Mathematical Model of Muscle Wasting in Cancer Cachexia



Abstract
1. Introduction
2. A Model of Healthy Muscle Tissue
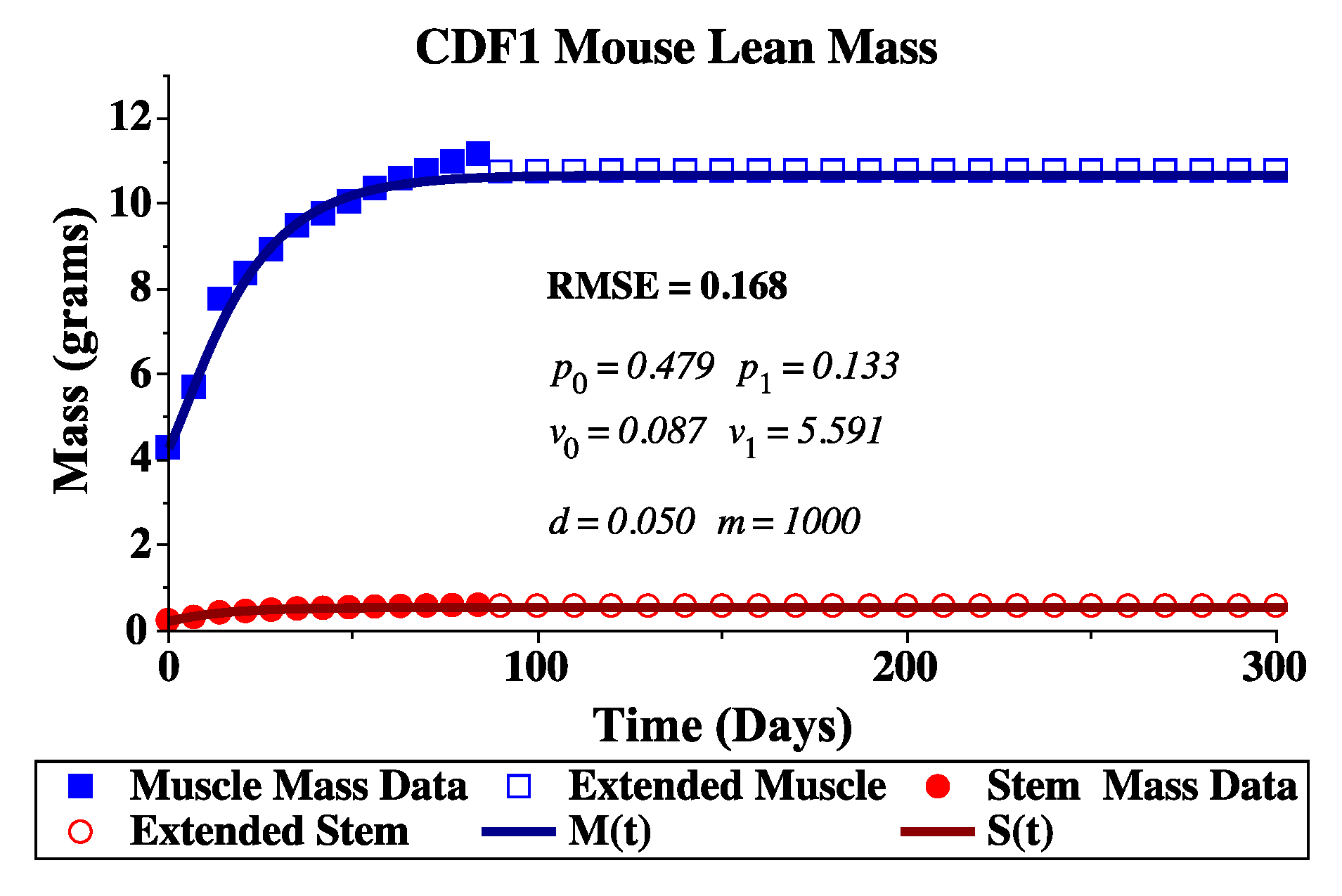
2.1. Model Parameterization
2.2. Healthy Experimental Data
Simulated Annealing for Parameter Fitting
3. Modeling Cancer Cachexia and Treatment
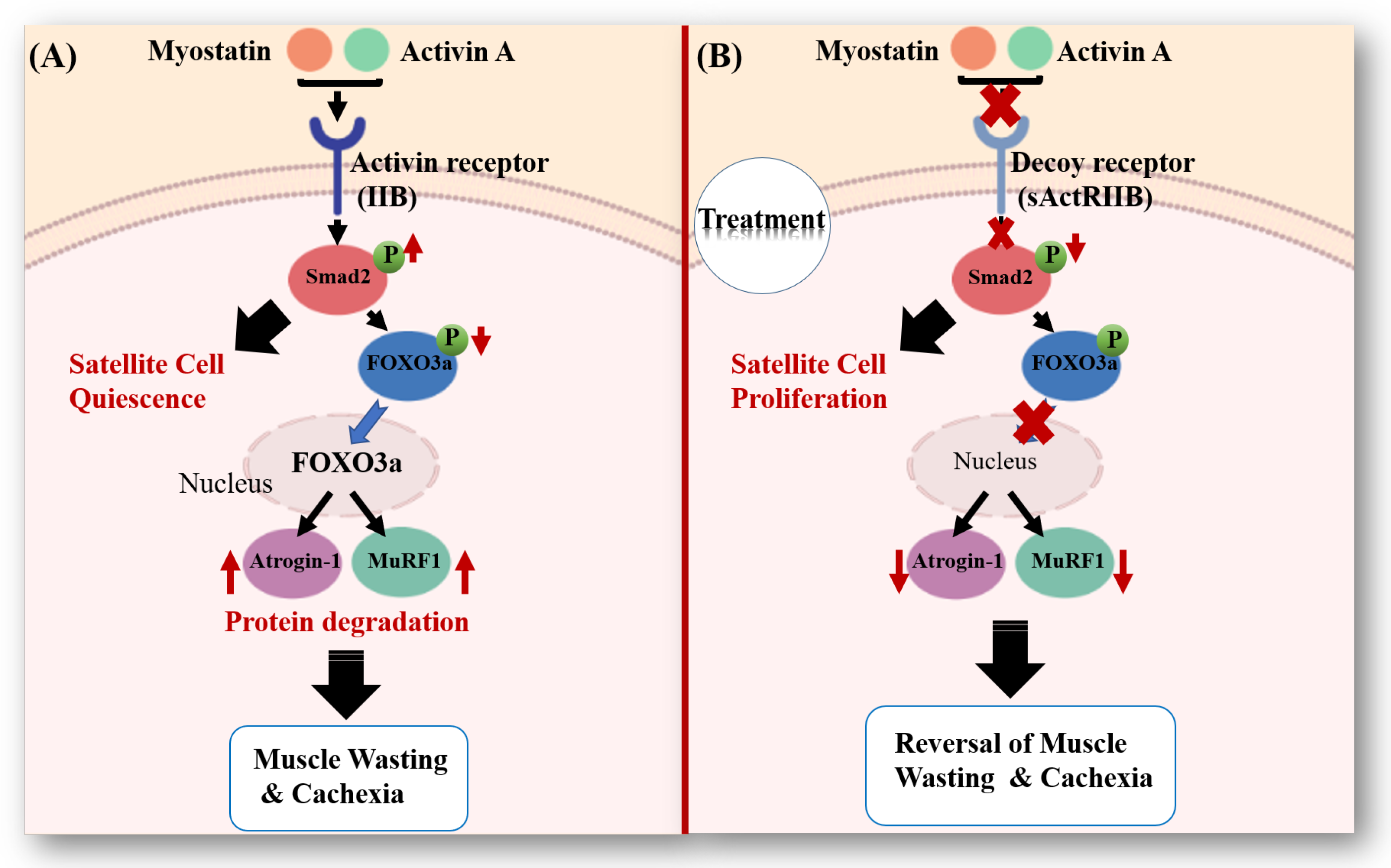
3.1. Muscle Loss in Cancer Cachexia
- muscle cells atrophy [85].
