Neutrophil Phenotypes in Coronary Artery Disease

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
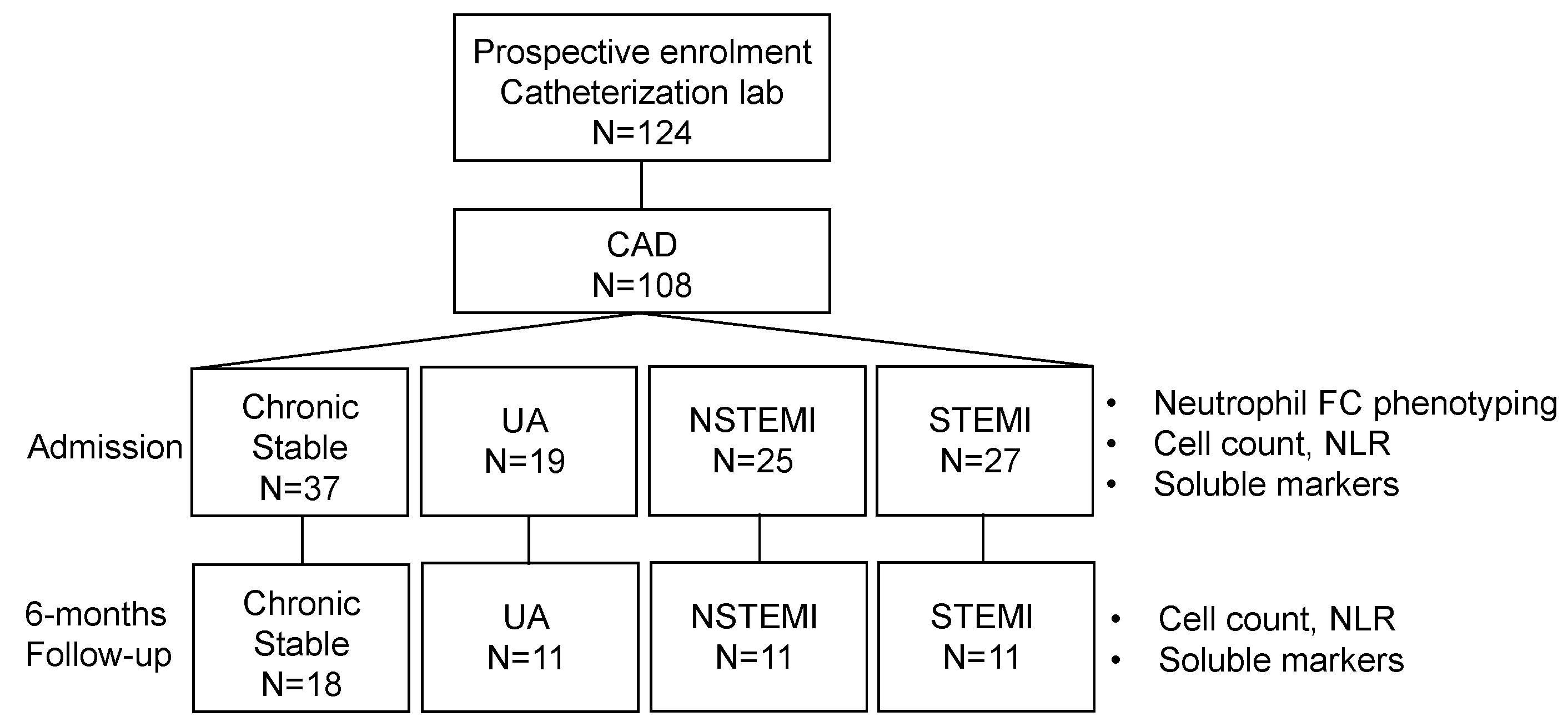
2.1. Patients and Study Design
2.2. Blood Samples, Sera and Platelet Poor Plasma (PPP) Preparation, Laboratory Measurements
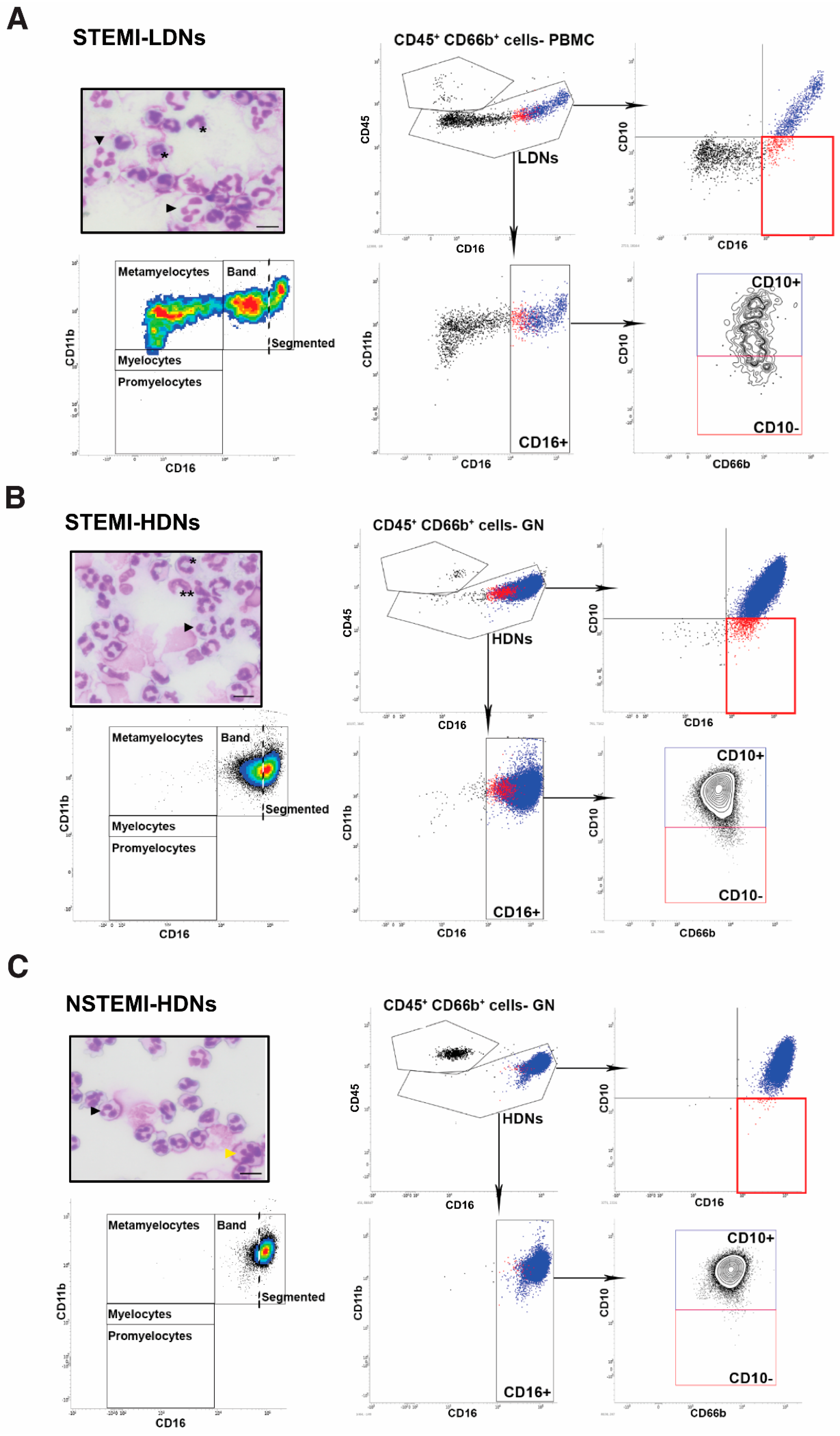
2.3. Normal and Low-Density Neutrophil Phenotyping
2.4. Statistics
3. Results
3.1. Patient Characteristics, Inflammatory and Conventional Neutrophil Markers
3.2. Neutrophil Phenotypes in ACS
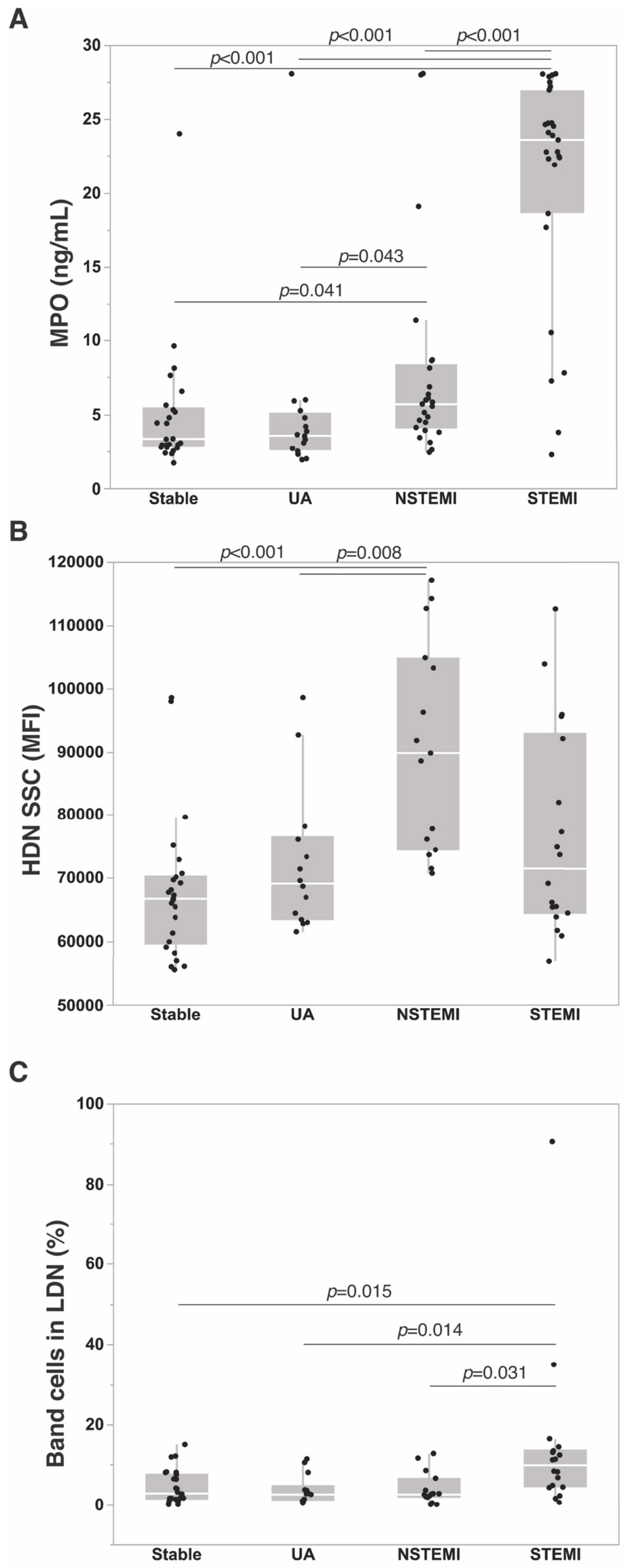
3.3. Neutrophil Markers in ACS
3.4. Evolution of Neutrophil Markers during Follow-Up
4. Discussion
4.1. Neutrophil Phenotypes in ACS
4.2. Soluble Neutrophil Markers in ACS
4.3. Neutrophil Dynamics after ACS
4.4. Clinical Implications
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koenig, W. Low-grade inflammation modifies cardiovascular risk even at very low ldl-c levels: Are we aiming for a dual target concept? Circulation 2018, 138, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Mani, P.; Puri, R.; Schwartz, G.G.; Nissen, S.E.; Shao, M.; Kastelein, J.J.P.; Menon, V.; Lincoff, A.M.; Nicholls, S.J. Association of initial and serial c-reactive protein levels with adverse cardiovascular events and death after acute coronary syndrome: A secondary analysis of the vista-16 trial. JAMA Cardiol. 2019, 4, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Bhat, T.; Teli, S.; Rijal, J.; Bhat, H.; Raza, M.; Khoueiry, G.; Meghani, M.; Akhtar, M.; Costantino, T. Neutrophil to lymphocyte ratio and cardiovascular diseases: A review. Expert Rev. Cardiovasc. Therapy 2013, 11, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Guasti, L.; Dentali, F.; Castiglioni, L.; Maroni, L.; Marino, F.; Squizzato, A.; Ageno, W.; Gianni, M.; Gaudio, G.; Grandi, A.M.; et al. Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34,000 subjects. Thromb. Haemost. 