A Novel Approach for the Identification of Pharmacogenetic Variants in MT-RNR1 through Next-Generation Sequencing Off-Target Data

 ,
, 

Abstract
1. Introduction
2. Experimental Section
2.1. Samples
2.2. Targeted Capture Next-Generation Sequencing
2.3. Bioinformatics Analysis
2.4. Genotyping and Sanger Sequencing of m.1555A>G
2.5. Statistical Methods
2.6. Data Availability
3. Results
3.1. Off-Target Sequencing Metrics
3.2. MT-RNR1 m.1555A>G Detection Using Off-Target Data
3.3. Clinical WES Samples for Off-Target MT-RNR1 Genotype Assignation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pacheu-Grau, D.; Gómez-Durán, A.; López-Pérez, M.J.; Montoya, J.; Ruiz-Pesini, E. Mitochondrial pharmacogenomics: Barcode for antibiotic therapy. Drug Discov. Today 2010, 15, 33–39. [Google Scholar] [CrossRef]
- Barbarino, J.M.; McGregor, T.L.; Altman, R.B.; Klein, T.E. PharmGKB summary: Very important pharmacogene information for MT-RNR1. Pharmacogenet. Genom. 2016, 26, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xing, G.; Yan, M.; Cao, X.; Liu, X.-Z.; Bu, X.; Guan, M.-X. Cosegregation of C-insertion at position 961 with the A1555G mutation of the mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. Am. J. Med. Genet. 2004, 124, 113–117. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, F.J. Heteroplasmy for the 1555A>G mutation in the mitochondrial 12S rRNA gene in six Spanish families with non-syndromic hearing loss. J. Med. Genet. 2003, 40, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Rydzanicz, M.; Wróbel, M.; Pollak, A.; Gawęcki, W.; Brauze, D.; Kostrzewska-Poczekaj, M.; Wojsyk-Banaszak, I.; Lechowicz, U.; Mueller-Malesińska, M.; Ołdak, M.; et al. Mutation analysis of mitochondrial 12S rRNA gene in Polish patients with non-syndromic and aminoglycoside-induced hearing loss. Biochem. Biophys. Res. Commun. 2010, 395, 116–121. [Google Scholar] [CrossRef]
- Nguyen, T.; Jeyakumar, A. Genetic susceptibility to aminoglycoside ototoxicity. Int. J. Pediatr. Otorhinolaryngol. 2019, 120, 15–19. [Google Scholar] [CrossRef]
- Jing, W.; Zongjie, H.; Denggang, F.; Na, H.; Bin, Z.; Aifen, Z.; Xijiang, H.; Cong, Y.; Yunping, D.; Ring, H.Z.; et al. Mitochondrial mutations associated with aminoglycoside ototoxicity and hearing loss susceptibility identified by meta-analysis. J. Med. Genet. 2015, 52, 95–103. [Google Scholar] [CrossRef]