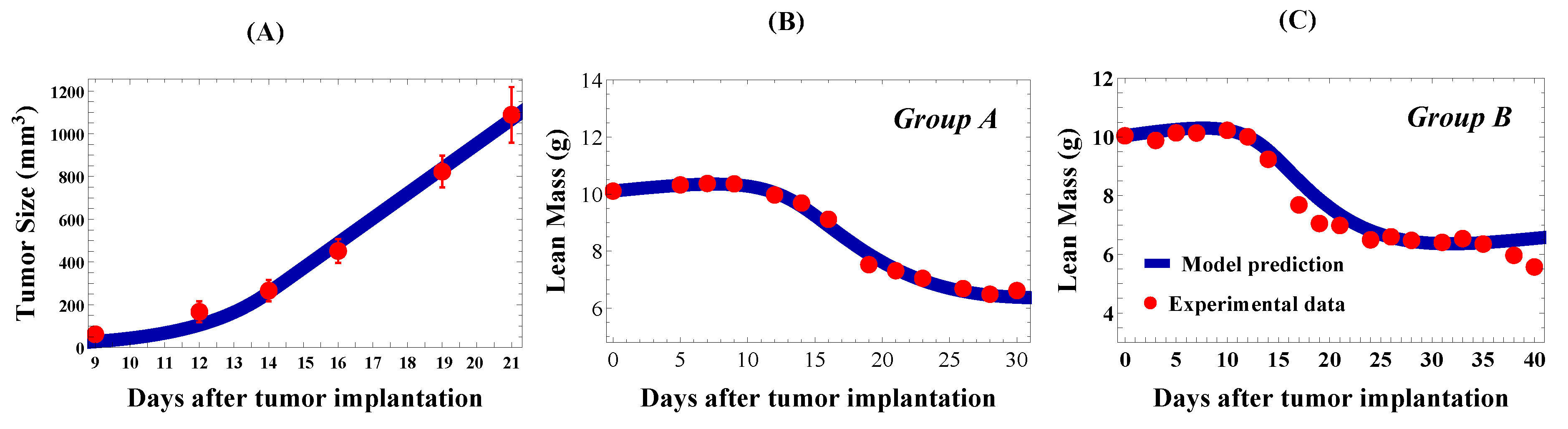
Parameterizing the Cachexia Model
3.2. Reversing Muscle Loss through Anti-Cachexia Treatment
Parameterizing the Anti-Cachexia Treatment Model
4. Results
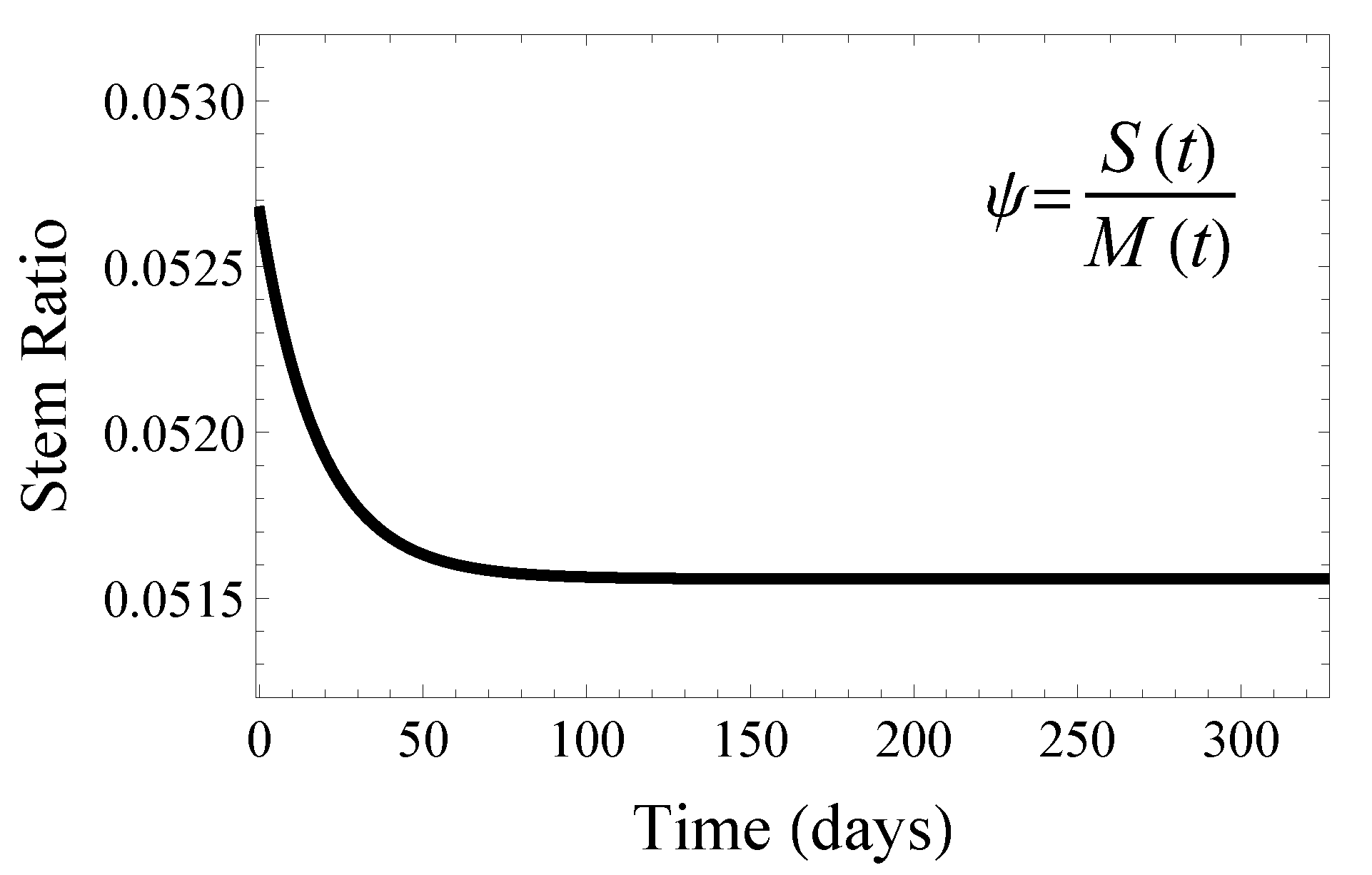
4.1. The Healthy Muscle Stem Cell Ratio
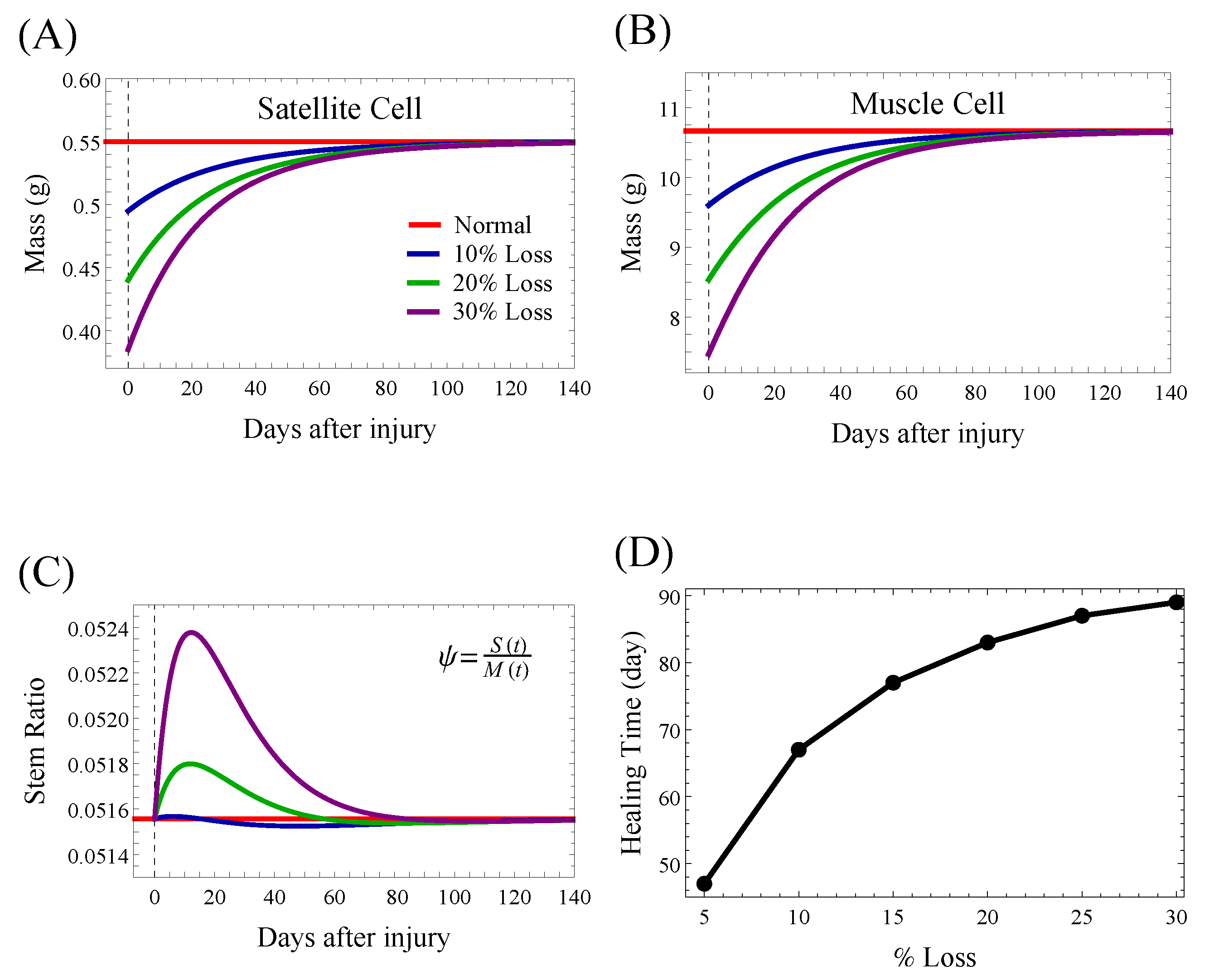
4.2. Wound Healing in the Healthy State
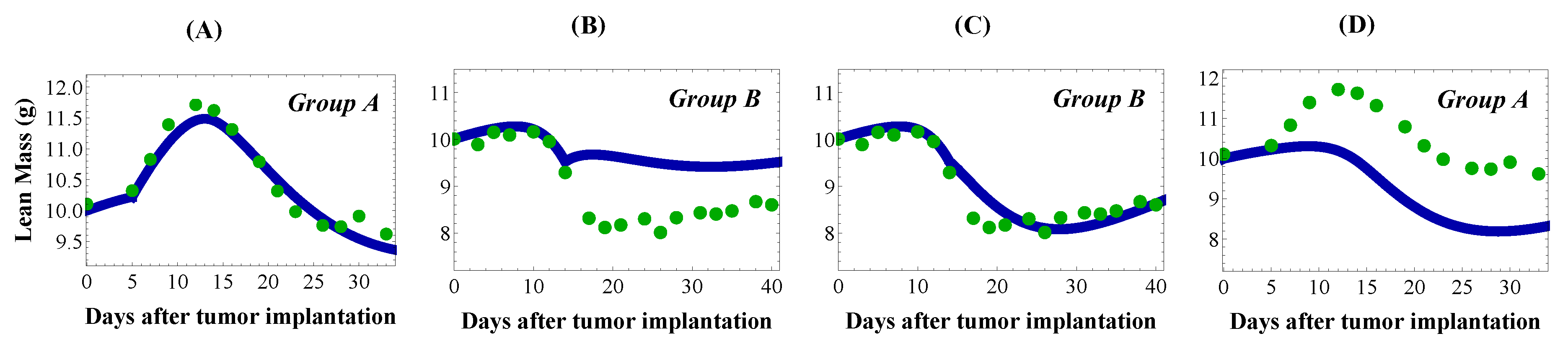
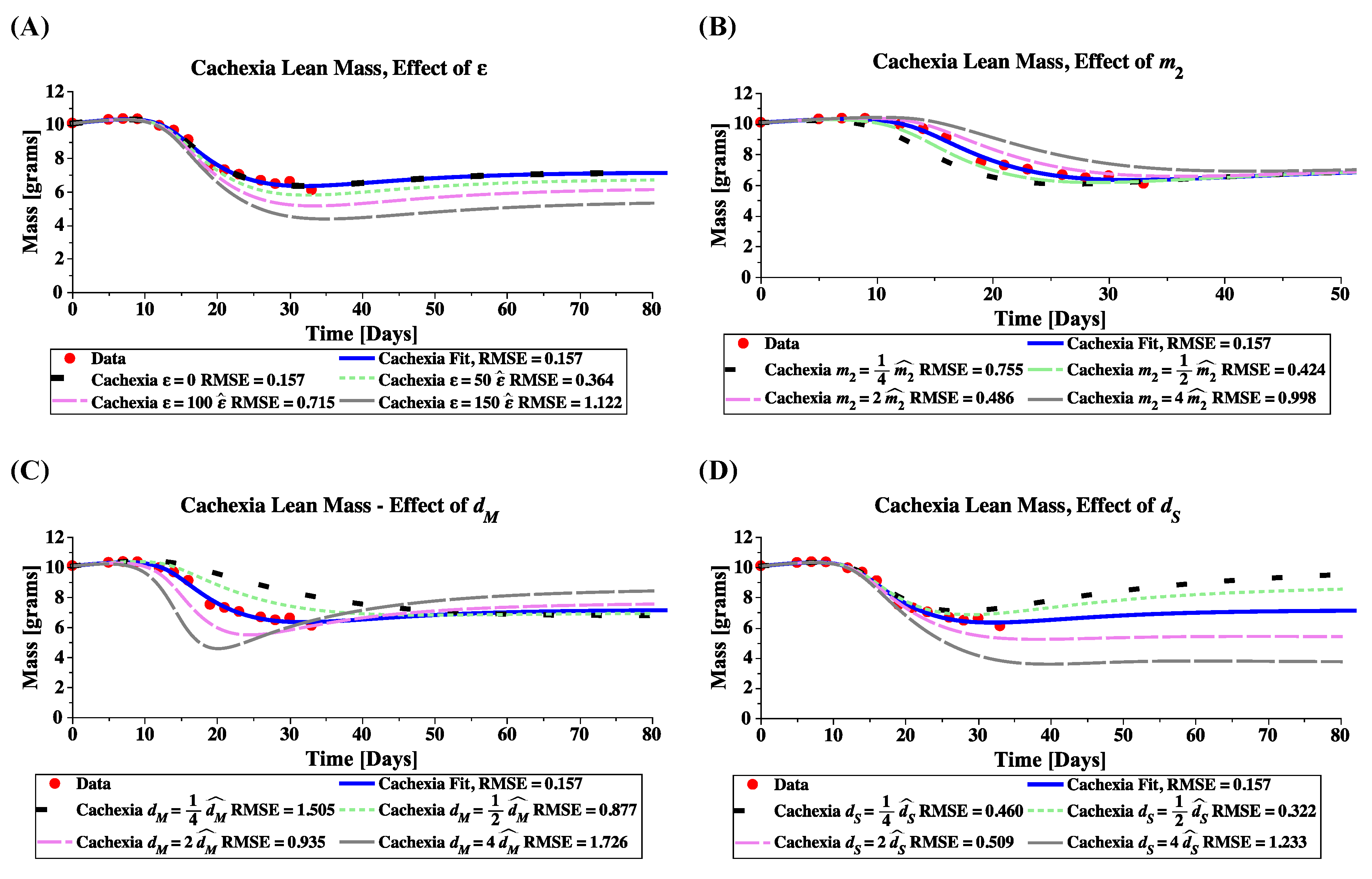
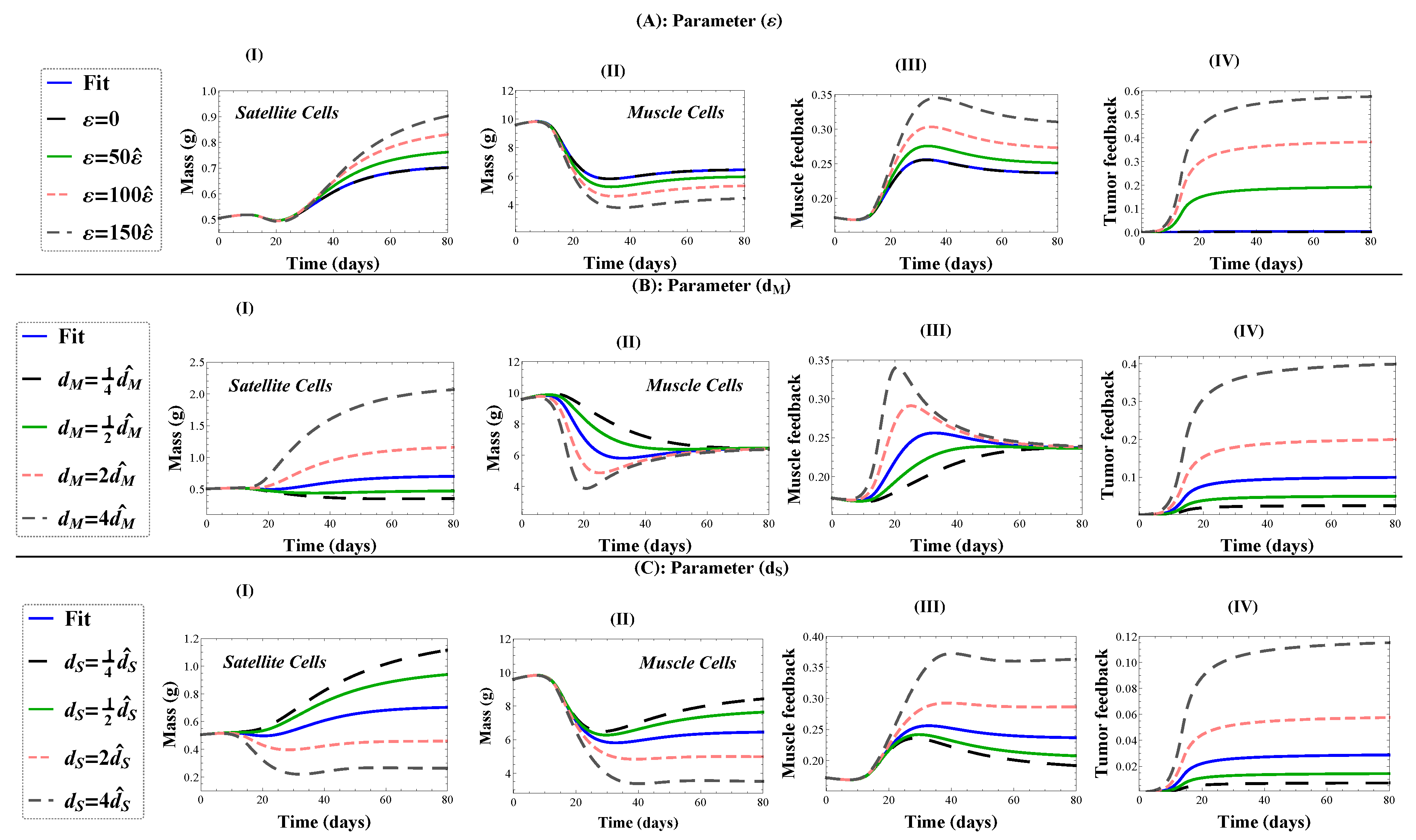
4.3. Sensitivity Analysis for Cachexia and Treatment
4.4. Mechanisms of Cachexia Target Muscle or Satellite Cells With Different Effects
4.5. Treatment Partially Restores Muscle Mass by Reactivating Satellite Cells
5. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ActRIIB | activin type-2B receptor |
| C26 | colon-26 adenocarcinoma mouse model |
| CDF1 mouse | a cross between female BALB/cAnNCrl and male DBA2NCrl mice |
| FOXO3a | forkhead box 03A |
| IL-1, IL-6, IL-10 | interleukin-1, interleukin-6, interleukin-10 |
| MAPK/ERK | mitogen-activated protein kinase/extracellular signal-regulated kinase |
| MuRF1 | muscle RING-finger protein-1 |
| NF-B | nuclear factor B |
| Pax7 | paired box 7 |
| RMSE | root-mean-squared-error |
| sActRIIB | soluble decoy Activin type-2B receptor |
| SMAD2 | mothers against decapentaplegic homolog 2 |
| STAT3 | signal transducer and activator of transcription 3 |
| TGF- | transforming growth factor |
| TNF | tumor necrosis factor |
Appendix A. Numerical Solution of Anti-Cachexia Treatment Model
References
- Tisdale, M.J. Cachexia in cancer patients. Nat. Rev. Cancer 2002, 2, 862. [Google Scholar] [CrossRef]
- Springer, J.; Von Haehling, S.; Anker, S.D. The need for a standardized definition for cachexia in chronic illness. Nat. Rev. Endocrinol. 2006, 2, 416–417. [Google Scholar] [CrossRef]
- Evans, W.J.; Morley, J.E.; Argilés, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. Espen 2008, 27, 793–799. [Google Scholar] [CrossRef]
- Donohoe, C.L.; Ryan, A.M.; Reynolds, J.V. Cancer cachexia: Mechanisms and clinical implications. Gastroent. Res. Pract. 2011, 2011, 1–13. [Google Scholar] [CrossRef]
- von Haehling, S.; Anker, S.D. Cachexia as a major underestimated and unmet medical need: Facts and numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O., Jr.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Am. J. Med. Sci. 1980, 69, 491–497. [Google Scholar]
- Fearon, K.C.; Voss, A.C.; Hustead, D.S. Definition of cancer cachexia: Effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am. J. Clin. Nutr. 2006, 83, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Kern, K.; Norton, J. Cancer cachexia. J. Parenter. Enter. Nutr. 1988, 12, 286–298. [Google Scholar] [CrossRef]
- Mathew, S.J. InACTIVatINg cancer cachexia. Dis. Model. Mech. 2011, 4, 283–285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Henderson, S.E.; Makhijani, N.; Mace, T.A. Pancreatic Cancer-Induced Cachexia and Relevant Mouse Models. Pancreas 2018, 47, 937–945. [Google Scholar] [CrossRef]