2011, 106, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Eliot, M.; Koestler, D.C.; Wu, W.C.; Kelsey, K.T. Association of neutrophil-to-lymphocyte ratio with mortality and cardiovascular disease in the jackson heart study and modification by the duffy antigen variant. JAMA Cardiol. 2018, 3, 455–462. [Google Scholar] [CrossRef]
- Templeton, A.J.; McNamara, M.G.; Seruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocana, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil diversity in health and disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Mackey, J.B.G.; Coffelt, S.B.; Carlin, L.M. Neutrophil maturity in cancer. Front. Immunol. 2019, 10, 1912. [Google Scholar] [CrossRef]
- Lancellotti, P.; Marechal, P.; Donis, N.; Oury, C. Inflammation, cardiovascular disease, and cancer: A common link with far-reaching implications. Eur. Heart J. 2019, 40, 3910–3912. [Google Scholar] [CrossRef]
- Franck, T.; Kohnen, S.; Boudjeltia, K.Z.; Van Antwerpen, P.; Bosseloir, A.; Niesten, A.; Gach, O.; Nys, M.; Deby-Dupont, G.; Serteyn, D. A new easy method for specific measurement of active myeloperoxidase in human biological fluids and tissue extracts. Talanta 2009, 80, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Marini, O.; Costa, S.; Bevilacqua, D.; Calzetti, F.; Tamassia, N.; Spina, C.; De Sabata, D.; Tinazzi, E.; Lunardi, C.; Scupoli, M.T.; et al. Mature cd10+ and immature cd10− neutrophils present in g-csf-treated donors display opposite effects on t cells. Blood 2017, 129, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Vergouwe, Y.; Royston, P.; Moons, K.G.; Altman, D.G. Development and validation of a prediction model with missing predictor data: A practical approach. J. Clin. Epidemiol. 