Abstract
Polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) are rare hematological conditions known as myeloproliferative neoplasms (MPNs). They are characterized for being BCR-ABL negative malignancies and affected patients often present with symptoms which can significantly impact their quality of life. MPNs are characterized by a clonal proliferation of an abnormal hematopoietic stem/progenitor cell. In MPNs; cells of all myeloid lineages; including those involved in the immune and inflammatory response; may belong to the malignant clone thus leading to an altered immune response and an overexpression of cytokines and inflammatory receptors; further worsening chronic inflammation. Many of these cytokines; in particular, IL-1β and IL-18; are released in active form by activating the inflammasome complexes which in turn mediate the inflammatory process. Despite this; little is known about the functional effects of stem cell-driven inflammasome signaling in MPN pathogenesis. In this review we focused on the role of inflammatory pathway and inflammasome in MPN diseases. A better understanding of the inflammatory-state-driving MPNs and of the role of the inflammasome may provide new insights on possible therapeutic strategies
1. Introduction
Myeloproliferative neoplasms (MPNs) are a closely related group of rare, but potentially life-threatening, diseases caused by hyperproliferation of bone marrow stem cells. In MPNs, the production of blood cells (hematopoiesis) in the bone marrow is defective, resulting in the production of an excessive number of blood cells. MPNs include three main conditions: polycythemia vera (PV), essential thrombocythemia (ET) and myelofibrosis (MF) [1], which are associated with frequent disease-related complications, such as venous and arterial thrombosis, hemorrhages and transformation to acute myeloid leukemia (AML) [2]. These neoplasms are characterized by a clonal proliferation of an abnormal hematopoietic stem/progenitor cell [3]. PV is characterized by an excess of erythrocytes and predominant erythroid lineage involvement and with a variable hyperplasia of the megakaryocytic/granulocytic lineages [3]. ET is characterized by an increased platelet count with a concomitant megakaryocytic hyperplasia, whereas PMF is a more heterogeneous disorder in terms of clinical and biologic characteristics, characterized by the presence of megakaryocytic hyperplasia and bone marrow fibrosis [3]. The MPNs are defined by a chronic inflammatory status contributing to microenvironmental transformation necessary for supporting tumor progression and severity of the disease [4]. inflammation plays an important and defined role in cancer development affecting all tumorigenesis stage, including initiation, promotion, malignant conversion, invasion and metastasis [5]. Indeed, cancer-related inflammation is triggered by a series of signals from immune system cells such as macrophages, dendritic cells (DCs), NK cells (natural killer), neutrophils and T and B lymphocytes. Several studies showed that some genes involved in inflammasome activation, were significantly overexpressed also in MPNs [6,7,8]. Therefore, MPN represents a useful model to assess the possible relationship between clonal development of a hematologic malignancy and chronic inflammation [1]. Chronic inflammation has been considered for long time a key element in the development of MPNs, which are maintained by a continuous release of proinflammatory cytokines, chemokines [9] and ROS accumulation leading to genetic instability and successively to the development of neoplasms and their progression [10,11,12]. In the inflammatory immune response, the activation of inflammasome is a key event that plays a dual role in cancer development and progression. The role of inflammasome in cancer growth and progression is still controversial and several lines of evidence showed different effects in various cancer types; therefore, the aim of the present review is to describe the possible relationship between inflammasome, related cytokines and myeloproliferative neoplasms.