- Lawson, D.H.; Richmond, A.; Nixon, D.W.; Rudman, D. Metabolic approaches to cancer cachexia. Annu. Rev. Nutr. 1982, 2, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet. Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, A.R.; Kamal, A.H.; LeBlanc, T.W.; Ma, J.D.; Baracos, V.E.; Roeland, E.J. Cancer cachexia: Beyond weight loss. J. Oncol. Pract. 2016, 12, 1163–1171. [Google Scholar] [CrossRef]
- Del Fabbro, E.; Inui, A.; Strasser, F. Overview of cancer cachexia. In Cancer Cachexia; Springer: Berlin, Germany, 2012; pp. 1–5. [Google Scholar]
- Tazi, E.; Errihani, H. Treatment of cachexia in oncology. Indian J. Palliat. Care 2010, 16, 129. [Google Scholar] [CrossRef]
- Naito, T. Emerging Treatment Options For Cancer-Associated Cachexia: A Literature Review. Ther. Clin. Risk. Manag. 2019, 15, 1253. [Google Scholar] [CrossRef]
- de Matos-Neto, E.M.; Lima, J.D.; de Pereira, W.O.; Figuerêdo, R.G.; Riccardi, D.M.; Radloff, K.; das Neves, R.X.; Camargo, R.G.; Maximiano, L.F.; Tokeshi, F.; et al. Systemic Inflammation in Cachexia—Is Tumor Cytokine Expression Profile the Culprit? Front. Immunol. 2015, 6, 629. [Google Scholar] [CrossRef]
- Moses, A.G.; Maingay, J.; Sangster, K.; Fearon, K.C.; Ross, J.A. Pro-inflammatory cytokine release by peripheral blood mononuclear cells from patients with advanced pancreatic cancer: Relationship to acute phase response and survival. Oncol. Rep. 2009, 21, 1091–1095. [Google Scholar]
- Carson, J.A.; Baltgalvis, K.A. Interleukin-6 as a key regulator of muscle mass during cachexia. Exerc. Sport Sci. Rev. 2010, 38, 168. [Google Scholar] [CrossRef]
- Inácio Pinto, N.; Carnier, J.; Oyama, L.M.; Otoch, J.P.; Alcântara, P.S.; Tokeshi, F.; Nascimento, C.M. Cancer as a proinflammatory environment: Metastasis and cachexia. Mediat. Inflamm. 2015, 2015, 791060. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.J.; Patel, B.M. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Markov, S.D.; Attri, K.S.; Vernucci, E.; King, R.J.; Dasgupta, A.; Grandgenett, P.M.; Hollingsworth, M.A.; Singh, P.K.; Yu, F.; et al. Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett. 2020, 484, 29–39. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-κB–mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [PubMed]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Huisman, C.; Buenafe, A.C.; Olson, B.; Michaelis, K.A.; Torres, E.R.S.; Jeng, S.; et al. Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. eLife 2020, 9, e54095. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef]
- Fearon, K.C.; Baracos, V.E. Cachexia in Pancreatic Cancer: New Treatment Options and Measures of Success. HPB 2010, 12, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Elkina, Y.; von Haehling, S.; Anker, S.D.; Springer, J. The role of myostatin in muscle wasting: An overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Mitch, W.E. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr. Opin. Support. Pa. 2011, 5, 334. [Google Scholar] [CrossRef] [PubMed]
