.; Iwata, Y.; Komura, K.; Ogawa, A.; Akiyama, Y.; Muroi, E.; Hara, T.; Ogawa, F.; Takenaka, M.; et al. Cell adhesion molecules regulate fibrotic process via Th1/Th2/Th17 cell balance in a bleomycin-induced scleroderma model. J. Immunol. 2010, 185, 2502–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, T.; Asano, Y.; Akamata, K.; Noda, S.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Trojanowska, M.; Sato, S. Fibrosis, vascular activation, and immune abnormalities resembling systemic sclerosis in bleomycin-treated fli-1-haploinsufficient mice. Arthritis Rheum. 2015, 67, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Higashi-Kuwata, N.; Jinnin, M.; Makino, T.; Fukushima, S.; Inoue, Y.; Muchemwa, F.C.; Yonemura, Y.; Komohara, Y.; Takeya, M.; Mitsuya, H.; et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res. Ther. 2010, 12, R128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yukawa, S.; Yamaoka, K.; Sawamukai, N.; Shimajiri, S.; Kubo, S.; Miyagawa, I.; Sonomoto, K.; Saito, K.; Tanaka, Y. Dermal mast cell density in fingers reflects severity of skin sclerosis in systemic sclerosis. Mod. Rheumatol 2013, 23, 1151–1157. [Google Scholar] [CrossRef]
- Matsushita, T.; Hasegawa, M.; Hamaguchi, Y.; Takehara, K.; Sato, S. Longitudinal analysis of serum cytokine concentrations in systemic sclerosis: Association of interleukin 12 elevation with spontaneous regression of skin sclerosis. J. Rheumatol. 2006, 33, 275–284. [Google Scholar]
- Yang, X.; Yang, J.; Xing, X.; Wan, L.; Li, M. Increased frequency of Th17 cells in systemic sclerosis is related to disease activity and collagen overproduction. Arthritis Res. Ther. 2014, 16, R4. [Google Scholar] [CrossRef] [Green Version]
- Tan, F.K.; Zhou, X.; Mayes, M.D.; Gourh, P.; Guo, X.; Marcum, C.; Jin, L.; Arnett, F.C. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology 2006, 45, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Kizu, A.; Medici, D.; Kalluri, R. Endothelial-mesenchymal transition as a novel mechanism for generating myofibroblasts during diabetic nephropathy. Am. J. Pathol. 2009, 175, 1371–1373. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir Cell Mol. Biol 2010, 43, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, S.A. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN Rheumatol 2013, 2013, 835948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, S.A.; Piera-Velazquez, S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol 2016, 51, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, F.A.; Piera-Velazquez, S.; Farber, J.L.; Feghali-Bostwick, C.; Jimenez, S.A. Endothelial Cells Expressing Endothelial and Mesenchymal Cell Gene Products in Lung Tissue From Patients With Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheum. 2016, 68, 210–217. [Google Scholar] [CrossRef]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; De Paulis, A.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef]
- Yamane, K.; Miyauchi, T.; Suzuki, N.; Yuhara, T.; Akama, T.; Suzuki, H.; Kashiwagi, H. Significance of plasma endothelin-1 levels in patients with systemic sclerosis. J. Rheumatol. 1992, 19, 1566–1571. [Google Scholar]
- Vancheeswaran, R.; Magoulas, T.; Efrat, G.; Wheeler-Jones, C.; Olsen, I.; Penny, R.; Black, C.M. Circulating endothelin-1 levels in systemic sclerosis subsets--a marker of fibrosis or vascular dysfunction? J. Rheumatol. 1994, 21, 1838–1844. [Google Scholar]
- Marvi, U.; Chung, L. Digital ischemic loss in systemic sclerosis. Int. J. Rheumatol. 2010, 2010. [Google Scholar] [CrossRef]
- Saigusa, R.; Asano, Y.; Yamashita, T.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Yoshizaki, A.; Miyagaki, T.; Sugaya, M.; et al. Fli1 deficiency contributes to the downregulation of endothelial protein C receptor in systemic sclerosis: A possible role in prothrombotic conditions. Br. J. Dermatol. 2016, 174, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Borg, J.Y.; Hellot, M.F.; Levesque, H. Plasma D-dimer concentration in patients with systemic sclerosis. Br. J. Dermatol. 2008, 158, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Volpe, A.; Caramaschi, P.; Salvagno, G.L.; Montagnana, M.; Guidi, G.C. Plasma D-dimer concentration in patients with systemic sclerosis. Thromb. J. 2006, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinnin, M.; Ihn, H.; Yamane, K.; Asano, Y.; Yazawa, N.; Tamaki, K. Plasma plasmin-alpha2-plasmin inhibitor complex levels are increased in systemic sclerosis patients with pulmonary hypertension. Rheumatology 2003, 42, 240–243. [Google Scholar] [CrossRef] [Green Version]
- Kahaleh, M.B. Raynaud’s phenomenon and vascular disease in scleroderma. Curr. Opin. Rheumatol. 1994, 6, 621–627. [Google Scholar] [CrossRef]
- Chung, L.; Fiorentino, D. Digital ulcers in patients with systemic sclerosis. Autoimmun. Rev. 2006, 5, 125–128. [Google Scholar] [CrossRef]
- Schiopu, E.; Impens, A.J.; Phillips, K. Digital ischemia in scleroderma spectrum of diseases. Int. J. Rheumatol. 2010, 2010. [Google Scholar] [CrossRef]
- MacDonald, K.G.; Dawson, N.A.; Huang, Q.; Dunne, J.V.; Levings, M.K.; Broady, R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J. Allergy Clin. Immunol. 2015, 135, e946–e949. [Google Scholar] [CrossRef]
- Bosello, S.; Angelucci, C.; Lama, G.; Alivernini, S.; Proietti, G.; Tolusso, B.; Sica, G.; Gremese, E.; Ferraccioli, G. Characterization of inflammatory cell infiltrate of scleroderma skin: B cells and skin score progression. Arthritis Res. Ther. 2018, 20, 75. [Google Scholar] [CrossRef]
- Lafyatis, R.; O’Hara, C.; Feghali-Bostwick, C.A.; Matteson, E. B cell infiltration in systemic sclerosis-associated interstitial lung disease. Arthritis Rheum. 2007, 56, 3167–3168. [Google Scholar] [CrossRef]