2010, 63, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, G.; Grauen Larsen, H.; Yndigegn, T.; Szabo, I.A.; Mares, R.G.; de Camp, L.; Weiland, M.; Tomas, L.; Goncalves, I.; Nilsson, J.; et al. Inhibition of pro-inflammatory myeloid cell responses by short-term s100a9 blockade improves cardiac function after myocardial infarction. Eur. Heart J. 2019, 40, 2713–2723. [Google Scholar] [CrossRef]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Munzel, T.; Simoons, M.L.; Hamm, C.W. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef]
- Brennan, M.L.; Penn, M.S.; Van Lente, F.; Nambi, V.; Shishehbor, M.H.; Aviles, R.J.; Goormastic, M.; Pepoy, M.L.; McErlean, E.S.; Topol, E.J.; et al. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 2003, 349, 1595–1604. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012, 110, 875–888. [Google Scholar] [CrossRef]
- Naruko, T.; Ueda, M.; Haze, K.; van der Wal, A.C.; van der Loos, C.M.; Itoh, A.; Komatsu, R.; Ikura, Y.; Ogami, M.; Shimada, Y.; et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002, 106, 2894–2900. [Google Scholar] [CrossRef]
- Pertiwi, K.R.; van der Wal, A.C.; Pabittei, D.R.; Mackaaij, C.; van Leeuwen, M.B.; Li, X.; de Boer, O.J. Neutrophil extracellular traps participate in all different types of thrombotic and haemorrhagic complications of coronary atherosclerosis. Thromb. Haemost. 2018, 118, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Zhang, Y.; Lee, C.; Li, L. Novel reprogramming of neutrophils modulates inflammation resolution during atherosclerosis. Sci. Adv. 2019, 5, eaav2309. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Scapini, P.; Marini, O.; Tecchio, C.; Cassatella, M.A. Human neutrophils in the saga of cellular heterogeneity: Insights and open questions. Immunol. Rev. 2016, 273, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Hacbarth, E.; Kajdacsy-Balla, A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986, 29, 1334–1342. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef]