- Xing, G.; Chen, Z.; Wei, Q.; Tian, H.; Li, X.; Zhou, A.; Bu, X.; Cao, X. Mitochondrial 12S rRNA A827G mutation is involved in the genetic susceptibility to aminoglycoside ototoxicity. Biochem. Biophys. Res. Commun. 2006, 346, 1131–1135. [Google Scholar] [CrossRef]
- Li, Z.; Li, R.; Chen, J.; Liao, Z.; Zhu, Y.; Qian, Y.; Xiong, S.; Heman-Ackah, S.; Wu, J.; Choo, D.I.; et al. Mutational analysis of the mitochondrial 12S rRNA gene in Chinese pediatric subjects with aminoglycoside-induced and non-syndromic hearing loss. Hum. Genet. 2005, 117, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Meza, G.; Torres-Ruíz, N.; Tirado-Gutiérrez, C.; Aguilera, P. mtDNA mutations, hearing loss and aminoglycoside treatment in Mexicans. Braz. J. Otorhinolaryngol. 2011, 77, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Bacino, C.; Prezant, T.R.; Bu, X.; Fournier, P.; Fischel-Ghodsian, N. Susceptibility mutations in the mitochondrial small ribosomal RNA gene in aminoglycoside induced deafness. Pharmacogenetics 1995, 5, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gürtler, N.; Schmuziger, N.; Kim, Y.; Mhatre, A.N.; Jungi, M.; Lalwani, A.K. Audiologic Testing and Molecular Analysis of 12S rRNA in Patients Receiving Aminoglycosides. Laryngoscope 2005, 115, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Bylstra, Y.; Davila, S.; Lim, W.K.; Wu, R.; Teo, J.X.; Kam, S.; Lysaght, T.; Rozen, S.; Teh, B.T.; Yeo, K.K.; et al. Implementation of genomics in medical practice to deliver precision medicine for an Asian population. NPJ Genom. Med. 2019, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Stark, Z.; Dolman, L.; Manolio, T.A.; Ozenberger, B.; Hill, S.L.; Caulfied, M.J.; Levy, Y.; Glazer, D.; Wilson, J.; Lawler, M.; et al. Integrating Genomics into Healthcare: A Global Responsibility. Am. J. Hum. Genet. 2019, 104, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, E.; Katsila, T.; Mitropoulou, C.; Tsermpini, E.-E.; Patrinos, G.P. Integrating Next-Generation Sequencing in the Clinical Pharmacogenomics Workflow. Front. Pharmacol. 2019, 10, 384. [Google Scholar] [CrossRef]