- Whitfield, M.L.; Finlay, D.R.; Murray, J.I.; Troyanskaya, O.G.; Chi, J.T.; Pergamenschikov, A.; McCalmont, T.H.; Brown, P.O.; Botstein, D.; Connolly, M.K. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc. Natl. Acad. Sci. USA 2003, 100, 12319–12324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Fujimoto, M.; Matsushita, T.; Hamaguchi, Y.; Hasegawa, M.; Takehara, K.; Komura, K.; Sato, S. Clinical association of serum interleukin-17 levels in systemic sclerosis: Is systemic sclerosis a Th17 disease? J. Dermatol. Sci. 2008, 50, 240–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Hou, W.; Xu, K.; Han, D.; Jiang, C.; Mou, K.; Li, Y.; Meng, L.; Lu, S. The elevated expression of Th17-related cytokines and receptors is associated with skin lesion severity in early systemic sclerosis. Hum. Immunol. 2015, 76, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Auffray, C.; Avouac, J.; Allanore, Y. Regulatory T Cells in Systemic Sclerosis. Front. Immunol. 2018, 9, 2356. [Google Scholar] [CrossRef]
- Sato, S.; Hasegawa, M.; Takehara, K. Serum levels of interleukin-6 and interleukin-10 correlate with total skin thickness score in patients with systemic sclerosis. J. Dermatol. Sci. 2001, 27, 140–146. [Google Scholar] [CrossRef]
- Nakashima, T.; Jinnin, M.; Yamane, K.; Honda, N.; Kajihara, I.; Makino, T.; Masuguchi, S.; Fukushima, S.; Okamoto, Y.; Hasegawa, M.; et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J. Immunol. 2012, 188, 3573–3583. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, A. B lymphocytes in systemic sclerosis: Abnormalities and therapeutic targets. J. Dermatol. 2016, 43, 39–45. [Google Scholar] [CrossRef]
- Sato, S.; Hasegawa, M.; Fujimoto, M.; Tedder, T.F.; Takehara, K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J. Immunol. 2000, 165, 6635–6643. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K. Altered blood B lymphocyte homeostasis in systemic sclerosis: Expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004, 50, 1918–1927. [Google Scholar] [CrossRef]
- Smith, V.; Van Praet, J.T.; Vandooren, B.; Van der Cruyssen, B.; Naeyaert, J.M.; Decuman, S.; Elewaut, D.; De Keyser, F. Rituximab in diffuse cutaneous systemic sclerosis: An open-label clinical and histopathological study. Ann. Rheum. Dis. 2010, 69, 193–197. [Google Scholar] [CrossRef]
- Daoussis, D.; Liossis, S.N.; Tsamandas, A.C.; Kalogeropoulou, C.; Kazantzi, A.; Sirinian, C.; Karampetsou, M.; Yiannopoulos, G.; Andonopoulos, A.P. Experience with rituximab in scleroderma: Results from a 1-year, proof-of-principle study. Rheumatology 2010, 49, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, S.; Distler, J.H.; Maurer, B.; Huscher, D.; van Laar, J.M.; Allanore, Y.; Distler, O. Effects and safety of rituximab in systemic sclerosis: An analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann. Rheum. Dis. 2015, 74, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Daoussis, D.; Melissaropoulos, K.; Sakellaropoulos, G.; Antonopoulos, I.; Markatseli, T.E.; Simopoulou, T.; Georgiou, P.; Andonopoulos, A.P.; Drosos, A.A.; Sakkas, L.; et al. A multicenter, open-label, comparative study of B-cell depletion therapy with Rituximab for systemic sclerosis-associated interstitial lung disease. Semin Arthritis Rheum. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bosello, S.L.; De Luca, G.; Rucco, M.; Berardi, G.; Falcione, M.; Danza, F.M.; Pirronti, T.; Ferraccioli, G. Long-term efficacy of B cell depletion therapy on lung and skin involvement in diffuse systemic sclerosis. Semin Arthritis Rheum. 2015, 44, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Ebata, S.; Yoshizaki, A.; Fukasawa, T.; Miura, S.; Takahashi, T.; Sumida, H.; Asano, Y.; Sato, S. Rituximab therapy is more effective than cyclophosphamide therapy for Japanese patients with anti-topoisomerase I-positive systemic sclerosis-associated interstitial lung disease. J. Dermatol. 2019. [Google Scholar] [CrossRef]
- Daoussis, D.; Antonopoulos, I.; Liossis, S.N.; Yiannopoulos, G.; Andonopoulos, A.P. Treatment of systemic sclerosis-associated calcinosis: A case report of rituximab-induced regression of CREST-related calcinosis and review of the literature. Semin Arthritis Rheum. 2012, 41, 822–829. [Google Scholar] [CrossRef]
- Khor, C.G.; Chen, X.L.; Lin, T.S.; Lu, C.H.; Hsieh, S.C. Rituximab for refractory digital infarcts and ulcers in systemic sclerosis. Clin. Rheumatol. 2014, 33, 1019–1020. [Google Scholar] [CrossRef]
- Maslyanskiy, A.L.; Lapin, S.V.; Kolesova, E.P.; Penin, I.N.; Cheshuina, M.D.; Feist, E.; Konradi, A.O. Effects of rituximab therapy on elastic properties of vascular wall in patients with progressive systemic sclerosis. Clin. Exp. Rheumatol. 2014, 32, S228. [Google Scholar]
- Hügle, T.; Hogan, V.; White, K.E.; van Laar, J.M. Mast cells are a source of transforming growth factor β in systemic sclerosis. Arthritis Rheum. 2011, 63, 795–799. [Google Scholar] [CrossRef]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
- Kim, D.; Peck, A.; Santer, D.; Patole, P.; Schwartz, S.M.; Molitor, J.A.; Arnett, F.C.; Elkon, K.B. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: Association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. 2008, 58, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Black, C.M.; Silman, A.J.; Herrick, A.I.; Denton, C.P.; Wilson, H.; Newman, J.; Pompon, L.; Shi-Wen, X. Interferon-alpha does not improve outcome at one year in patients with diffuse cutaneous scleroderma: Results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 1999, 42, 299–305. [Google Scholar] [CrossRef]
- Hügle, T.; Gratzl, S.; Daikeler, T.; Frey, D.; Tyndall, A.; Walker, U.A. Sclerosing skin disorders in association with multiple sclerosis. Coincidence, underlying autoimmune pathology or interferon induced? Ann. Rheum. Dis. 2009, 68, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Pelidou, S.H.; Tsifetaki, N.; Giannopoulos, S.; Deretzi, G.; Voulgari, P.; Kyritsis, A. Multiple sclerosis associated with systemic sclerosis. Rheumatol. Int. 2007, 27, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, A.; Sensi, F.; Barrella, M.; Francia, A. Systemic sclerosis and multiple sclerosis. J. Neurol. 1999, 246, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Kojima, A.; Hirokawa, T.; Oyama, T.; Naganuma, A.; Maruta, S.; Okada, K.; Ban, S.; Yoshida, K.; Takagi, H.; et al. Systemic sclerosis after interferon alphacon-1 therapy for hepatitis C. Intern. Med. 2007, 46, 473–476. [Google Scholar] [CrossRef] [Green Version]
- Solans, R.; Bosch, J.A.; Esteban, I.; Vilardell, M. Systemic sclerosis developing in association with the use of interferon alpha therapy for chronic viral hepatitis. Clin. Exp. Rheumatol. 2004, 22, 625–628. [Google Scholar]