- McCroskery, S.; Thomas, M.; Maxwell, L.; Sharma, M.; Kambadur, R. Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 2003, 162, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Klimek, M.E.B.; Aydogdu, T.; Link, M.J.; Pons, M.; Koniaris, L.G.; Zimmers, T.A. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem. Biophys. Res. Commun. 2010, 391, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.T.; Koopman, R.; Naim, T.; Léger, B.; Trieu, J.; Ibebunjo, C.; Lynch, G.S. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010, 24, 4433–4442. [Google Scholar] [CrossRef] [PubMed]
- Busquets, S.; Toledo, M.; Orpí, M.; Massa, D.; Porta, M.; Capdevila, E.; Padilla, N.; Frailis, V.; López-Soriano, F.J.; Han, H.; et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J. Cachexia Sarcopenia Muscle 2012, 3, 37–43. [Google Scholar] [CrossRef]
- Mathews, L.S.; Vale, W.W. Expression cloning of an activin receptor, a predicted transmembrane serine kinase. Cell 1991, 65, 973–982. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L.; Cheifetz, S.; Massague, J. Novel activin receptors: Distinct genes and alternative mRNA splicing generate a repertoire of serine/threonine kinase receptors. Cell 1992, 68, 97–108. [Google Scholar] [CrossRef]
- Oh, S.P.; Li, E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes Dev. 1997, 11, 1812–1826. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Reversing cachexia. Cell 2010, 142, 511–512. [Google Scholar] [CrossRef]
- Gatenby, R. Mathematical models of tumor-host interactions. Cancer J. 1998, 11, 289–293. [Google Scholar]
- Gatenby, R.A.; Maini, P.K. Mathematical oncology: Cancer summed up. Nature 2003, 421, 321. [Google Scholar] [CrossRef]
- Araujo, R.P.; McElwain, D.S. A history of the study of solid tumour growth: The contribution of mathematical modelling. Bull. Math. Biol. 2004, 66, 1039–1091. [Google Scholar] [CrossRef]
- Wheldon, T. Mathematical Models in Cancer Research; CRC Press: Baco Raton, FL, USA, 1988. [Google Scholar]
- Thomas, R.M.; Van Dyke, T.; Merlino, G.; Day, C.P. Concepts in cancer modeling: A brief history. Cancer Res. 2016, 76, 5921–5925. [Google Scholar] [CrossRef]
- Zhan, W.; Alamer, M.; Xu, X.Y. Computational modelling of drug delivery to solid tumour: Understanding the interplay between chemotherapeutics and biological system for optimised delivery systems. Adv. Drug Deliv. Rev. 2018, 132, 81–103. [Google Scholar] [CrossRef]
- Rockne, R.C.; Hawkins-Daarud, A.; Swanson, K.R.; Sluka, J.P.; Glazier, J.A.; Macklin, P.; Hormuth, D.A., II; Jarrett, A.M.; Lima, E.A.; Oden, J.T.; et al. The 2019 mathematical oncology roadmap. Phys. Biol. 2019, 16, 041005. [Google Scholar] [CrossRef]