Inflammasome
The term inflammasome was firstly used by Tschoppand co-workers in 2002 [13] when describing a high-molecular-weight cytosolic complex in stimulated immune cells responsible for the activation of the inflammatory caspase-1. inflammasomes are indispensable mediators of the innate immune response to infection [13]; they are constituted of large multimeric intracellular complexes that are capable of controlling activation of the proteolytic enzyme caspase-1 [13], which in turn regulates the proteolytic maturation of Interleukin-1β (IL-1β) and Interleukin-18 (IL-18), as well as a rapid, noxious, inflammatory form of cell death termed pyroptosis through cleavage of gasdermin D (GSDMD) [14,15]. Activation of inflammasome leads to an inflammatory response resulting either anti- and/or pro-tumor growth. inflammasomes are generally formed of a pattern recognition receptor (PRR), an apoptosis-associated speck-like protein (ASC) and the cysteine protease caspase-1 [16]. Five different families of PRRs have been described so far including nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs), Toll-like receptors (TLRs), Rig-I-like receptors (RLRs) and C-type lectin receptors (CLRs) [17,18]. Previous data suggest that other NLR and ALR family proteins (i.e., NLRP6, NLRP9b and pyrin) may also form functional inflammasomes [19,20,21,22] (Figure 1A). Assembly of inflammasome complexes is triggered by cytosolic sensing of pathogen-associated molecular patterns (PAMPs) or host-derived signals associated with cell stress (danger-associated molecular patterns, DAMPs) [23,24]. All together these complex series of biochemical events lead to the activation of the inflammasome and induce an immune response inhibiting pathogen replication [23,25,26,27] (Figure 1B). Intracellular danger signals (danger-associated molecular patterns) released from damaged or dying cells are also able to activate inflammasomes, but impaired inflammasome signaling causes and hyperinflammatory state, leading to the development of autoimmune and neurodegenerative diseases and cancer progression [28,29,30,31,32,33].
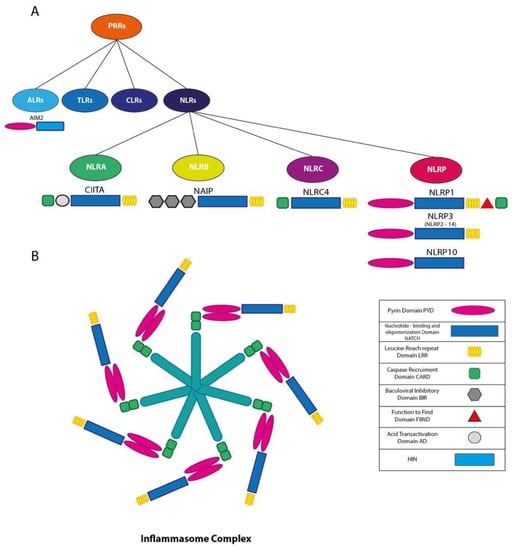
Figure 1.
(A) Schematic representation of Pattern Recognition receptors (PRR) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs); (B) schematic representation of inflammasome complex.
Several studies showed that aberrant inflammasome activation is often associated with the development and progression of cancer. In particular, tumor associated inflammation is triggered by a variety of immune cells, including neutrophils, T and B lymphocytes, macrophages, NK cells and DC cells. Other studies indicate that inflammasome may have ambivalent effect in tumors: it can contribute to the pathogenesis, development and neoplastic progression and maintenance of the tumor microenvironment or on the contrary suppress tumor growth through the phenomenon of pyroptosis and death of pre-malignant cells. Such different effect can in part be explained by the heterogenicity of cancer cells and the fact that different inflammasomes have different roles. [34,35,36,37]. Consistently, mutations in genes encoding inflammasome components often lead to susceptibility to cancer, infection and autoinflammatory diseases in humans [35,38,39,40,41,42,43,44,45]. Taken together, these results suggest that the pro-tumorigenic or antitumorigenic properties of inflammasomes and related cytokines are largely determined by the types of cells, tissues and organs involved (Figure 2).
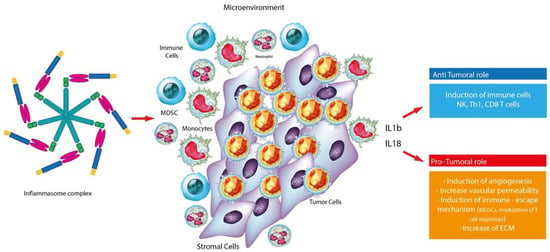
Figure 2.