- Beretta, L.; Caronni, M.; Vanoli, M.; Scorza, R. Systemic sclerosis after interferon-alfa therapy for myeloproliferative disorders. Br. J. Dermatol. 2002, 147, 385–386. [Google Scholar] [CrossRef]
- Pammer, J.; Reinisch, C.; Birner, P.; Pogoda, K.; Sturzl, M.; Tschachler, E. Interferon-alpha prevents apoptosis of endothelial cells after short-term exposure but induces replicative senescence after continuous stimulation. Lab. Investig. 2006, 86, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Ah Kioon, M.D.; Tripodo, C.; Fernandez, D.; Kirou, K.A.; Spiera, R.F.; Crow, M.K.; Gordon, J.K.; Barrat, F.J. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Tourkina, E.; Bonner, M.; Oates, J.; Hofbauer, A.; Richard, M.; Znoyko, S.; Visconti, R.P.; Zhang, J.; Hatfield, C.M.; Silver, R.M.; et al. Altered monocyte and fibrocyte phenotype and function in scleroderma interstitial lung disease: Reversal by caveolin-1 scaffolding domain peptide. Fibrogenesis Tissue Repair 2011, 4, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitorowicz-Buniak, J.; Denton, C.P.; Abraham, D.; Stratton, R. Partially Evoked Epithelial-Mesenchymal Transition (EMT) Is Associated with Increased TGFβ Signaling within Lesional Scleroderma Skin. PLoS ONE 2015, 10, e0134092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheum. 2015, 67, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Gruschwitz, M.; Müller, P.U.; Sepp, N.; Hofer, E.; Fontana, A.; Wick, G. Transcription and expression of transforming growth factor type beta in the skin of progressive systemic sclerosis: A mediator of fibrosis? J. Investig. Dermatol. 1990, 94, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Eckes, B.; Huerkamp, C.; Krieg, T.; Sollberg, S. Expression of TGF-beta 1, -beta 2 and -beta 3 in localized and systemic scleroderma. J. Dermatol. Sci. 1999, 21, 13–22. [Google Scholar] [CrossRef]
- Kulozik, M.; Hogg, A.; Lankat-Buttgereit, B.; Krieg, T. Co-localization of transforming growth factor beta 2 with alpha 1(I) procollagen mRNA in tissue sections of patients with systemic sclerosis. J. Clin. Investig. 1990, 86, 917–922. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y.; Ihn, H.; Yamane, K.; Kubo, M.; Tamaki, K. Impaired Smad7-Smurf-mediated negative regulation of TGF-beta signaling in scleroderma fibroblasts. J. Clin. Investig. 2004, 113, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y.; Ihn, H.; Yamane, K.; Kubo, M.; Tamaki, K. Increased expression levels of integrin alphavbeta5 on scleroderma fibroblasts. Am. J. Pathol. 2004, 164, 1275–1292. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Involvement of alphavbeta5 integrin-mediated activation of latent transforming growth factor beta1 in autocrine transforming growth factor beta signaling in systemic sclerosis fibroblasts. Arthritis Rheum. 2005, 52, 2897–2905. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Tamaki, K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am. J. Pathol. 2006, 168, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, Y.; Ihn, H.; Jinnin, M.; Asano, Y.; Yamane, K.; Tamaki, K. Constitutive thrombospondin-1 overexpression contributes to autocrine transforming growth factor-beta signaling in cultured scleroderma fibroblasts. Am. J. Pathol. 2005, 166, 1451–1463. [Google Scholar] [CrossRef]
- Chizzolini, C.; Rezzonico, R.; Ribbens, C.; Burger, D.; Wollheim, F.A.; Dayer, J.M. Inhibition of type I collagen production by dermal fibroblasts upon contact with activated T cells: Different sensitivity to inhibition between systemic sclerosis and control fibroblasts. Arthritis Rheum. 1998, 41, 2039–2047. [Google Scholar] [CrossRef]
- Chizzolini, C.; Parel, Y.; De Luca, C.; Tyndall, A.; Akesson, A.; Scheja, A.; Dayer, J.M. Systemic sclerosis Th2 cells inhibit collagen production by dermal fibroblasts via membrane-associated tumor necrosis factor alpha. Arthritis Rheum. 2003, 48, 2593–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Asano, Y.; Akamata, K.; Noda, S.; Taniguchi, T.; Takahashi, T.; Toyama, T.; Tada, Y.; Sugaya, M.; Sato, S.; et al. Progranulin Overproduction Due to Fli-1 Deficiency Contributes to the Resistance of Dermal Fibroblasts to Tumor Necrosis Factor in Systemic Sclerosis. Arthritis Rheum. 2015, 67, 3245–3255. [Google Scholar] [CrossRef]
- Saigusa, R.; Asano, Y.; Nakamura, K.; Hirabayashi, M.; Miura, S.; Yamashita, T.; Taniguchi, T.; Ichimura, Y.; Takahashi, T.; Yoshizaki, A.; et al. Systemic Sclerosis Dermal Fibroblasts Suppress Th1 Cytokine Production via Galectin-9 Overproduction due to Fli1 Deficiency. J. Investig. Dermatol. 2017, 137, 1850–1859. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.S.; Santillo, M.; Bevilacqua, F.; Luchetti, M.; Spadoni, T.; Mancini, M.; Fraticelli, P.; Sambo, P.; Funaro, A.; Kazlauskas, A.; et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N. Engl J. Med. 2006, 354, 2667–2676. [Google Scholar] [CrossRef] [Green Version]
- Riemekasten, G.; Philippe, A.; Näther, M.; Slowinski, T.; Müller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirják, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 530–536. [Google Scholar] [CrossRef]
- Kill, A.; Tabeling, C.; Undeutsch, R.; Kuhl, A.A.; Gunther, J.; Radic, M.; Becker, M.O.; Heidecke, H.; Worm, M.; Witzenrath, M.; et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res. Ther. 2014, 16, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, J.; Kill, A.; Becker, M.O.; Heidecke, H.; Rademacher, J.; Siegert, E.; Radić, M.; Burmester, G.R.; Dragun, D.; Riemekasten, G. Angiotensin receptor type 1 and endothelin receptor type A on immune cells mediate migration and the expression of IL-8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res. Ther. 2014, 16, R65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kühl, A.A.; Heidecke, H.; Ghofrani, H.A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Vancheeswaran, R.; Azam, A.; Black, C.; Dashwood, M.R. Localization of endothelin-1 and its binding sites in scleroderma skin. J. Rheumatol. 1994, 21, 1268–1276. [Google Scholar] [PubMed]