- Teague, H.L.; Varghese, N.J.; Tsoi, L.C.; Dey, A.K.; Garshick, M.S.; Silverman, J.I.; Baumer, Y.; Harrington, C.L.; Stempinski, E.; Elnabawi, Y.A.; et al. Neutrophil subsets, platelets, and vascular disease in psoriasis. JACC Basic Transl. Sci. 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Crea, F.; Libby, P. Acute coronary syndromes: The way forward from mechanisms to precision treatment. Circulation 2017, 136, 1155–1166. [Google Scholar] [CrossRef]
- Libby, P. Superficial erosion and the precision management of acute coronary syndromes: Not one-size-fits-all. Eur. Heart J. 2017, 38, 801–803. [Google Scholar] [CrossRef][Green Version]
- Brezinski, M.E. Comparing the risk factors of plaque rupture and failed plaque healing in acute coronary syndrome. JAMA Cardiol. 2019, 4, 329–331. [Google Scholar] [CrossRef]
- Ferrante, G.; Nakano, M.; Prati, F.; Niccoli, G.; Mallus, M.T.; Ramazzotti, V.; Montone, R.A.; Kolodgie, F.D.; Virmani, R.; Crea, F. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: A clinicopathological study. Circulation 2010, 122, 2505–2513. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in st-elevation acute coronary syndrome are predictors of st-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.J.; Robertson, S.; Barraclough, J.; Xia, Q.; Mallat, Z.; Bursill, C.; Celermajer, D.S.; Patel, S. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J. Am. Heart Assoc. 2015, 4, e002128. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular neutrophil proteases are efficient regulators of il-1, il-33, and il-36 cytokine activity but poor effectors of microbial killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Monserrat-Mesquida, M.; Quetglas-Llabres, M.; Capo, X.; Bouzas, C.; Mateos, D.; Pons, A.; Tur, J.A.; Sureda, A. Metabolic syndrome is associated with oxidative stress and proinflammatory state. Antioxidants 2020, 9, 236. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; Infusino, F.; Maestrini, V.; Mancone, M.; Fedele, F. Myocardial ischemia and diabetes mellitus: Role of oxidative stress in the connection between cardiac metabolism and coronary blood flow. J. Diabetes Res. 2019, 2019, 9489826. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Stable n = 37 | UA n = 19 | NSTEMI n = 25 | STEMI n = 27 | p | |