Graphic representation of the dual role of the inflammasome complex in cancer.
2. Inflammation and Myeloproliferative Neoplasms (MPNs)
2.1. Mutation in MPNs
MPNs are characterized by abnormal proliferation of bone marrow stem cells accompanied by changes in the number of platelets, red blood cells and white blood cells; PMF also presents with an increase in bone marrow fibrosis, splenomegaly and extra-medullary hematopoiesis (EMH). EMH is observed not only in the liver and spleen, but also in the lymph nodes, in the urogenital system, in the serum surfaces and in the epidural and paraspinal spaces because of modification of the CXCL12/CXCR4 axis [46]. In most cases MPNs are secondary to genetic defects regarding pluripotent stem cell populations leading to cell proliferation during the disease [47]. Such genetic defects mainly concern mutations involving MPL, JAK2 and more recently, CALR genes, which are involved in the JAK/STAT pathway with its consequent dysregulation [48]. Altogether these mechanisms induce the abnormal signaling of STAT transcription factors with consequent cell growth and proliferation. STAT3, in particular, is closely linked to the development of cancer through the activation of immunomodulatory cytokines (IL-6, IL-10 and IL-17), growth factors (FGF, VEGF) and matrix metalloproteinases [49]. STAT3 deletion result in lower white blood counts, lower spleen weights and a reduced degree of reticulin fibrosis, reduced disease severity and cytokine-mediated inflammation, similar to the effects observed with ruxolitinib therapy [50]. JAK2V617F is the best characterized mutations observed in these disorders with a prevalence of more than 95% in PV, 50%–70% in ET and 40%–50% in PMF [48,51]. JAK2V617F mutation negative cases of PV may present JAK2 mutations in exon 12 present at a frequency of approximately 2%–3% [48,52]. JAK2V617F negative cases of ET and PMF can contain MPL mutations occurring at the frequency of 5% to 10% [48,53]. As far as concern CALR mutation, this was found to occur in most MPN patients with non-mutated JAK2 or MPL [48,53,54]. Although JAK2 is the most frequent mutation, there are patients without any of the three mutations and are therefore called triple negatives [48]. In investigations of large cohorts of MPN, JAK2V617F can be detected in 95% of patients with PV and JAK2 exon12 mutation in the remaining 5% of patients [1]. Among ET patients, JAK2V617F can be detected in 60% and 65% of patients, MPLW515 L/K in approximately 5% and CALR mutation in approximately 20% and 25%. Very recently, several other mutations have been associated with MPN [48], including TET2, ASXL1, IDH1/2 and SRSF2 [55]. Regardless of the molecular state, all patients present a deregulation in JAK/STAT signaling. In all MPNs, megakaryocytes proliferate, acquire multilobulate nuclei and exhibit clustering in the bone marrow [56]. These cells are characterized by an abnormal localization of P selectin in their intracytoplasmic vacuoles and in the membrane demarcation system (DMS) which leads to an increased emperipolesis of neutrophils which release their enzymes into megakaryocytes and thus releasing other cytokines such as the transformation of the growth factor beta (TGF-β), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) from their alpha granules [57].
2.2. Treatment in MPNs
Despite therapies targeting clones that support myeloproliferation, PMF is still considered an incurable disease, except for patients who successfully undergo an allogeneic stem cell transplantation [58]. To date, the only effective pharmacological therapy includes JAK inhibitors (JAKi). The prototypical compound of this class is ruxolitinib. However, while ruxolitinib is able to provide significant improvements in splenomegaly, associated clinical manifestations and constitutional symptoms related to the disease, at least part of its clinical benefits have been associated with a marked downregulation in serum proinflammatory cytokines [59] produced in particular by immunological and hematopoietic cells, both after four weeks of treatment [60] and after 24 months [61] demonstrating the importance of inflammation in the pathologic process [62].