- Rudnicka, L.; Varga, J.; Christiano, A.M.; Iozzo, R.V.; Jimenez, S.A.; Uitto, J. Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. Regulation by transforming growth factor-beta. J. Clin. Invest. 1994, 93, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Pap, T.; Kowal-Bielecka, O.; Meyringer, R.; Guiducci, S.; Landthaler, M.; Scholmerich, J.; Michel, B.A.; Gay, R.E.; Matucci-Cerinic, M.; et al. Overexpression of monocyte chemoattractant protein 1 in systemic sclerosis: Role of platelet-derived growth factor and effects on monocyte chemotaxis and collagen synthesis. Arthritis Rheum. 2001, 44, 2665–2678. [Google Scholar] [CrossRef]
- Davies, C.A.; Jeziorska, M.; Freemont, A.J.; Herrick, A.L. The differential expression of VEGF, VEGFR-2, and GLUT-1 proteins in disease subtypes of systemic sclerosis. Hum. Pathol. 2006, 37, 190–197. [Google Scholar] [CrossRef]
- Distler, J.H.; Jungel, A.; Kowal-Bielecka, O.; Michel, B.A.; Gay, R.E.; Sprott, H.; Matucci-Cerinic, M.; Chilla, M.; Reich, K.; Kalden, J.R.; et al. Expression of interleukin-21 receptor in epidermis from patients with systemic sclerosis. Arthritis Rheum. 2005, 52, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Aden, N.; Nuttall, A.; Shiwen, X.; de Winter, P.; Leask, A.; Black, C.M.; Denton, C.P.; Abraham, D.J.; Stratton, R.J. Epithelial cells promote fibroblast activation via IL-1alpha in systemic sclerosis. J. Investig. Dermatol. 2010, 130, 2191–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitorowicz-Buniak, J.; Shiwen, X.; Denton, C.P.; Abraham, D.; Stratton, R. Abnormally differentiating keratinocytes in the epidermis of systemic sclerosis patients show enhanced secretion of CCN2 and S100A9. J. Investig. Dermatol. 2014, 134, 2693–2702. [Google Scholar] [CrossRef] [Green Version]
- McCoy, S.S.; Reed, T.J.; Berthier, C.C.; Tsou, P.S.; Liu, J.; Gudjonsson, J.E.; Khanna, D.; Kahlenberg, J.M. Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta. Rheumatology 2017, 56, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Asano, Y.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Miura, S.; Ichimura, Y.; Toyama, T.; Hirabayashi, M.; Taniguchi, T.; et al. A potential contribution of psoriasin to vascular and epithelial abnormalities and inflammation in systemic sclerosis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Yamashita, T.; Miura, S.; Hirabayashi, M.; Nakamura, K.; Miyagawa, T.; Fukui, Y.; Yoshizaki, A.; Sato, S.; Asano, Y. A potential contribution of decreased galectin-7 expression in stratified epithelia to the development of cutaneous and oesophageal manifestations in systemic sclerosis. Exp. Dermatol. 2019, 28, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Asano, Y.; Sugawara, K.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Akamata, K.; et al. Epithelial Fli1 deficiency drives systemic autoimmunity and fibrosis: Possible roles in scleroderma. J. Exp. Med. 2017, 214, 1129–1151. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, P.S.; Kahaleh, B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006, 54, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Markiewicz, M.; Kubo, M.; Szalai, G.; Watson, D.K.; Trojanowska, M. Transcription factor Fli1 regulates collagen fibrillogenesis in mouse skin. Mol. Cell Biol. 2009, 29, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujor, A.M.; El Adili, F.; Parvez, A.; Marden, G.; Trojanowska, M. Fli1 Downregulation in Scleroderma Myeloid Cells Has Profibrotic and Proinflammatory Effects. Front Immunol. 2020, 11, 800. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Miyagawa, T.; Toyama, S.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Yoshizaki, A.; et al. CXCL13 produced by macrophages due to Fli1 deficiency may contribute to the development of tissue fibrosis, vasculopathy and immune activation in systemic sclerosis. Exp. Dermatol. 2018, 27, 1030–1037. [Google Scholar] [CrossRef]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [Green Version]
- Mathis, D.; Benoist, C. Aire. Annu. Rev. Immunol. 2009, 27, 287–312. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Johnson, M.E.; Franks, J.M.; Cai, G.; Mehta, B.K.; Wood, T.A.; Archambault, K.; Pioli, P.A.; Simms, R.W.; Orzechowski, N.; Arron, S.; et al. Microbiome dysbiosis is associated with disease duration and increased inflammatory gene expression in systemic sclerosis skin. Arthritis Res. Ther. 2019, 21, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Wei, J.; Varga, J. Understanding fibrosis in systemic sclerosis: Shifting paradigms, emerging opportunities. Nat. Rev. Rheumatol. 2012, 8, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Shao, M.; Hepler, C.; Zi, Z.; Zhao, S.; An, Y.A.; Zhu, Y.; Ghaben, A.; Wang, M.Y.; Li, N.; et al. Dermal adipose tissue has high plasticity and undergoes reversible dedifferentiation in mice. J. Clin. Investig. 2019. [Google Scholar] [CrossRef]
- Varga, J.; Marangoni, R.G. Systemic sclerosis in 2016: Dermal white adipose tissue implicated in SSc pathogenesis. Nat. Rev. Rheumatol. 2017, 13, 71–72. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Shimomura, I.; Kihara, S.; Funahashi, T. Importance of adipocytokines in obesity-related diseases. Horm. Res. 2003, 60, 56–59. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Shibata, S.; Noda, S.; Aozasa, N.; Akamata, K.; Yamada, D.; Tamaki, Z.; Tada, Y.; Sugaya, M.; et al. Serum adiponectin levels inversely correlate with the activity of progressive skin sclerosis in patients with diffuse cutaneous systemic sclerosis. J. Eur. Acad. Dermatol. Venereol 2012, 26, 354–360. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Takahashi, T.; Shibata, S.; Akamata, K.; Aozasa, N.; Noda, S.; Taniguchi, T.; Ichimura, Y.; Toyama, T.; et al. Clinical significance of monitoring serum adiponectin levels during intravenous pulse cyclophosphamide therapy in interstitial lung disease associated with systemic sclerosis. Mod. Rheumatol. 2013, 23, 323–329. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Shibata, S.; Noda, S.; Akamata, K.; Aozasa, N.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; et al. A possible contribution of visfatin to the resolution of skin sclerosis in patients with diffuse cutaneous systemic sclerosis via a direct anti-fibrotic effect on dermal fibroblasts and Th1 polarization of the immune response. Rheumatology 2013, 52, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Masui, Y.; Asano, Y.; Akamata, K.; Aozasa, N.; Noda, S.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; et al. Serum resistin levels: A possible correlation with pulmonary vascular involvement in patients with systemic sclerosis. Rheumatol. Int. 2014, 34, 1165–1170. [Google Scholar] [CrossRef]