|---|---|---|---|---|---|
| Age (yrs) | 69 ± 9 | 67 ± 11 | 63 ± 12 | 64 ± 10 | 0.081 |
| Male gender, n (%) | 25 (67.6) | 17 (89.5) | 19 (76) | 20 (74.1) | 0.356 |
| Smoking, n (%) | 24 (64.9) | 12 (63.2) | 15 (60) | 22 (81.5) | 0.343 |
| Body mass index | 27.8 (25.3–31.0) | 28.7 (24.6–34.3) | 26.8 (25.4–29.7) | 26.9 (24.2–31.0) | 0.616 |
| Hypertension, n (%) | 29 (78.4) | 16 (84.2) | 14 (56) | 18 (66.7) | 0.133 |
| Hypercholesterolemia, n (%) | 25 (67.6) | 13 (68.4) | 13 (52) | 13 (48.1) | 0.302 |
| Diabetes, n (%) | 13 (35.1) | 8 (42.1) | 5 (20) | 8 (29.6) | 0.425 |
| Chronic renal failure, n (%) | 5 (13.5) | 3 (15.8) | 1 (4) | 4 (14.8) | 0.530 |
| Chronic inflammatory disease, n (%) | 7 (18.9) | 2 (10.5) | 0 (0) | 4 (14.8) | 0.107 |
| Active cancer, n (%) | 0 (0) | 2 (10.5) | 2 (8) | 4 (14.8) | 0.069 |
| History of DVT, n (%) | 2 (5.4) | 1 (5.3) | 0 (0) | 2 (7.4) | 0.669 |
| History of stroke, n (%) | 4 (10.8) | 2 (10.5) | 0 (0) | 0 (0) | 0.086 |
| History of MI, n (%) | 11 (29.7) | 4 (21.1) | 1 (4) | 7 (25.9) | 0.095 |
| History of CABG, n (%) | 6 (16.2) | 2 (10.5) | 3 (12) | 1 (3.7) | 0.475 |
| History of PCI, n (%) | 12 (32.4) | 10 (52.6) | 5 (20) | 8 (29.6) | 0.145 |
| Aspirin, n (%) | 31 (83.8) | 15 (78.9) | 9 (36) | 15 (55.6) | 0.0005 |
| DAPT, n (%) | 5 (13.5) | 1 (5.3) | 3 (12) | 3 (11.1) | 0.886 |
| Anticoagulant, n (%) | 1 (2.7) | 1 (5.3) | 0 (0) | 1 (3.7) | 0.878 |
| Lipid-lowering drug, n (%) | 28 (75.7) | 14 (73.7) | 8 (32) | 12 (44.4) | 0.001 |
| hs-cTnT (ng/L) | 13 (8–23) | 16 (10–20) | 689 (303–1304) a,b | 293 (19–1438) a,b | <0.0001 |
| CK-MB (μg/L) | 2.80 (2.00–4.50) | 2.50 (1.80–3.50) | 22.50 (14.80-60.00) a,b | 4.94 (2.50–47.56) a,b | <0.0001 |