2.3. Inflammation in MPNs
In recent years it has emerged that the tumor microenvironment (TME) and in particular the inflammatory microenvironment [63], plays a fundamental role in the pathogenesis of MPN. This progressively transformed tumor microenvironment (TME) is infiltrated by different types of immune cells that are likely to kill cancer cells in the initial stages, but over time the inability of immune cells to inhibit the abnormal growth of cancer cells prevails depending on a variety of events occurring at the tumor site [36]. Hence, MPNs, that are initiated by the appearance of a myelostimulatory mutation, propagate through an evolving cascade of inflammatory conduits that entail dramatic symptoms and impairment of the quality of life of affected patients [47]. Taken all together these evidences suggest that MPN is as a chronic tumor model driven by inflammation [64], in which chronic inflammation plays a critical role in the development and maintenance of MPN itself [49]. Consistently, numerous studies have been carried out over the years confirmed this theory and it is now clear that there is a close relationship between chronic inflammation and the pathogenesis of MPN. An evaluation of abnormal cytokine expression determined that primary myelofibrosis (PMF) patients had significantly increased levels of IL-1 β, IL-1RA, IL-2R, IL-6, IL-8, IL-10, IL-12, IL-13, IL-15, TNF-alpha, G-CSF, IFN-α, MIP-1α, HGF, IFN-γ and VEGF in addition to reduced IFN-γ levels while MIP-1β gave conflicting results. In PV, patients increased levels of IL-1RA, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL- 13, IFN-γ, GM-CSF, MCP-1, MIP-1α, MIP-1β, HGF, IP-10, MIG, MCP-1, PDGF, TNF-α, IFN-γ and VEGF were measured [65,66,67]. An analysis of ET patients reported the presence of elevated levels of IL-1 β, IL-4, IL-6, IL-8, IL-10, IL-12, HGF, GM-CSF, IFN-γ, MCP-1, PDGF, TNF-α and VEGF. Taken together this evidenceline, levels of reported cytokines were higher compared to healthy controls, however some of these differences were not significant [65]. Interestingly, IL-4, IL-8, GM- CSF, IFN-γ, MCP-1, PDGF and VEGF appeared to be significantly higher in ET patients when compared to PV populations and may be used as markers to distinguish the two disorders [67] (Figure 3).
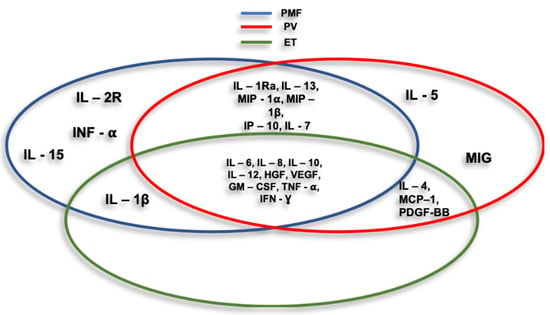
Figure 3.
Cycles represent cytokines and chemokines associated with myeloproliferative neoplasms (MPNs) main entities.