- Bris, C.; Goudenege, D.; Desquiret-Dumas, V.; Charif, M.; Colin, E.; Bonneau, D.; Amati-Bonneau, P.; Lenaers, G.; Reynier, P.; Procaccio, V. Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front. Genet. 2018, 9, 632. [Google Scholar] [CrossRef]
- Diroma, M.A.; Calabrese, C.; Simone, D.; Santorsola, M.; Calabrese, F.M.; Gasparre, G.; Attimonelli, M. Extraction and annotation of human mitochondrial genomes from 1000 Genomes Whole Exome Sequencing data. BMC Genom. 2014, 15, S2. [Google Scholar] [CrossRef] [PubMed]
- Vdauwera GATK—mtDNA Analysis with Mutect2. Available online: https://github.com/broadinstitute/gatk-docs/blob/master/blog-2012-to-2019/2019-03-05-New!_Mitochondrial_Analysis_with_Mutect2.md?id=23598 (accessed on 24 June 2020).
- Puttick, C.; Kumar, K.R.; Davis, R.L.; Pinese, M.; Thomas, D.M.; Dinger, M.E.; Sue, C.M.; Cowley, M.J. mity: A Highly Sensitive Mitochondrial Variant Analysis Pipeline for Whole Genome Sequencing Data. BioRxiv 2019. [Google Scholar] [CrossRef]
- Genome Aggregation Database Consortium; Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Usami, S.; Nishio, S. Nonsyndromic Hearing Loss and Deafness, Mitochondrial. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2004; p. 23. [Google Scholar]
- Ye, F.; Samuels, D.C.; Clark, T.; Guo, Y. High-throughput sequencing in mitochondrial DNA research. Mitochondrion 2014, 17, 157–163. [Google Scholar] [CrossRef]
- Lévêque, M.; Marlin, S.; Jonard, L.; Procaccio, V.; Reynier, P.; Amati-Bonneau, P.; Baulande, S.; Pierron, D.; Lacombe, D.; Duriez, F.; et al. Whole mitochondrial genome screening in maternally inherited non-syndromic hearing impairment using a microarray resequencing mitochondrial DNA chip. Eur. J. Hum. Genet. 2007, 15, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Tuna, S.; Keogh, M.J.; Smith, K.R.; Aitman, T.J.; Beales, P.L.; Bennett, D.L.; Gale, D.P.; Bitner-Glindzicz, M.A.K.; Black, G.C.; et al. Germline selection shapes human mitochondrial DNA diversity. Science 2019, 364, eaau6520. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed]