| Creatinine (mg/dL) | 1.08 (0.90–1.30) | 1.00 (0.88–1.23) | 0.94 (0.81–1.09) | 0.95 (0.86–1.13) | 0.207 |
| Total cholesterol (mg/dL) | 155 (138–181) | 142 (126–208) | 170 (143–211) | 181 (142–195) | 0.299 |
| LDL (mg/dL) | 87 (68–106) | 78 (72–127) | 109 (91–137) | 111 (73–143) | 0.052 |
| HDL (mg/dL) | 44 (36–53) | 35 (29–45) | 39 (34–48) | 39 (34–49) | 0.162 |
| Triglycerides (mg/dL) | 113 (88–180) | 139 (100–197) | 136 (101–165) | 82 (62–114) b,c | 0.003 |
| Apo A-I (g/dL) | 1.32 (1.11–1.48) | 1.14 (1.03–1.40) | 1.29 (1.14–1.44) | 1.21 (1.08–1.40) | 0.497 |
| Apo B (g/dL) | 0.77 (0.61–0.90) | 0.79 (0.74–1.05) | 0.95 (0.85–1.08) | 0.91 (0.70–1.07) | 0.081 |
| Lipoprotein (a) (nmol/L) | 15 (8–70) | 35 (10–118) | 33 (10–135) | 25 (9–116) | 0.727 |
| PCSK9 (pg/mL) | 45514 (29768–85118) | 55279 (23275–74669) | 58512 (27008–78619) | 76762 (33957–127092) | 0.307 |
| Stable n = 37 | UA n = 19 | NSTEMI n = 25 | STEMI n = 27 | p | |
|---|---|---|---|---|---|
| Lymphocyte count (1000/μL) | 1.41 (1.06–1.77) | 1.82 (0.95–2.24) | 1.79 (1.39–2.20) | 1.54 (1.20-2.13) | 0.185 |
| Monocyte count (1000/μL) | 0.46 (0.33–0.58) | 0.53 (0.39–0.76) | 0.73 (0.59–0.92) a | 0.64 (0.46-0.86) | <0.0001 |
| Neutrophil count (1000/μL) | 3.0 (2.1–4.5) | 3.9 (2.5–5.2) | 5.8 (4.6–8.3) a,b | 7.4 (6.4-9.7) a,b | <0.0001 |
| Eosinophil count (1000/μL) | 0.12 (0.06–0.25) | 0.12 (0.05–0.24) | 0.06 (0.04–0.13) | 0.07 (0.04-0.13) | 0.094 |
| Basophil count (1000/μL) | 0.05 (0.04–0.08) | 0.06 (0.04–0.07) | 0.06 (0.04–0.08) | 0.06 (0.04–0.07) | 0.98 |
| Haematocrit (%) | 42 (41–47) | 44 (41–48) | 45 (42–48) | 42 (38–47) | 0.346 |
| Platelet count (1000/μL) | 255 ± 71 | 243 ± 63 | 247 ± 81 | 279 ± 72 | 0.309 |
| Mean platelet volume (fL) | 7.8 (7.4–8.3) | 7.8 (7.6–8.8) | 7.8 (7.1–8.3) | 7.6 (7.0–8.5) | 0.455 |
| NLR | 2.3 (1.8–3.2) | 2.2 (1.7–3.1) | 3.7 (2.3–5.3) a | 4.9 (2.7–7.5) a,b | <0.0001 |
| PLR | 157 (141–244) | 138 (112–239) | 144 (119–170) | 174 (122–209) | 0.212 |