- Toyama, T.; Asano, Y.; Takahashi, T.; Aozasa, N.; Akamata, K.; Noda, S.; Taniguchi, T.; Ichimura, Y.; Sumida, H.; Tamaki, Z.; et al. Clinical significance of serum retinol binding protein-4 levels in patients with systemic sclerosis. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Aozasa, N.; Asano, Y.; Akamata, K.; Noda, S.; Masui, Y.; Yamada, D.; Tamaki, Z.; Tada, Y.; Sugaya, M.; Kadono, T.; et al. Serum apelin levels: Clinical association with vascular involvements in patients with systemic sclerosis. J. Eur. Acad. Dermatol. Venereol 2013, 27, 37–42. [Google Scholar] [CrossRef]
- Takahashi, T.; Asano, Y.; Noda, S.; Aozasa, N.; Akamata, K.; Taniguchi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; Kuwano, Y.; et al. A possible contribution of lipocalin-2 to the development of dermal fibrosis, pulmonary vascular involvement and renal dysfunction in systemic sclerosis. Br. J. Dermatol. 2015, 173, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Asano, Y.; Saigusa, R.; Yamashita, T.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Tamaki, Z.; Tada, Y.; et al. Serum omentin levels: A possible contribution to vascular involvement in patients with systemic sclerosis. J. Dermatol. 2015, 42, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Asano, Y.; Saigusa, R.; Yamashita, T.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Tamaki, Z.; Tada, Y.; et al. Serum vaspin levels: A possible correlation with digital ulcers in patients with systemic sclerosis. J. Dermatol. 2015, 42, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Lakota, K.; Wei, J.; Carns, M.; Hinchcliff, M.; Lee, J.; Whitfield, M.L.; Sodin-Semrl, S.; Varga, J. Levels of adiponectin, a marker for PPAR-gamma activity, correlate with skin fibrosis in systemic sclerosis: Potential utility as biomarker? Arthritis Res. Ther. 2012, 14, R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomcik, M.; Arima, K.; Hulejova, H.; Kuklova, M.; Filkova, M.; Braun, M.; Belacek, J.; Novak, M.; Becvar, R.; Vencovsky, J.; et al. Adiponectin relation to skin changes and dyslipidemia in systemic sclerosis. Cytokine 2012, 58, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Jinnin, M.; Muchemwa, F.C.; Makino, T.; Kajihara, I.; Makino, K.; Honda, N.; Sakai, K.; Fukushima, S.; Ihn, H. Adiponectin expression is decreased in the involved skin and sera of diffuse cutaneous scleroderma patients. Exp. Dermatol. 2011, 20, 764–766. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Masui, Y.; Fang, F.; Korman, B.; Lord, G.; Lee, J.; Lakota, K.; Wei, J.; Scherer, P.E.; Otvos, L.; et al. Adiponectin is an endogenous anti-fibrotic mediator and therapeutic target. Sci. Rep. 2017, 7, 4397. [Google Scholar] [CrossRef]
- Yamashita, T.; Lakota, K.; Taniguchi, T.; Yoshizaki, A.; Sato, S.; Hong, W.; Zhou, X.; Sodin-Semrl, S.; Fang, F.; Asano, Y.; et al. An orally-active adiponectin receptor agonist mitigates cutaneous fibrosis, inflammation and microvascular pathology in a murine model of systemic sclerosis. Sci. Rep. 2018, 8, 11843. [Google Scholar] [CrossRef] [Green Version]
- Sjogren, R.W. Gastrointestinal features of scleroderma. Curr. Opin. Rheumatol. 1996, 8, 569–575. [Google Scholar] [CrossRef]
- Poirier, T.J.; Rankin, G.B. Gastrointestinal manifestations of progressive systemic scleroderma based on a review of 364 cases. Am. J. Gastroenterol. 1972, 58, 30–44. [Google Scholar] [PubMed]
- Young, M.A.; Rose, S.; Reynolds, J.C. Gastrointestinal manifestations of scleroderma. Rheum. Dis. Clin. N. Am. 1996, 22, 797–823. [Google Scholar] [CrossRef]
- Emmanuel, A. Current management of the gastrointestinal complications of systemic sclerosis. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, G.; Holstege, A.; Lang, B.; Scholmerich, J. Gastrointestinal manifestations of progressive systemic sclerosis. Am. J. Gastroenterol 1997, 92, 763–771. [Google Scholar] [PubMed]
- Malandrini, A.; Selvi, E.; Villanova, M.; Berti, G.; Sabadini, L.; Salvadori, C.; Gambelli, S.; De Stefano, R.; Vernillo, R.; Marcolongo, R.; et al. Autonomic nervous system and smooth muscle cell involvement in systemic sclerosis: Ultrastructural study of 3 cases. J. Rheumatol. 2000, 27, 1203–1206. [Google Scholar] [PubMed]
- Lepri, G.; Guiducci, S.; Bellando-Randone, S.; Giani, I.; Bruni, C.; Blagojevic, J.; Carnesecchi, G.; Radicati, A.; Pucciani, F.; Marco, M.C. Evidence for oesophageal and anorectal involvement in very early systemic sclerosis (VEDOSS): Report from a single VEDOSS/EUSTAR centre. Ann. Rheum. Dis. 2015, 74, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Weston, S.; Thumshirn, M.; Wiste, J.; Camilleri, M. Clinical and upper gastrointestinal motility features in systemic sclerosis and related disorders. Am. J. Gastroenterol. 1998, 93, 1085–1089. [Google Scholar] [CrossRef]
- Roman, S.; Hot, A.; Fabien, N.; Cordier, J.F.; Miossec, P.; Ninet, J.; Mion, F. Esophageal dysmotility associated with systemic sclerosis: A high-resolution manometry study. Dis. Esophagus 2011, 24, 299–304. [Google Scholar] [CrossRef]
- Clements, P.J.; Becvar, R.; Drosos, A.A.; Ghattas, L.; Gabrielli, A. Assessment of gastrointestinal involvement. Clin. Exp. Rheumatol. 2003, 21, S15–S18. [Google Scholar]
- Weber, P.; Ganser, G.; Frosch, M.; Roth, J.; Hulskamp, G.; Zimmer, K.P. Twenty-four hour intraesophageal pH monitoring in children and adolescents with scleroderma and mixed connective tissue disease. J. Rheumatol. 2000, 27, 2692–2695. [Google Scholar] [PubMed]