- Griffin, H.R.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Duff, J.; Hudson, G.; Horvath, R.; Wilson, I.J.; Santibanez-Koref, M.; Taylor, R.W.; et al. Accurate mitochondrial DNA sequencing using off-target reads provides a single test to identify pathogenic point mutations. Genet. Med. 2014, 16, 962–971. [Google Scholar] [CrossRef]
- Samuels, D.C.; Han, L.; Li, J.; Quanghu, S.; Clark, T.A.; Shyr, Y.; Guo, Y. Finding the lost treasures in exome sequencing data. Trends Genet. 2013, 29, 593–599. [Google Scholar] [CrossRef]
- Wagner, M.; Berutti, R.; Lorenz-Depiereux, B.; Graf, E.; Eckstein, G.; Mayr, J.A.; Meitinger, T.; Ahting, U.; Prokisch, H.; Strom, T.M.; et al. Mitochondrial DNA mutation analysis from exome sequencing—A more holistic approach in diagnostics of suspected mitochondrial disease. J. Inherit. Metab. Dis. 2019, 42, 909–917. [Google Scholar] [CrossRef]
- Conrad, D.J.; Stenbit, A.E.; Zettner, E.M.; Wick, I.; Eckhardt, C.; Hardiman, G. Frequency of mitochondrial 12S ribosomal RNA variants in an adult cystic fibrosis population. Pharmacogenet. Genom. 2008, 18, 1095–1102. [Google Scholar] [CrossRef]
- CPIC. Available online: https://cpicpgx.org/ (accessed on 25 May 2020).
- PharmGKB. Available online: https://www.pharmgkb.org/ (accessed on 25 May 2020).
- Zhang, P.; Samuels, D.C.; Lehmann, B.; Stricker, T.; Pietenpol, J.; Shyr, Y.; Guo, Y. Mitochondria sequence mapping strategies and practicability of mitochondria variant detection from exome and RNA sequencing data. Brief. Bioinform. 2016, 17, 224–232. [Google Scholar] [CrossRef]
- Shen, S.-S.; Liu, C.; Xu, Z.-Y.; Hu, Y.-H.; Gao, G.-F.; Wang, S.-Y. Heteroplasmy levels of mtDNA1555A>G mutation is positively associated with diverse phenotypes and mutation transmission in a Chinese family. Biochem. Biophys. Res. Commun. 2012, 420, 907–912. [Google Scholar] [CrossRef]
- Trost, B.; Walker, S.; Haider, S.A.; Sung, W.W.L.; Pereira, S.; Phillips, C.L.; Higginbotham, E.J.; Strug, L.J.; Nguyen, C.; Raajkumar, A.; et al. Impact of DNA source on genetic variant detection from human whole-genome sequencing data. J. Med. Genet. 2019, 56, 809–817. [Google Scholar] [CrossRef]
- Goode, M.R.; Cheong, S.Y.; Li, N.; Ray, W.C.; Bartlett, C.W. Collection and Extraction of Saliva DNA for Next Generation Sequencing. J. Vis. Exp. 2014, 51697. [Google Scholar] [CrossRef] [PubMed]