| hs-CRP (mg/L) | 2.93 (0.84–6.87) | 1.26 (0.58–4.51) | 6.09 (2.62–19.55) | 2.51 (0.68–13.22) | 0.082 |
| IL-6 (pg/mL) | 1.7 (0.4–3.4) | 0.7 (0.2–1.5) | 2.8 (0.9–13.0) | 3.0 (0.8–12.3) | 0.033 |
| S100A9 (pg/mL) | 213 (142–399) | 250 (126–361) | 273 (213–466) | 431 (292–621) a,b | 0.008 |
| Active MPO (ng/mL) | 1.5 (1.2–3.2) | 1.4 (1.1–1.9) | 2.0 (1.5–3.2) | 8.4 (4.9–13.2) a,b,c | <0.0001 |
| Total MPO (ng/mL) | 4.1 (2.9–7.2) | 3.7 (2.6–5.5) | 5.7 (4.1–8.1) | 23.6 (18.6–27.0) a,b,c | <0.0001 |
| Nucleosomes (AU) | 0.04 (0.02–0.11) | 0.03 (0.02–0.06) | 0.06 (0.04–0.15) | 0.09 (0.05–0.22) a,b | 0.006 |
| Stable (n = 37) | UA (n = 19) | NSTEMI (n = 25) | STEMI (n = 27) | p | |
|---|---|---|---|---|---|
| HDN | |||||
| SSC | 67,704 (61,896–74,737) | 68,632 (63,191–76,788) | 83,690 (72,344–99,563) a,b | 72,985 (64,586–92,739) | 0.034 |
| FSC | 100,264 (90,863–111,559) | 98,243 (92,717–108,761) | 119,603 (104,467–130,478) | 105,721 (90,881–120,638) | 0.108 |
| CD11b (MFI) | 12,108 ± 4070 | 12,236 ± 4527 | 13,798 ± 4300 | 14,150 ± 3541 | 0.215 |
| CD10 (MFI) | 12,254 ± 3501 | 12,840 ± 3353 | 12,170 ± 3931 | 11,756 ± 4358 | 0.463 |
| CD16 (MFI) | 94,082 (83,307–137,384) | 97,458 (74,226–113,011) | 82,009 (65,544–102,078) | 83,194 (65,209–108,398) | 0.344 |
| Band cells (%) | 0.03 (0.01–0.08) | 0.01 (0.00–0.06) | 0.02 (0.01–0.09) | 0.11 (0.03–0.36) a,b | 0.019 |
| LDN | |||||
| % in PBMC | 0.86 (0.36–1.80) | 0.88 (0.39–1.77) | 0.95 (0.38–2.10) | 1.53 (0.57–6.69) | 0.272 |
| Band cells (%) | 2.5 (1.2–6.8) | 2.7 (1.0–6.5) | 2.7 (1.8–7.3) | 9.5 (4.0–13.7) a,b,c | 0.007 |
| Variable | Comparison | Unit | Odds Ratio (95% CI) | p |
|---|---|---|---|---|
| hs-cTnT (ng/L) | NSTEMI vs. Stable | 10 | 1.062 (1.020–1.105) | 0.003 |
| STEMI vs. Stable | 10 | 1.061 (1.019–1.104) | 0.004 | |
| Unstable vs. Stable | 10 | 0.953 (0.824–1.103) | 0.288 | |
| NSTEMI vs. Unstable | 10 | 1.114 (0.961–1.291) | 0.152 | |