- Eaker, E.Y.; Kuldau, J.G.; Verne, G.N.; Ross, S.O.; Sallustio, J.E. Myenteric neuronal antibodies in scleroderma: Passive transfer evokes alterations in intestinal myoelectric activity in a rat model. J. Lab. Clin. Med. 1999, 133, 551–556. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Nakamura, Y.; Matsumoto, I.; Nishimagi, E.; Satoh, T.; Kuwana, M.; Sumida, T.; Hara, M. Muscarinic-3 acetylcholine receptor autoantibody in patients with systemic sclerosis: Contribution to severe gastrointestinal tract dysmotility. Ann. Rheum. Dis. 2009, 68, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Mehendiratta, V.; Del Galdo, F.; Jimenez, S.A.; Cohen, S.; DiMarino, A.J.; Rattan, S. Immunoglobulins from scleroderma patients inhibit the muscarinic receptor activation in internal anal sphincter smooth muscle cells. Am. J. Physiol. Gastrointest Liver Physiol. 2009, 297, G1206–G1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafaja, S.; Valera, I.; Divekar, A.A.; Saggar, R.; Abtin, F.; Furst, D.E.; Khanna, D.; Singh, R.R. pDCs in lung and skin fibrosis in a bleomycin-induced model and patients with systemic sclerosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Dantas, R.O.; Aprile, L.R. Esophageal striated muscle contractions in patients with gastroesophageal reflux symptoms. Dig. Dis. Sci. 2002, 47, 2586–2590. [Google Scholar] [CrossRef]
- Miwa, H.; Kondo, T.; Oshima, T. Gastroesophageal reflux disease-related and functional heartburn: Pathophysiology and treatment. Curr. Opin. Gastroenterol. 2016, 32, 344–352. [Google Scholar] [CrossRef]
- Dessein, P.H.; Joffe, B.I.; Metz, R.M.; Millar, D.L.; Lawson, M.; Stanwix, A.E. Autonomic dysfunction in systemic sclerosis: Sympathetic overactivity and instability. Am. J. Med. 1992, 93, 143–150. [Google Scholar] [CrossRef]
- Zhong, D.; Wu, C.; Zeng, X.; Wang, Q. The role of gut microbiota in the pathogenesis of rheumatic diseases. Clin. Rheumatol. 2018, 37, 25–34. [Google Scholar] [CrossRef]
- De Luca, F.; Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 2019, 195, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Rosser, E.C.; Mauri, C. A clinical update on the significance of the gut microbiota in systemic autoimmunity. J. Autoimmun 2016, 74, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Bellocchi, C.; Volkmann, E.R. Update on the Gastrointestinal Microbiome in Systemic Sclerosis. Curr. Rheumatol. Rep. 2018, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, E.R. Intestinal microbiome in scleroderma: Recent progress. Curr. Opin. Rheumatol. 2017, 29, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.D.; Medsger, T.A. Changes in causes of death in systemic sclerosis, 1972-2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhai, M.; Meune, C.; Boubaya, M.; Avouac, J.; Hachulla, E.; Balbir-Gurman, A.; Riemekasten, G.; Airo, P.; Joven, B.; Vettori, S.; et al. Mapping and predicting mortality from systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1897–1905. [Google Scholar] [CrossRef]
- Steen, V.; Medsger, T.A. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum. 2003, 48, 516–522. [Google Scholar] [CrossRef]
- Daraban, A.M.; Enache, R.; Predescu, L.; Platon, P.; Constantinescu, T.; Mihai, C.; Coman, I.M.; Ginghina, C.; Jurcut, R. Pulmonary veno-occlusive disease: A rare cause of pulmonary hypertension in systemic sclerosis. Case presentation and review of the literature. Rom. J. Intern. Med. 2015, 53, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.C.; Cordeiro, A.; Loureiro, M.J.; Ferreira, F. Pulmonary veno-occlusive disease: A probably underdiagnosed cause of pulmonary hypertension in systemic sclerosis. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef]
- Steele, R.; Hudson, M.; Lo, E.; Baron, M. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res. 2012, 64, 519–524. [Google Scholar] [CrossRef] [Green Version]
- White, B. Interstitial lung disease in scleroderma. Rheum. Dis. Clin. N. Am. 2003, 29, 371–390. [Google Scholar] [CrossRef]
- Nihtyanova, S.I.; Schreiber, B.E.; Ong, V.H.; Rosenberg, D.; Moinzadeh, P.; Coghlan, J.G.; Wells, A.U.; Denton, C.P. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheum. 2014, 66, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.; Domsic, R.T.; Lucas, M.; Fertig, N.; Medsger, T.A., Jr. A clinical and serologic comparison of African American and Caucasian patients with systemic sclerosis. Arthritis Rheum. 2012, 64, 2986–2994. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, V.K.; Wirz, E.G.; Allanore, Y.; Rossbach, P.; Riemekasten, G.; Hachulla, E.; Distler, O.; Airo, P.; Carreira, P.E.; Balbir Gurman, A.; et al. Incidences and Risk Factors of Organ Manifestations in the Early Course of Systemic Sclerosis: A Longitudinal EUSTAR Study. PLoS ONE 2016, 11, e0163894. [Google Scholar] [CrossRef] [PubMed]
- Ostojic, P.; Damjanov, N. Different clinical features in patients with limited and diffuse cutaneous systemic sclerosis. Clin. Rheumatol. 2006, 25, 453–457. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Kubo, M.; Tamaki, K. The prevalence and clinical significance of anti-U1 RNA antibodies in patients with systemic sclerosis. J. Investig. Dermatol. 2003, 120, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Man, A.; Davidyock, T.; Ferguson, L.T.; Ieong, M.; Zhang, Y.; Simms, R.W. Changes in forced vital capacity over time in systemic sclerosis: Application of group-based trajectory modelling. Rheumatology 2015, 54, 1464–1471. [Google Scholar] [CrossRef] [Green Version]
- Steen, V.D.; Conte, C.; Owens, G.R.; Medsger, T.A., Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994, 37, 1283–1289. [Google Scholar] [CrossRef]
- Morgan, C.; Knight, C.; Lunt, M.; Black, C.M.; Silman, A.J. Predictors of end stage lung disease in a cohort of patients with scleroderma. Ann. Rheum. Dis. 2003, 62, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Bouros, D.; Wells, A.U.; Nicholson, A.G.; Colby, T.V.; Polychronopoulos, V.; Pantelidis, P.; Haslam, P.L.; Vassilakis, D.A.; Black, C.M.; du Bois, R.M. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am. J. Respir. Crit. Care Med. 2002, 165, 1581–1586. [Google Scholar] [CrossRef]
- Kim, D.S.; Yoo, B.; Lee, J.S.; Kim, E.K.; Lim, C.M.; Lee, S.D.; Koh, Y.; Kim, W.S.; Kim, W.D.; Colby, T.V.; et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc. Diffus. Lung Dis. 2002, 19, 121–127. [Google Scholar]