Some studies showed that among the myriad of cytokines evaluated in the MPN, could be prognostic for patients [64], such us IL-1 β which mediates inflammation both at tissue and systemic level; IL-6 that induces the activation of JAK/STAT pathway; IL-8 that influences the tumor microenvironment by retrieving MDSCs; VEGF which promotes angiogenesis; TGF-β which in primary myelofibrosis is associated with bone marrow fibrosis. In healthy subjects the inflammatory cascade is driven by the interaction finely regulated by cellular responses and stimulating factors/cytokines. Dysregulation of this balance can lead to chronic inflammation, which is characteristic of several pathologies including MPNs. An Italian study, conducted by Sollazzo et al. showed significantly higher IL-1β levels in PMF patients than healthy controls and observed an increase in the proliferation of CD34+ cells treated with culture media containing IL-1 β, suggesting that IL-1 β plays an important role in the progression of myelofibrosis [63]. In MF, the combination of TNF-α and TIMP-1 has been shown to promote survival of CD34+ stem cells, whereas the combination of ATP and TNF-α has been shown to reduce proliferation [68]. Previous reports showed that JAK2V617F positive patients have significantly higher levels of IL-8, IL1B, IL-17A and IFN versus triple-negative (JAK2, MPL negative) patients [69]. TGF-β, a cytokine involved in normal hematopoiesis and in hematological malignancies, plays a key role in the progression of MPNs, so many studies have focused on the its role. Campanelli et al. showed, through CCL64 cell line tests, that PV, ET and PMF patients had significantly higher levels of TGF-β both in peripheral blood (PB) and bone marrow compared to healthy controls [70]. In addition to myeloproliferation, PMF is characterized by bone marrow fibrosis, neoangiogenesis and osteosclerosis [58]. Megakaryocytes and monocytes derived from malignant clones produce high levels of TGF- β other than PDGF, FGF and VEGF. In particular, TGF- β exerts pro-fibrotic effects on fibroblasts and is considered to be the main driver of fibrosis, as is able to induce collagen production. The inhibition of TGF- β in different experimental models resulted in a reduction of fibrosis [71].
2.4. Inflammasomes and Myeloproliferative Neoplasms
As described above, chronic inflammation is considered a driving force for the development of MPN, intensified by the continuous release of pro-inflammatory cytokines and chemokines that in the aging bone marrow niche can predispose patients to leukemic transformation. Many of these cytokines, in particular IL-1 β and IL-18, are released in active form by activating the inflammasome complexes which in turn mediate the inflammatory process. Despite this, little is known about the functional effects of stem cell-driven inflammasome signaling in MPN pathogenesis. Wang et al. showed that JAK2V617F positive macrophages expressed greater production of cytokines and chemokines has been demonstrated in CD11+ splenic cells, which in turn has led to an increase in the secretion of IL-1 β and an increase in plasma levels of IL-18, which could contribute to increasing the production and activation of neutrophils and the entry of leukocytes into the lesion [7]. Another study by Shinar et al. has shown that a mutation in the Mediterranean fever gene (MEFV), highly expressed in myeloid cells and coding for pyrin, a cytoplasmic protein that regulates the maturation and secretion of the pro-inflammatory cytokines IL-1β and IL-18 in the complex of the inflammasome, resulted in Mediterranean fever disease in JAK2V617F patient post-polycythemia myelofibrosis (PPV-MF) [72]. The microarray analysis conducted by Liew et al. identified numerous genes involved in the activation of the inflammasome, such as AIM2, IL-1 β, CASP1, which were significantly upregulated in the cells where JAK2V617F was induced. In particular, induction of JAK2V617F leads to a significant inflammatory response consistent with recent studies demonstrating the involvement of IL-1β in the development of myelofibrosis in a JAK2V617F mouse model. Furthermore, these evidences further suggest that AIM2 is a downstream target of JAK2V617F in D9 cell line [6]. Given the crucial role of AIM2 in combination with CASP1, in converting pro-IL1B to its active form [73,74], the induction of the AIM2, CASP1 and IL1B mRNAs in JAK2V617F-induced cells suggests that IL1B activation is linked to MPN development. Consistently, JAK2V617F-positive hematopoietic stem cells secrete IL1β inducing inflammation and promoting bone marrow myelofibrosis also in animal models [75].
3. Conclusions
Available data are not sufficient to fully establish the role of inflammasome in myeloproliferative neoplasms (MPN) and further studies are needed in order to clarify such a role. However, it was shown that there is an inflammatory-state-driving MPNs and that the available drugs (i.e., JAKi), are effective and improve symptoms through the immune system regulation although they do not present curative potential when used as a single agent. A better understanding on the role of the inflammasome may provide new insights on possible therapeutic strategies.