- Hadjicharalambous, M.; Wijeratne, P.A.; Vavourakis, V. From tumour perfusion to drug delivery and clinical translation of in silico cancer models. Methods 2020. [Google Scholar] [CrossRef] [PubMed]
- Altrock, P.M.; Liu, L.L.; Michor, F. The mathematics of cancer: Integrating quantitative models. Nat. Rev. Cancer 2015, 15, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.T.; Cameron, A.N.; Gutierrez, J.B. A review of mathematical models for muscular dystrophy: A systems biology approach. PLoS Curr. Muscular Dystrophy 2018, 1. [Google Scholar] [CrossRef]
- Stephenson, E.R.; Kojouharov, H.V. A mathematical model of skeletal muscle regeneration. Math. Method Appl. Sci. 2018, 41, 8589–8602. [Google Scholar] [CrossRef]
- Hall, K.D.; Baracos, V.E. Computational Modeling of Cancer Cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2009, 11, 214–221. [Google Scholar] [CrossRef]
- Friesen, D.E.; Baracos, V.E.; Tuszynski, J.A. Modeling the energetic cost of cancer as a result of altered energy metabolism: Implications for cachexia. Theor. Biol. Med. Model. 2015, 12, 17. [Google Scholar] [CrossRef]
- Wilkie, K.P.; Aktar, F. Mathematically modelling inflammation as a promoter of tumour growth. Math. Med. Biol. 2020. [Google Scholar] [CrossRef]
- Rodriguez-Brenes, I.A.; Komarova, N.L.; Wodarz, D. Evolutionary dynamics of feedback escape and the development of stem-cell–driven cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 18983–18988. [Google Scholar] [CrossRef]
- Janssen, I.; Heymsfield, S.B.; Wang, Z.; Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J. Appl. Physiol. 2000, 89, 81–88. [Google Scholar] [CrossRef]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493. [Google Scholar] [CrossRef]
- Allbrook, D.; Han, M.; Hellmuth, A. Population of muscle satellite cells in relation to age and mitotic activity. J. Pathol 1971, 3, 233–243. [Google Scholar]
- Hellmuth, A.; Allbrook, D. Muscle satellite cell numbers during the postnatal period. J. Anat. 1971, 110, 503. [Google Scholar] [PubMed]
- Schultz, E. A quantitative study of the satellite cell population in postnatal mouse lumbrical muscle. Anat. Rec. 1974, 180, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.; Le Grand, F.; McKinnell, I.; Kuang, S. The molecular regulation of muscle stem cell function. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 323–331. [Google Scholar] [CrossRef]
- Snow, M.H. Myogenic cell formation in regenerating rat skeletal muscle injured by mincing II. An autoradiographic study. Anat. Rec. 1977, 188, 201–217. [Google Scholar] [CrossRef]
- Bischoff, R. A satellite cell mitogen from crushed adult muscle. Dev. Biol. 1986, 115, 140–147. [Google Scholar] [CrossRef]
- Schultz, E. Changes in the satellite cells of growing muscle following denervation. Anat. Rec. 1978, 190, 299–311. [Google Scholar] [CrossRef]