- Fischer, A.; Swigris, J.J.; Groshong, S.D.; Cool, C.D.; Sahin, H.; Lynch, D.A.; Curran-Everett, D.; Gillis, J.Z.; Meehan, R.T.; Brown, K.K. Clinically significant interstitial lung disease in limited scleroderma: Histopathology, clinical features, and survival. Chest 2008, 134, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Winstone, T.A.; Assayag, D.; Wilcox, P.G.; Dunne, J.V.; Hague, C.J.; Leipsic, J.; Collard, H.R.; Ryerson, C.J. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: A systematic review. Chest 2014, 146, 422–436. [Google Scholar] [CrossRef] [PubMed]
- Beon, M.; Harley, R.A.; Wessels, A.; Silver, R.M.; Ludwicka-Bradley, A. Myofibroblast induction and microvascular alteration in scleroderma lung fibrosis. Clin. Exp. Rheumatol. 2004, 22, 733–742. [Google Scholar] [PubMed]
- Savarino, E.; Bazzica, M.; Zentilin, P.; Pohl, D.; Parodi, A.; Cittadini, G.; Negrini, S.; Indiveri, F.; Tutuian, R.; Savarino, V.; et al. Gastroesophageal reflux and pulmonary fibrosis in scleroderma: A study using pH-impedance monitoring. Am. J. Respir Crit Care Med. 2009, 179, 408–413. [Google Scholar] [CrossRef]
- Appel, J.Z., 3rd; Lee, S.M.; Hartwig, M.G.; Li, B.; Hsieh, C.C.; Cantu, E., 3rd; Yoon, Y.; Lin, S.S.; Parker, W.; Davis, R.D. Characterization of the innate immune response to chronic aspiration in a novel rodent model. Respir. Res. 2007, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Christmann, R.B.; Wells, A.U.; Capelozzi, V.L.; Silver, R.M. Gastroesophageal reflux incites interstitial lung disease in systemic sclerosis: Clinical, radiologic, histopathologic, and treatment evidence. Semin Arthritis Rheum. 2010, 40, 241–249. [Google Scholar] [CrossRef]
- de Souza, R.B.; Borges, C.T.; Capelozzi, V.L.; Parra, E.R.; Jatene, F.B.; Kavakama, J.; Kairalla, R.A.; Bonfa, E. Centrilobular fibrosis: An underrecognized pattern in systemic sclerosis. Respiration 2009, 77, 389–397. [Google Scholar] [CrossRef]
- Venalis, P.; Kumánovics, G.; Schulze-Koops, H.; Distler, A.; Dees, C.; Zerr, P.; Palumbo-Zerr, K.; Czirják, L.; Mackevic, Z.; Lundberg, I.E.; et al. Cardiomyopathy in murine models of systemic sclerosis. Arthritis Rheum. 2015, 67, 508–516. [Google Scholar] [CrossRef]
- Allanore, Y.; Meune, C. Primary myocardial involvement in systemic sclerosis: Evidence for a microvascular origin. Clin. Exp. Rheumatol. 2010, 28, S48–S53. [Google Scholar]
- Hachulla, A.L.; Launay, D.; Gaxotte, V.; de Groote, P.; Lamblin, N.; Devos, P.; Hatron, P.Y.; Beregi, J.P.; Hachulla, E. Cardiac magnetic resonance imaging in systemic sclerosis: A cross-sectional observational study of 52 patients. Ann. Rheum. Dis. 2009, 68, 1878–1884. [Google Scholar] [CrossRef]
- Hesselstrand, R.; Scheja, A.; Akesson, A. Mortality and causes of death in a Swedish series of systemic sclerosis patients. Ann. Rheum. Dis. 1998, 57, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.; Vlachoyiannopoulos, P.G.; Haidich, A.B.; Medsger, T.A., Jr.; Lucas, M.; Michet, C.J.; Kuwana, M.; Yasuoka, H.; van den Hoogen, F.; Te Boome, L.; et al. Mortality in systemic sclerosis: An international meta-analysis of individual patient data. Am. J. Med. 2005, 118, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.D.; Medsger, T.A. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000, 43, 2437–2444. [Google Scholar] [CrossRef]
- Kahan, A.; Coghlan, G.; McLaughlin, V. Cardiac complications of systemic sclerosis. Rheumatology 2009, 48, iii45–iii48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allanore, Y.; Meune, C.; Kahan, A. Outcome measures for heart involvement in systemic sclerosis. Rheumatology 2008, 47, v51–v53. [Google Scholar] [CrossRef] [Green Version]
- Ferri, C.; Valentini, G.; Cozzi, F.; Sebastiani, M.; Michelassi, C.; La Montagna, G.; Bullo, A.; Cazzato, M.; Tirri, E.; Storino, F.; et al. Systemic sclerosis: Demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine 2002, 81, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Bulkley, B.H.; Ridolfi, R.L.; Salyer, W.R.; Hutchins, G.M. Myocardial lesions of progressive systemic sclerosis. A cause of cardiac dysfunction. Circulation 1976, 53, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Belloli, L.; Carlo-Stella, N.; Ciocia, G.; Chiti, A.; Massarotti, M.; Marasini, B. Myocardial involvement in systemic sclerosis. Rheumatology 2008, 47, 1070–1072. [Google Scholar] [CrossRef] [Green Version]
- Desai, C.S.; Lee, D.C.; Shah, S.J. Systemic sclerosis and the heart: Current diagnosis and management. Curr. Opin. Rheumatol. 2011, 23, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Tzelepis, G.E.; Kelekis, N.L.; Plastiras, S.C.; Mitseas, P.; Economopoulos, N.; Kampolis, C.; Gialafos, E.J.; Moyssakis, I.; Moutsopoulos, H.M. Pattern and distribution of myocardial fibrosis in systemic sclerosis: A delayed enhanced magnetic resonance imaging study. Arthritis Rheum. 2007, 56, 3827–3836. [Google Scholar] [CrossRef]
- Meune, C.; Allanore, Y.; Devaux, J.Y.; Dessault, O.; Duboc, D.; Weber, S.; Kahan, A. High prevalence of right ventricular systolic dysfunction in early systemic sclerosis. J. Rheumatol. 2004, 31, 1941–1945. [Google Scholar] [PubMed]
- Kahan, A.; Devaux, J.Y.; Amor, B.; Menkes, C.J.; Weber, S.; Venot, A.; Strauch, G. The effect of captopril on thallium 201 myocardial perfusion in systemic sclerosis. Clin. Pharmacol. Ther. 1990, 47, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Duboc, D.; Kahan, A.; Maziere, B.; Loc’h, C.; Crouzel, C.; Menkes, C.J.; Amor, B.; Strauch, G.; Guerin, F.; Syrota, A. The effect of nifedipine on myocardial perfusion and metabolism in systemic sclerosis. A positron emission tomographic study. Arthritis Rheum. 1991, 34, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Kahan, A.; Devaux, J.Y.; Amor, B.; Menkes, C.J.; Weber, S.; Guerin, F.; Venot, A.; Strauch, G. Pharmacodynamic effect of nicardipine on left ventricular function in systemic sclerosis. J. Cardiovasc. Pharmacol. 1990, 15, 249–253. [Google Scholar] [CrossRef]
- Lekakis, J.; Mavrikakis, M.; Emmanuel, M.; Prassopoulos, V.; Papazoglou, S.; Papamichael, C.; Moulopoulou, D.; Kostamis, P.; Stamatelopoulos, S.; Moulopoulos, S. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin. Exp. Rheumatol. 1998, 16, 135–140. [Google Scholar]