Author Contributions
Conceptualization, L.L., D.T., C.G., M.D.R., G.L.V. and G.A.P.; validation, A.R., I.B., M.D.R., M.S., G.A.P., D.T., L.L.; writing—original draft preparation, D.T., C.G., G.A.P., G.L.V., M.D.R.; supervision, D.T., G.A.P., I.B., R.A. and G.L.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. The chronic myeloproliferative disorders: Clonality and clinical heterogeneity. Semin. Hematol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediators Inflamm. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gouravani, M.; Khalili, N.; Razi, S.; Keshavarz-Fathi, M.; Khalili, N.; Rezaei, N. The NLRP3 inflammasome: A therapeutic target for inflammation-associated cancers. Expert Rev. Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Liew, E.L.; Araki, M.; Hironaka, Y.; Mori, S.; Tan, T.Z.; Morishita, S.; Edahiro, Y.; Ohsaka, A.; Komatsu, N. Identification of AIM2 as a downstream target of JAK2V617F. Exp. Hematol. Oncol. 2016. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in JAK2V617F mice. Circ. Res. 2018. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011. [Google Scholar] [CrossRef]
- Balaian, L.; Crews, L.A.; Mayson, C.; Holm, F.; Diep, R.H.; Leu, H.; Kulidjian, A.; Ball, E.D.; Jamieson, C. Comparative Inflammasome Analysis of Bone Marrow Stroma in Aging and Myelofibrosis. Blood 2018. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008. [Google Scholar] [CrossRef]
- Barresi, V.; Romano, A.; Musso, N.; Capizzi, C.; Consoli, C.; Martelli, M.P.; Palumbo, G.; Raimondo, F.D.; Condorelli, D.F. Broad copy neutral-loss of heterozygosity regions and rare recurring copy number abnormalities in normal karyotype-acute myeloid leukemia genomes. Genes Chromosom. Cancer 2010. [Google Scholar] [CrossRef]
- Canestraro, M.; Galimberti, S.; Savli, H.; Palumbo, G.A.; Tibullo, D.; Nagy, B.; Guerrini, F.; Piaggi, S.; Cine, N.; Metelli, M.R.; et al. Synergistic antiproliferative effect of arsenic trioxide combined with bortezomib in HL60 cell line and primary blasts from patients affected by myeloproliferative disorders. Cancer Genet. Cytogenet. 2010. [Google Scholar] [CrossRef]
- Jurg, T.; Martinon, F.; Burns, K. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-NL. Mol. Cell 2002. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M.; Núñez, G. Intracellular NOD-like Receptors in Host Defense and Disease. Immunity 2007. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yi, F. Implication of Pattern-Recognition Receptors in Cardiovascular Diseases. Antioxidants Redox Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Juliana, C.; Solorzano, L.; Kang, S.; Wu, J.; Datta, P.; McCormick, M.; Huang, L.; McDermott, E.; et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Newton, K.; Lamkanfi, M.; Mariathasan, S.; Dixit, V.M.; Monack, D.M. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 2010. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Strowig, T.; Flavell, R.A. Inflammasomes: Far beyond inflammation. Nat. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Maltez, V.I.; Miao, E.A. Reassessing the Evolutionary Importance of Inflammasomes. J. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Brodsky, I.E. The inflammasome: Learning from bacterial evasion strategies. Semin. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; La Cava, P.; Branca, A.; Conticello, C.; Stagno, F.; Chiarenza, A.; Palumbo, G.A.; Raimondo, F. Di Effects of second-generation tyrosine kinase inhibitors towards osteogenic differentiation of human mesenchymal cells of healthy donors. Hematol. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Bernot, A.; Clepet, C.; Dasilva, C.; Devaud, C.; Petit, J.L.; Caloustian, C.; Cruaud, C.; Samson, D.; Pulcini, F.; Weissenbach, J.; et al. A candidate gene for familial Mediterranean fever. Nat. Genet. 1997. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I. The autoinflammatory syndromes. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 511–516. [Google Scholar] [CrossRef]