- Dhawan, J.; Rando, T.A. Stem cells in postnatal myogenesis: Molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005, 15, 666–673. [Google Scholar] [CrossRef]
- Pallafacchina, G.; Blaauw, B.; Schiaffino, S. Role of satellite cells in muscle growth and maintenance of muscle mass. Nutr. Metab. Carbiovasc. Dis. 2013, 23, S12–S18. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- Lander, A.D.; Gokoffski, K.K.; Wan, F.Y.; Nie, Q.; Calof, A.L. Cell lineages and the logic of proliferative control. PLoS Biol. 2009, 7, e1000015. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Gillespie, M.A.; Rudnicki, M.A. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2008, 2, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Charles River CD2F1 (CDF1) Mouse Growth Chart. 2018. Available online: https://www.criver.com/products-services/find-model/cd2f1-cdf1-mouse (accessed on 20 September 2018).
- Corana, A.; Marchesi, M.; Martini, C.; Ridella, S. Minimizing multimodal functions of continuous variables with the “simulated annealing” algorithm Corrigenda for this article is available here. ACM Trans. Math. Softw. 1987, 13, 262–280. [Google Scholar] [CrossRef]
- Bertsimas, D.; Tsitsiklis, J. Simulated annealing. Stat. Sci. 1993, 8, 10–15. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of cachexia in chronic disease states. Am. J. Med. Sci. 2015, 350, 250–256. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef]
- Bondulich, M.K.; Jolinon, N.; Osborne, G.F.; Smith, E.J.; Rattray, I.; Neueder, A.; Sathasivam, K.; Ahmed, M.; Ali, N.; Benjamin, A.C.; et al. Myostatin inhibition prevents skeletal muscle pathophysiology in Huntington’s disease mice. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Jejurikar, S.S.; Marcelo, C.L.; Kuzon, J.W. Skeletal muscle denervation increases satellite cell susceptibility to apoptosis. Plast. Reconstr. Surg. 2002, 110, 160–168. [Google Scholar] [CrossRef]
- Jejurikar, S.S.; Kuzon, W.M. Satellite cell depletion in degenerative skeletal muscle. Apoptosis 2003, 8, 573–578. [Google Scholar] [CrossRef]
- Ferreira, R.; Neuparth, M.J.; Ascensão, A.; Magalhães, J.; Vitorino, R.; Duarte, J.A.; Amado, F. Skeletal muscle atrophy increases cell proliferation in mice gastrocnemius during the first week of hindlimb suspension. Eur. J. Appl. Physiol. 2006, 97, 340–346. [Google Scholar] [CrossRef]
- Guo, B.S.; Cheung, K.K.; Yeung, S.S.; Zhang, B.T.; Yeung, E.W. Electrical stimulation influences satellite cell proliferation and apoptosis in unloading-induced muscle atrophy in mice. PLoS ONE 2012, 7, e30348. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Guttridge, D.C. Impaired regeneration: A role for the muscle microenvironment in cancer cachexia. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 54, pp. 82–91. [Google Scholar]