| STEMI vs. Unstable | 10 | 1.113 (0.960-1.290) | 0.156 | |
| NSTEMI vs. STEMI | 10 | 0.999 (0.994–1.004) | 0.722 | |
| Total MPO (ng/mL) | NSTEMI vs. Stable | 1 | 1.182 (0.919–1.519) | 0.193 |
| STEMI vs. Stable | 1 | 1.434 (1.119–1.837) | <0.0001 | |
| Unstable vs. Stable | 1 | 0.975 (0.857–1.11) | 0.703 | |
| NSTEMI vs. Unstable | 1 | 1.212 (0.942–1.559) | 0.135 | |
| STEMI vs. Unstable | 1 | 1.47 (1.146–1.886) | 0.002 | |
| STEMI vs. NSTEMI | 1 | 1.213 (1.100–1.338) | 0.0001 | |
| HDN SSC | NSTEMI vs. Stable | 10000 | 3.828 (1.033–14.184) | 0.045 |
| STEMI vs. Stable | 10000 | 3.029 (0.899–10.202) | 0.074 | |
| Unstable vs. Stable | 10000 | 1.101 (0.603–2.007) | 0.755 | |
| NSTEMI vs. Unstable | 10000 | 3.478 (0.990–12.217) | 0.052 | |
| STEMI vs. Unstable | 10000 | 2.752 (0.843–8.977) | 0.094 | |
| STEMI vs. NSTEMI | 10000 | 0.791 (0.423–1.478) | 0.462 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maréchal, P.; Tridetti, J.; Nguyen, M.-L.; Wéra, O.; Jiang, Z.; Gustin, M.; Donneau, A.-F.; Oury, C.; Lancellotti, P. Neutrophil Phenotypes in Coronary Artery Disease. J. Clin. Med. 2020, 9, 1602. https://doi.org/10.3390/jcm9051602
Maréchal P, Tridetti J, Nguyen M-L, Wéra O, Jiang Z, Gustin M, Donneau A-F, Oury C, Lancellotti P. Neutrophil Phenotypes in Coronary Artery Disease. Journal of Clinical Medicine. 2020; 9(5):1602. https://doi.org/10.3390/jcm9051602
Chicago/Turabian StyleMaréchal, Patrick, Julien Tridetti, Mai-Linh Nguyen, Odile Wéra, Zheshen Jiang, Maxime Gustin, Anne-Françoise Donneau, Cécile Oury, and Patrizio Lancellotti. 2020. "Neutrophil Phenotypes in Coronary Artery Disease" Journal of Clinical Medicine 9, no. 5: 1602. https://doi.org/10.3390/jcm9051602
APA StyleMaréchal, P., Tridetti, J., Nguyen, M.-L., Wéra, O., Jiang, Z., Gustin, M., Donneau, A.-F., Oury, C., & Lancellotti, P. (2020). Neutrophil Phenotypes in Coronary Artery Disease. Journal of Clinical Medicine, 9(5), 1602. https://doi.org/10.3390/jcm9051602