- Savic, S.; Dickie, L.J.; Wittmann, M.; McDermott, M.F. Autoinflammatory syndromes and cellular responses to stress: Pathophysiology, diagnosis and new treatment perspectives. Best Pract. Res. Clin. Rheumatol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Pasquinucci, L.; Spitale, F.M.; Chiechio, S.; Turnaturi, R.; Caraci, F.; Tibullo, D.; Avola, R.; Gulino, R.; Parenti, R.; et al. Simultaneous Activation of Mu and Delta Opioid Receptors Reduces Allodynia and Astrocytic Connexin 43 in an Animal Model of Neuropathic Pain. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Man, S.M.; Kanneganti, T.D. Inflammasomes and cancer. Cancer Immunol. Res. 2017. [Google Scholar] [CrossRef]
- Karan, D. Inflammasomes: Emerging Central Players in Cancer Immunology and Immunotherapy. Front. Immunol. 2018. [Google Scholar] [CrossRef]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.-D. IL-18 Production Downstream of the Nlrp3 Inflammasome Confers Protection against Colorectal Tumor Formation. J. Immunol. 2010. [Google Scholar] [CrossRef]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerbäck, C.; Rosdahl, I.; Söderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 2012. [Google Scholar] [CrossRef]
- Girardelli, M.; Maestri, I.; Rinaldi, R.R.; Tognon, M.; Boldorini, R.; Bovenzi, M.; Crovella, S.; Comar, M. NLRP1 polymorphisms in patients with asbestos-associated mesothelioma. Infect. Agent. Cancer 2012. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016. [Google Scholar] [CrossRef]
- Ungerbäck, J.; Belenki, D.; Jawad Ul-Hassan, A.; Fredrikson, M.; Fransén, K.; Elander, N.; Verma, D.; Söderkvist, P. Genetic variation and alterations of genes involved in NFκB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 2012. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Laird, P.W.; Park, P.J. XThe landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017. [Google Scholar] [CrossRef]
- Zaki, M.H.; Vogel, P.; Malireddi, R.K.S.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. The NOD-Like Receptor NLRP12 Attenuates Colon Inflammation and Tumorigenesis. Cancer Cell 2011. [Google Scholar] [CrossRef]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.T.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 Suppresses Colon Inflammation and Tumorigenesis through the Negative Regulation of Noncanonical NF-κB Signaling. Immunity 2012. [Google Scholar] [CrossRef]
- Bogani, C.; Ponziani, V.; Guglielmelli, P.; Desterke, C.; Rosti, V.; Bosi, A.; Le Bousse-Kerdilès, M.-C.; Barosi, G.; Vannucchi, A.M. Hypermethylation of CXCR4 Promoter in CD34 + Cells from Patients with Primary Myelofibrosis. Stem Cells 2008. [Google Scholar] [CrossRef]
- Geyer, H.L.; Dueck, A.C.; Scherber, R.M.; Mesa, R.A. Impact of Inflammation on Myeloproliferative Neoplasm Symptom Development. Mediators Inflamm. 2015. [Google Scholar] [CrossRef]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The role of new technologies in myeloproliferative neoplasms. Front. Oncol. 2019. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2012. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015. [Google Scholar] [CrossRef]
- Them, N.C.C.; Kralovics, R. Genetic basis of MPN: Beyond JAK2-V617F. Curr. Hematol. Malig. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013. [Google Scholar] [CrossRef]