- Simeoni, M.; Magni, P.; Cammia, C.; De Nicolao, G.; Croci, V.; Pesenti, E.; Germani, M.; Poggesi, I.; Rocchetti, M. Predictive pharmacokinetic-pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 2004, 64, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Schmierer, B.; Hill, C.S. TGFβ–SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Li, Z.B.; Zhang, J.; Wagner, K.R. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J. Cell Sci. 2012, 125, 3957–3965. [Google Scholar]
- George Carlson, C.; Bruemmer, K.; Sesti, J.; Stefanski, C.; Curtis, H.; Ucran, J.; Lachey, J.; Seehra, J.S. Soluble activin receptor type IIB increases forward pulling tension in the mdx mouse. Muscle Nerve 2011, 43, 694–699. [Google Scholar] [CrossRef]
- Morine, K.J.; Bish, L.T.; Selsby, J.T.; Gazzara, J.A.; Pendrak, K.; Sleeper, M.M.; Barton, E.R.; Lee, S.J.; Sweeney, H.L. Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve 2010, 42, 722–730. [Google Scholar] [CrossRef]
- Pistilli, E.E.; Bogdanovich, S.; Goncalves, M.D.; Ahima, R.S.; Lachey, J.; Seehra, J.; Khurana, T. Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am. J. Pathol. 2011, 178, 1287–1297. [Google Scholar] [CrossRef]
- Akpan, I.; Goncalves, M.D.; Dhir, R.; Yin, X.; Pistilli, E.; Bogdanovich, S.; Khurana, T.; Ucran, J.; Lachey, J.; Ahima, R.S. The effects of a soluble activin type IIB receptor on obesity and insulin sensitivity. Int. J. Obes. 2009, 33, 1265–1273. [Google Scholar] [CrossRef]
- Zhang, C.; McFarlane, C.; Lokireddy, S.; Masuda, S.; Ge, X.; Gluckman, P.D.; Sharma, M.; Kambadur, R. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 2012, 55, 183–193. [Google Scholar] [CrossRef]
- Koncarevic, A.; Cornwall-Brady, M.; Pullen, A.; Davies, M.; Sako, D.; Liu, J.; Kumar, R.; Tomkinson, K.; Baker, T.; Umiker, B.; et al. A soluble activin receptor type IIb prevents the effects of androgen deprivation on body composition and bone health. Endocrinology 2010, 151, 4289–4300. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Read, B.P.; Edelstein, R.; Yang, N.; Pierson, C.R.; Stein, M.J.; Wermer-Colan, A.; Buj-Bello, A.; Lachey, J.L.; Seehra, J.S.; et al. Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am. J. Pathol. 2011, 178, 784–793. [Google Scholar] [CrossRef]
- Morrison, B.M.; Lachey, J.L.; Warsing, L.C.; Ting, B.L.; Pullen, A.E.; Underwood, K.W.; Kumar, R.; Sako, D.; Grinberg, A.; Wong, V.; et al. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2009, 217, 258–268. [Google Scholar] [CrossRef]
- Ohsawa, Y.; Hagiwara, H.; Nakatani, M.; Yasue, A.; Moriyama, K.; Murakami, T.; Tsuchida, K.; Noji, S.; Sunada, Y. Muscular atrophy of caveolin-3–deficient mice is rescued by myostatin inhibition. J. Clin. Investig. 2006, 116, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Morvan, F.; Rondeau, J.M.; Zou, C.; Minetti, G.; Scheufler, C.; Scharenberg, M.; Jacobi, C.; Brebbia, P.; Ritter, V.; Toussaint, G.; et al. Blockade of activin type II receptors with a dual anti-ActRIIA/IIB antibody is critical to promote maximal skeletal muscle hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 12448–12453. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef]