- Bitner-Glindzicz, M.; Pembrey, M.; Duncan, A.; Heron, J.; Ring, S.M.; Hall, A.; Rahman, S. Prevalence of Mitochondrial 1555A→G Mutation in European Children. N. Engl. J. Med. 2009, 360, 640–642. [Google Scholar] [CrossRef]
- Vandebona, H.; Mitchell, P.; Manwaring, N.; Griffiths, K.; Gopinath, B.; Wang, J.J.; Sue, C.M. Prevalence of Mitochondrial 1555A→G Mutation in Adults of European Descent. N. Engl. J. Med. 2009, 360, 642–644. [Google Scholar] [CrossRef] [PubMed]
- Preste, R.; Vitale, O.; Clima, R.; Gasparre, G.; Attimonelli, M. HmtVar: A new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res. 2019, 47, D1202–D1210. [Google Scholar] [CrossRef]
- Fang, H.; Wu, Y.; Narzisi, G.; Ronemus, M.; Iossifov, I.; Schatz, M.C.; Lyon, G.J. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014, 6, 89. [Google Scholar] [CrossRef]
- Kobayashi, K.; Oguchi, T.; Asamura, K.; Miyagawa, M.; Horai, S.; Abe, S.; Usami, S. Genetic features, clinical phenotypes, and prevalence of sensorineural hearing loss associated with the 961delT mitochondrial mutation. Auris. Nasus. Larynx 2005, 32, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Genomics England Research Consortium; NIHR BioResource; Wei, W.; Pagnamenta, A.T.; Gleadall, N.; Sanchis-Juan, A.; Stephens, J.; Broxholme, J.; Tuna, S.; Odhams, C.A.; et al. Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat. Commun. 2020, 11, 1740. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
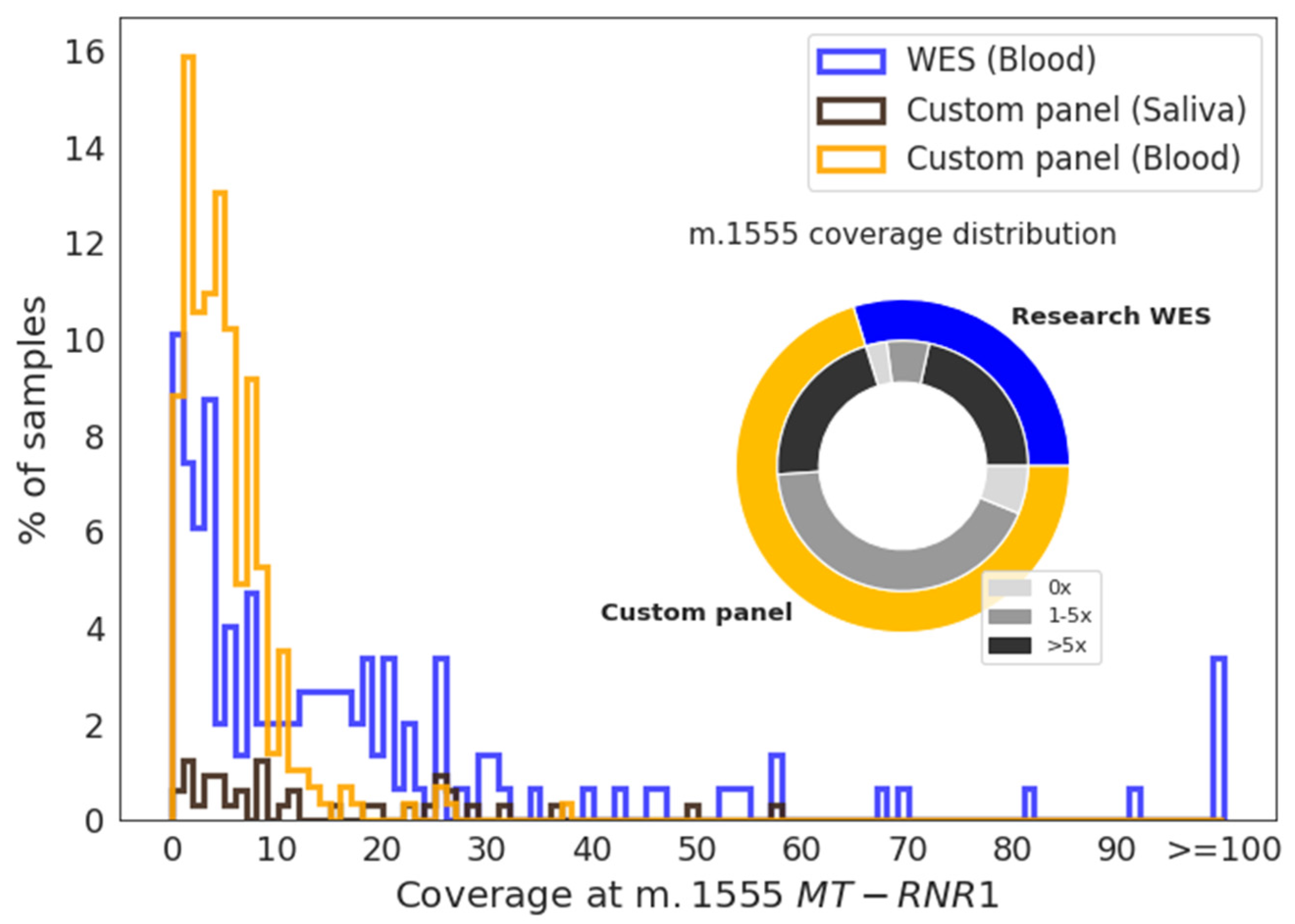
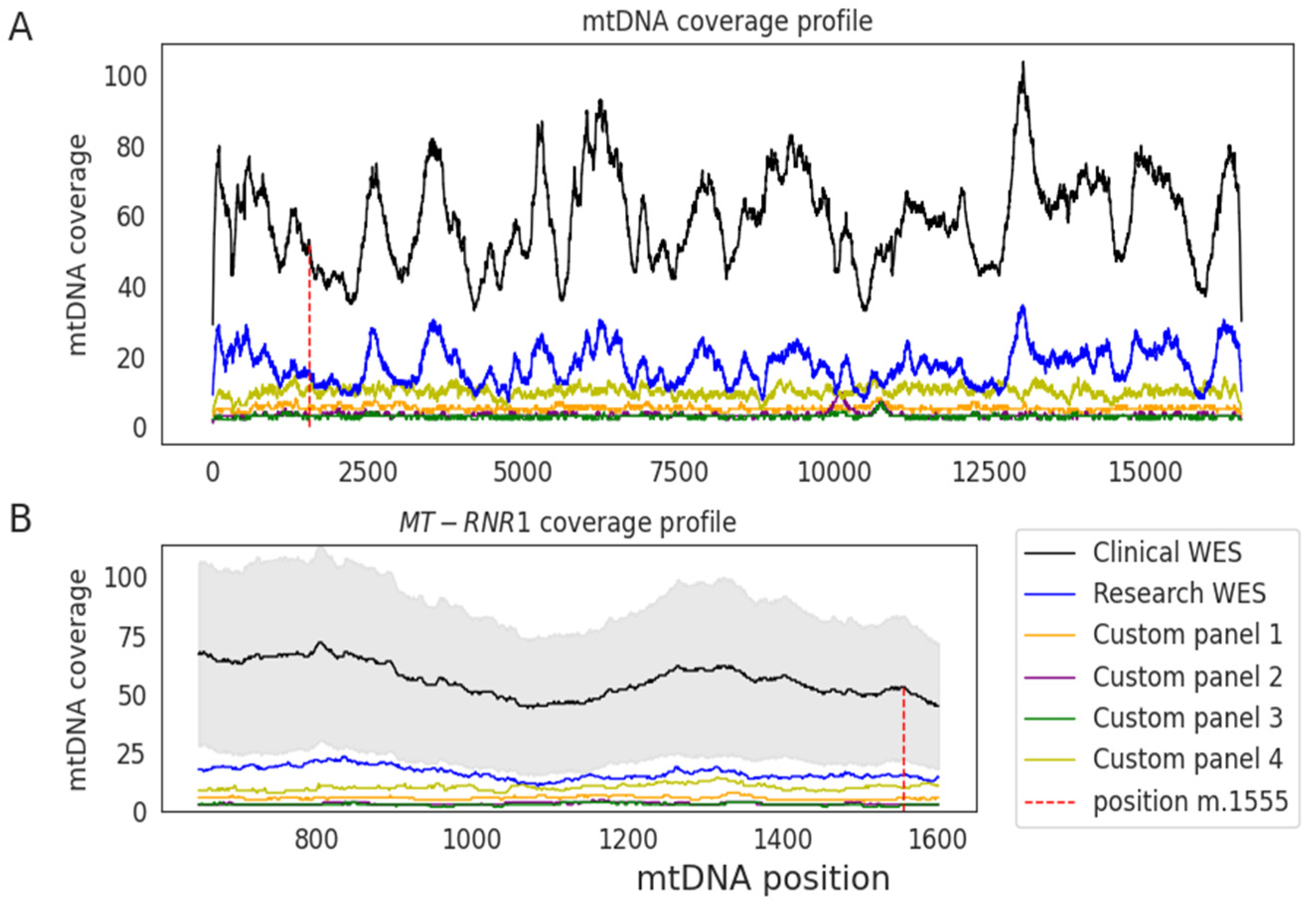
| Sample Type | Number of Samples | Median Coverage | |
|---|---|---|---|
| Mitochondria | m.1555 | ||
| Research samples-custom panel | |||
| Blood | 283 | 5 | 4 |
| Saliva | 38 | 11 | 14 |
| Kidney FFPE | 4 | 93 | 96 |
| Research samples-WES | |||
| Blood | 120 | 21 | 20 |
| Kidney frozen | 23 | 5 | 2 |
| Kidney FFPE | 5 | 18 | 40 |
| Clinical samples-WES | |||
| Blood | 1245 | 59 | 53 |
| CHROM 1 | POS 2 | REF 3 | ALT 4 | N. 5 | Heteroplasmy 6, Sample (VAF 7) | Association with Aminoglycoside-Induced Ototoxicity | (References) |
|---|---|---|---|---|---|---|---|
| chrM | 1555 | A | G | 3 | - | Strong | [2,3,4,5] |
| chrM | 1494 | C | T | 1 | - | [2] | |
| chrM | 669 | T | C | 2 | - | Further studies needed | [5] |