- Tefferi, A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010. [Google Scholar] [CrossRef]
- Briere, J.; Kiladjian, J.J.; Peynaud-Debayle, E. Megakaryocytes and platelets in myeloproliferative disorders. Baillieres. Clin. Haematol. 1997. [Google Scholar] [CrossRef]
- Schmitt, A.; Jouault, H.; Guichard, J.; Wendling, F.; Drouin, A.; Cramer, E.M. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood 2000. [Google Scholar] [CrossRef]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdilès, M.C. Inflammation as a keystone of bone marrow stroma alterations in primary myelofibrosis. Mediators Inflamm. 2015. [Google Scholar] [CrossRef]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010. [Google Scholar] [CrossRef]
- Kvasnicka, H.M.; Thiele, J.; Bueso-Ramos, C.E.; Kamalanabhaiah, S.; Cortes, J.E.; Kantarjian, H.; Verstovsek, S. Changes in Activated Bone Marrow Macrophages and Mast Cells in Patients with Myelofibrosis Following Ruxolitinib Therapy. Blood 2014. [Google Scholar] [CrossRef]
- Agarwal, A.; Morrone, K.; Bartenstein, M.; Zhao, Z.J.; Verma, A.; Goel, S. Bone marrow fibrosis in primary myelofibrosis: Pathogenic mechanisms and the role of TGF-β. Stem Cell Investig. 2016. [Google Scholar] [CrossRef]
- Sollazzo, D.; Forte, D.; Polverelli, N.; Romano, M.; Perricone, M.; Rossi, L.; Ottaviani, E.; Luatti, S.; Martinelli, G.; Vianelli, N.; et al. Crucial factors of the inflammatory microenvironment (IL-1β/TNF-α/TIMP-1) promote the maintenance of the malignant hemopoietic clone of myelofibrosis: An in vitro study. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zuo, X. Cytokines frequently implicated in myeloproliferative neoplasms. Cytokine X 2019. [Google Scholar] [CrossRef]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma cytokines in polycythemia vera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am. J. Hematol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014. [Google Scholar] [CrossRef]
- Catani, L.; Rossi, L.; Sollazzo, D.; Franchini, E.; Romano, M.; Zuffa, E.; Barone, M.; Perricone, M.; Polverelli, N.; Ottaviani, E.; et al. Crucial Factors of the Inflammatory Microenvironment Promote Maintenance of the Malignant Hemopoietic Clone of Myelofibrosis By Stimulating Survival and Inhibiting Proliferation of CD34+ stem/Progenitor Cells. Blood 2014. [Google Scholar] [CrossRef]
- Tabarroki, A.; Rogers, H.J.; Visconte, V.; Hasrouni, E.; Advani, A.; Sekeres, M.A.; Duong, H.K.; Kalaycio, M.; Copelan, E.A.; Stein, B.L.; et al. The Molecular and Cytokine Profile of Triple-Negative (JAK2 V617F, JAK2 exon 12, MPL negative) Myelofibrosis, a Myeloproliferative Neoplasm with Distinct Clinico-Pathologic Characteristics. Blood 2012. [Google Scholar] [CrossRef]
- Campanelli, R.; Rosti, V.; Villani, L.; Castagno, M.; Moretti, E.; Bonetti, E.; Bergamaschi, G.; Balduini, A.; Barosi, G.; Massa, M. Evaluation of the bioactive and total transforming growth factor β1 levels in primary myelofibrosis. Cytokine 2011. [Google Scholar] [CrossRef]
- Shah, M.; Foreman, D.M.; Ferguson, M.W.J. Neutralization of TGF-beta(1) and TGF-beta(2) or exogenous addition of TGF-beta(3) to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995, 108, 985–1002. [Google Scholar] [PubMed]
- Shinar, Y.; Tohami, T.; Livneh, A.; Schiby, G.; Hirshberg, A.; Nagar, M.; Goldstein, I.; Cohen, R.; Kukuy, O.; Shubman, O.; et al. Acquired familial Mediterranean fever associated with a somatic MEFV mutation in a patient with JAK2 associated post-polycythemia myelofibrosis. Orphanet J. Rare Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009. [Google Scholar] [CrossRef] [PubMed]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).