- Rigamonti, C.; Bogdanos, D.P.; Mytilinaiou, M.G.; Smyk, D.S.; Rigopoulou, E.I.; Burroughs, A.K. Primary biliary cirrhosis associated with systemic sclerosis: Diagnostic and clinical challenges. Int. J. Rheumatol. 2011, 2011, 976427. [Google Scholar] [CrossRef] [Green Version]
- Smyk, D.S.; Mytilinaiou, M.G.; Milkiewicz, P.; Rigopoulou, E.I.; Invernizzi, P.; Bogdanos, D.P. Towards systemic sclerosis and away from primary biliary cirrhosis: The case of PTPN22. Auto. Immun. Highlights 2012, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Imura-Kumada, S.; Hasegawa, M.; Matsushita, T.; Hamaguchi, Y.; Encabo, S.; Shums, Z.; Norman, G.L.; Takehara, K.; Fujimoto, M. High prevalence of primary biliary cirrhosis and disease-associated autoantibodies in Japanese patients with systemic sclerosis. Mod. Rheumatol. 2012, 22, 892–898. [Google Scholar] [CrossRef]
- Vaiphei, K.; Bhatia, A.; Sinha, S.K. Liver pathology in collagen vascular disorders highlighting the vascular changes within portal tracts. Indian J. Pathol. Microbiol 2011, 54, 25–31. [Google Scholar] [CrossRef]
- Hartleb, M.; Gutkowski, K.; Milkiewicz, P. Nodular regenerative hyperplasia: Evolving concepts on underdiagnosed cause of portal hypertension. World J. Gastroenterol. 2011, 17, 1400–1409. [Google Scholar] [CrossRef]
- Arvanitaki, M.; Adler, M. Nodular regenerative hyperplasia of the liver. A review of 14 cases. Hepatogastroenterology 2001, 48, 1425–1429. [Google Scholar]
- Graf, L.; Dobrota, R.; Jordan, S.; Wildi, L.M.; Distler, O.; Maurer, B. Nodular Regenerative Hyperplasia of the Liver: A Rare Vascular Complication in Systemic Sclerosis. J. Rheumatol. 2018, 45, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Soumura, M.; Danno, K.; Kaji, K.; Kondo, M.; Hirata, K.; Nakazawa, J.; Uzu, T.; Nishio, Y.; Kashiwagi, A. Scleroderma renal crisis in a patient with anticentromere antibody-positive limited cutaneous systemic sclerosis. Mod. Rheumatol. 2006, 16, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Sanada, M.; Kashiwagi, A. Is scleroderma renal crisis with anti-centromere antibody-positive limited cutaneous systemic sclerosis overlooked in patients with hypertension and/or renal dysfunction? Nephrology 2008, 13, 179–180. [Google Scholar] [CrossRef]
- Chang, M.; Wang, R.J.; Yangco, D.T.; Sharp, G.C.; Komatireddy, G.R.; Hoffman, R.W. Analysis of autoantibodies against RNA polymerases using immunoaffinity-purifed RNA polymerase I, II, and III antigen in an enzyme-linked immunosorbent assay. Clin. Immunol. Immunopathol. 1998, 89, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.; Baron, M.; Hudson, M.; Burlingame, R.W.; Fritzler, M.J. Antibodies to RNA polymerase III in systemic sclerosis detected by ELISA. J. Rheumatol. 2007, 34, 1528–1534. [Google Scholar] [PubMed]
- Steen, V.D. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005, 35, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Lapadula, G.; Mouthon, L.; Muller-Ladner, U. Renal complications and scleroderma renal crisis. Rheumatology 2009, 48, iii32–iii35. [Google Scholar] [CrossRef] [Green Version]
- Steen, V.D.; Medsger, T.A., Jr. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998, 41, 1613–1619. [Google Scholar] [CrossRef]
- Montanelli, G.; Beretta, L.; Santaniello, A.; Scorza, R. Effect of dihydropyridine calcium channel blockers and glucocorticoids on the prevention and development of scleroderma renal crisis in an Italian case series. Clin. Exp. Rheumatol. 2013, 31, 135–139. [Google Scholar]
- Hudson, M.; Baron, M.; Tatibouet, S.; Furst, D.E.; Khanna, D. Exposure to ACE inhibitors prior to the onset of scleroderma renal crisis-results from the International Scleroderma Renal Crisis Survey. Semin Arthritis Rheum. 2014, 43, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Haviv, Y.S.; Safadi, R. Normotensive scleroderma renal crisis: Case report and review of the literature. Ren. Fail. 1998, 20, 733–736. [Google Scholar] [CrossRef]
- Lewandowski, B.; Domyslawska, I.; Klimiuk, P.A.; Sierakowski, S. Kidney crisis in systemic sclerosis. Rocz. Akad. Med. Bialymst. 2005, 50, 294–296. [Google Scholar] [PubMed]
- Helfrich, D.J.; Banner, B.; Steen, V.D.; Medsger, T.A., Jr. Normotensive renal failure in systemic sclerosis. Arthritis Rheum. 1989, 32, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Woodworth, T.G.; Suliman, Y.A.; Li, W.; Furst, D.E.; Clements, P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat. Rev. Nephrol. 2016, 12, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Batal, I.; Domsic, R.T.; Shafer, A.; Medsger, T.A., Jr.; Kiss, L.P.; Randhawa, P.; Bastacky, S. Renal biopsy findings predicting outcome in scleroderma renal crisis. Hum. Pathol. 2009, 40, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.R.; Rodnan, G.P. Pathologic observations concerning the kidney in progressive systemic sclerosis. AMA Arch. Pathol. 1958, 65, 29–39. [Google Scholar]
- Mouthon, L.; Berezne, A.; Bussone, G.; Noel, L.H.; Villiger, P.M.; Guillevin, L. Scleroderma renal crisis: A rare but severe complication of systemic sclerosis. Clin. Rev. Allergy Immunol. 2011, 40, 84–91. [Google Scholar] [CrossRef]
- Trostle, D.C.; Bedetti, C.D.; Steen, V.D.; Al-Sabbagh, M.R.; Zee, B.; Medsger, T.A. Renal vascular histology and morphometry in systemic sclerosis. A case-control autopsy study. Arthritis Rheum. 1988, 31, 393–400. [Google Scholar] [CrossRef]
- Steen, V.D. Kidney involvement in systemic sclerosis. Presse. Med. 2014, 43, e305–e314. [Google Scholar] [CrossRef]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asano, Y. The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies. J. Clin. Med. 2020, 9, 2687. https://doi.org/10.3390/jcm9092687
Asano Y. The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies. Journal of Clinical Medicine. 2020; 9(9):2687. https://doi.org/10.3390/jcm9092687
Chicago/Turabian StyleAsano, Yoshihide. 2020. "The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies" Journal of Clinical Medicine 9, no. 9: 2687. https://doi.org/10.3390/jcm9092687
APA StyleAsano, Y. (2020). The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies. Journal of Clinical Medicine, 9(9), 2687. https://doi.org/10.3390/jcm9092687