| chrM | 827 | A | G | 107 | MT384 (0.71); MT1358 (0.12) | [8,29] | |
| chrM | 896 | A | G | 1 | - | ||
| chrM | 930 | G | A | 19 | MT1043 8 (0.93) | ||
| chrM | 961 | T | C | 7 | - | ||
| chrM | 961 | T | G | 2 | - | ||
| chrM | 988 | G | A | 1 | - | [5] | |
| chrM | 1005 | T | C | 1 | - | [9,29] | |
| chrM | 1048 | C | T | 6 | - | [9] | |
| chrM | 1189 | T | C | 54 | MT1370 (0.86); MT368 (0.86); MT392 (0.89) | [10] | |
| chrM | 1243 | T | C | 11 | - | [11] | |
| chrM | 1438 | G | A | 26 | - | [12] | |
| chrM | 1462 | G | A | 8 | MT652 (0.719) | ||
| chrM | 1537 | C | T | 4 | - | [23] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanillos, J.; Santos, M.; Carcajona, M.; Roldan-Romero, J.M.; Martinez, A.M.; Calsina, B.; Monteagudo, M.; Leandro-García, L.J.; Montero-Conde, C.; Cascón, A.; et al. A Novel Approach for the Identification of Pharmacogenetic Variants in MT-RNR1 through Next-Generation Sequencing Off-Target Data. J. Clin. Med. 2020, 9, 2082. https://doi.org/10.3390/jcm9072082
Lanillos J, Santos M, Carcajona M, Roldan-Romero JM, Martinez AM, Calsina B, Monteagudo M, Leandro-García LJ, Montero-Conde C, Cascón A, et al. A Novel Approach for the Identification of Pharmacogenetic Variants in MT-RNR1 through Next-Generation Sequencing Off-Target Data. Journal of Clinical Medicine. 2020; 9(7):2082. https://doi.org/10.3390/jcm9072082
Chicago/Turabian StyleLanillos, Javier, María Santos, Marta Carcajona, Juan María Roldan-Romero, Angel M. Martinez, Bruna Calsina, María Monteagudo, Luis Javier Leandro-García, Cristina Montero-Conde, Alberto Cascón, and et al. 2020. "A Novel Approach for the Identification of Pharmacogenetic Variants in MT-RNR1 through Next-Generation Sequencing Off-Target Data" Journal of Clinical Medicine 9, no. 7: 2082. https://doi.org/10.3390/jcm9072082
APA StyleLanillos, J., Santos, M., Carcajona, M., Roldan-Romero, J. M., Martinez, A. M., Calsina, B., Monteagudo, M., Leandro-García, L. J., Montero-Conde, C., Cascón, A., Maietta, P., Alvarez, S., Robledo, M., & Rodriguez-Antona, C. (2020). A Novel Approach for the Identification of Pharmacogenetic Variants in MT-RNR1 through Next-Generation Sequencing Off-Target Data. Journal of Clinical Medicine, 9(7), 2082. https://doi.org/10.3390/jcm9072082