- Amthor, H.; Otto, A.; Vulin, A.; Rochat, A.; Dumonceaux, J.; Garcia, L.; Mouisel, E.; Hourdé, C.; Macharia, R.; Friedrichs, M.; et al. Muscle hypertrophy driven by myostatin blockade does not require stem/precursor-cell activity. Proc. Natl. Acad. Sci. USA 2009, 106, 7479–7484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; McPherron, A.C. Myostatin inhibition induces muscle fibre hypertrophy prior to satellite cell activation. J. Physiol. 2012, 590, 2151–2165. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Huynh, T.V.; Lee, Y.S.; Sebald, S.M.; Wilcox-Adelman, S.A.; Iwamori, N.; Lepper, C.; Matzuk, M.M.; Fan, C.M. Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proc. Natl. Acad. Sci. USA 2012, 109, E2353–E2360. [Google Scholar] [CrossRef] [PubMed]
- Myrianthefs, P.; Batistaki, C. Cancer cachexia and immunomodulation. J. BUON. 2005, 10, 181–188. [Google Scholar] [PubMed]
- Porporato, P. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, G.W.P.d.; Farkas, J.; Dora, E.; von Haehling, S.; Lainscak, M. Cancer Cachexia and Related Metabolic Dysfunction. Int. J. Mol. Sci. 2020, 21, 2321. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Parameters | ||
| day | day | |
| day | mm | |
| Initial Conditions | ||
| g | g | g/mm |
| mm | mm | |
| Model Equations | Fitted Parameter Values | ||||
|---|---|---|---|---|---|
| 1. | Exp-linear model (5) | days | mmdays | mm | |
| 2. | Cancer-Cachexia (6) and (7) | days | days | mm | |
| 3. | Group A ICs | mm | mm | ||
| 4. | Group B ICs | mm | mm | ||
| 5. | sActRIIB Treatment (9) and (10) Group A | ||||
| 6. | Group B | ||||
| Parameter | Relative Change Satellite Cells | Relative Change Muscle Cells | ||
|---|---|---|---|---|
| Decrease | Increase | Decrease | Increase | |
| 22,541.6% | ||||
| Cachexia Parameters | ||||
| Parameter | Relative Change Satellite Cells | Relative Change Muscle Cells | ||
| decrease | increase | decrease | increase | |
| Treatment Parameters (Fit to Group A) | ||||
| parameter | decrease | increase | decrease | increase |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farhang-Sardroodi, S.; Wilkie, K.P. Mathematical Model of Muscle Wasting in Cancer Cachexia. J. Clin. Med. 2020, 9, 2029. https://doi.org/10.3390/jcm9072029
Farhang-Sardroodi S, Wilkie KP. Mathematical Model of Muscle Wasting in Cancer Cachexia. Journal of Clinical Medicine. 2020; 9(7):2029. https://doi.org/10.3390/jcm9072029
Chicago/Turabian StyleFarhang-Sardroodi, Suzan, and Kathleen P. Wilkie. 2020. "Mathematical Model of Muscle Wasting in Cancer Cachexia" Journal of Clinical Medicine 9, no. 7: 2029. https://doi.org/10.3390/jcm9072029
APA StyleFarhang-Sardroodi, S., & Wilkie, K. P. (2020). Mathematical Model of Muscle Wasting in Cancer Cachexia. Journal of Clinical Medicine, 9(7), 2029. https://doi.org/10.3390/jcm9072029