Interactions between Amyloid-Β Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease

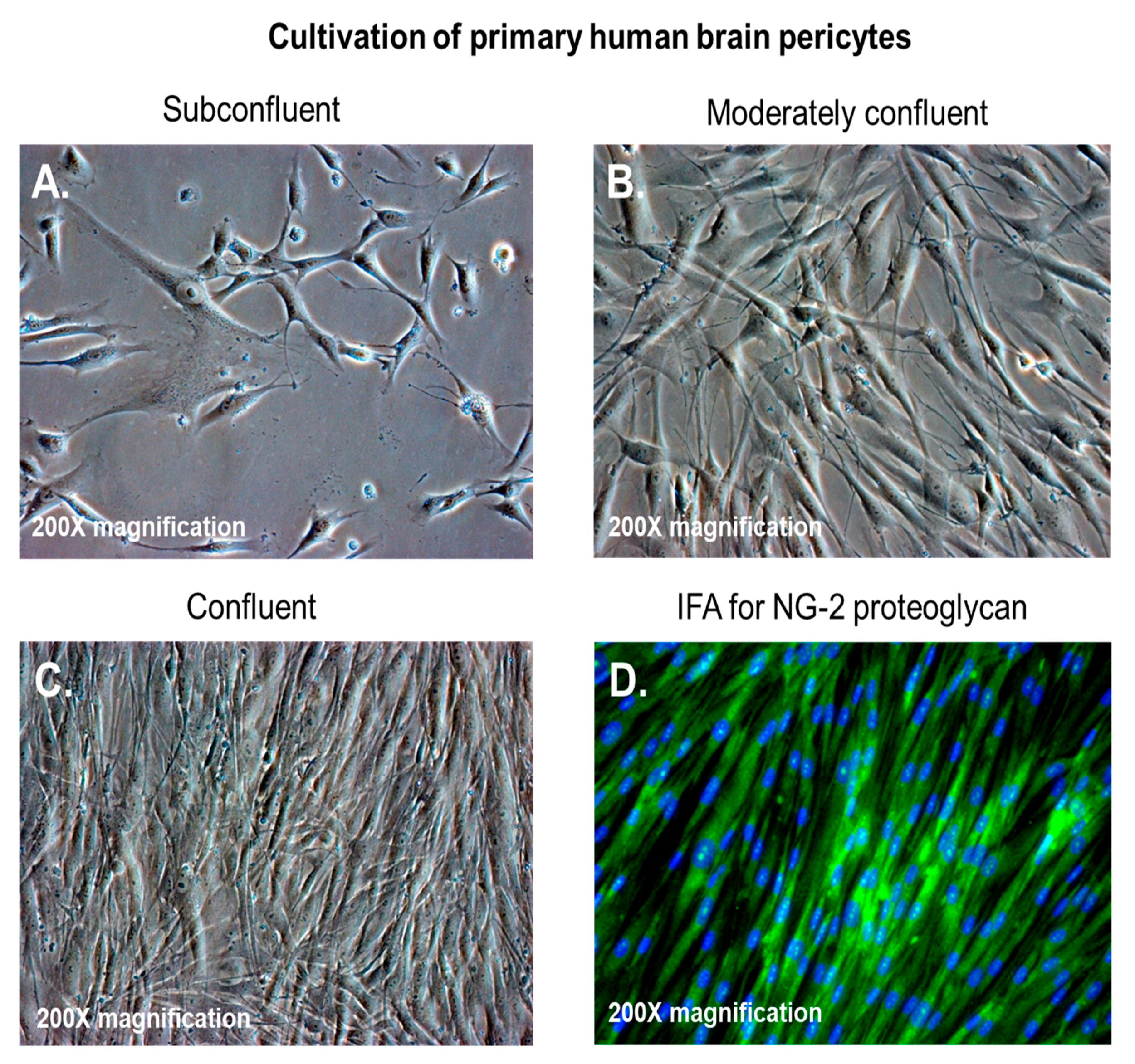{kind=link}
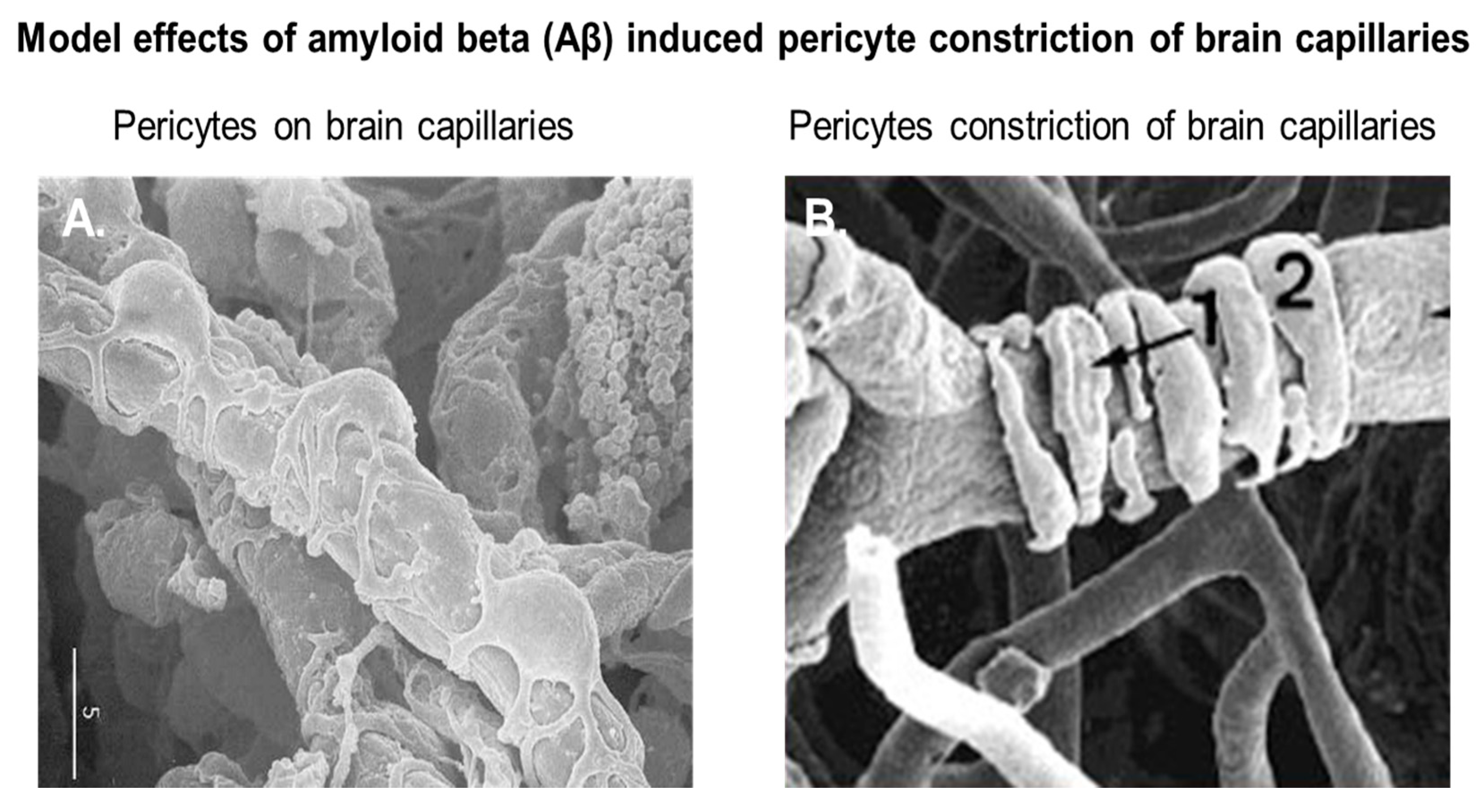{kind=link}
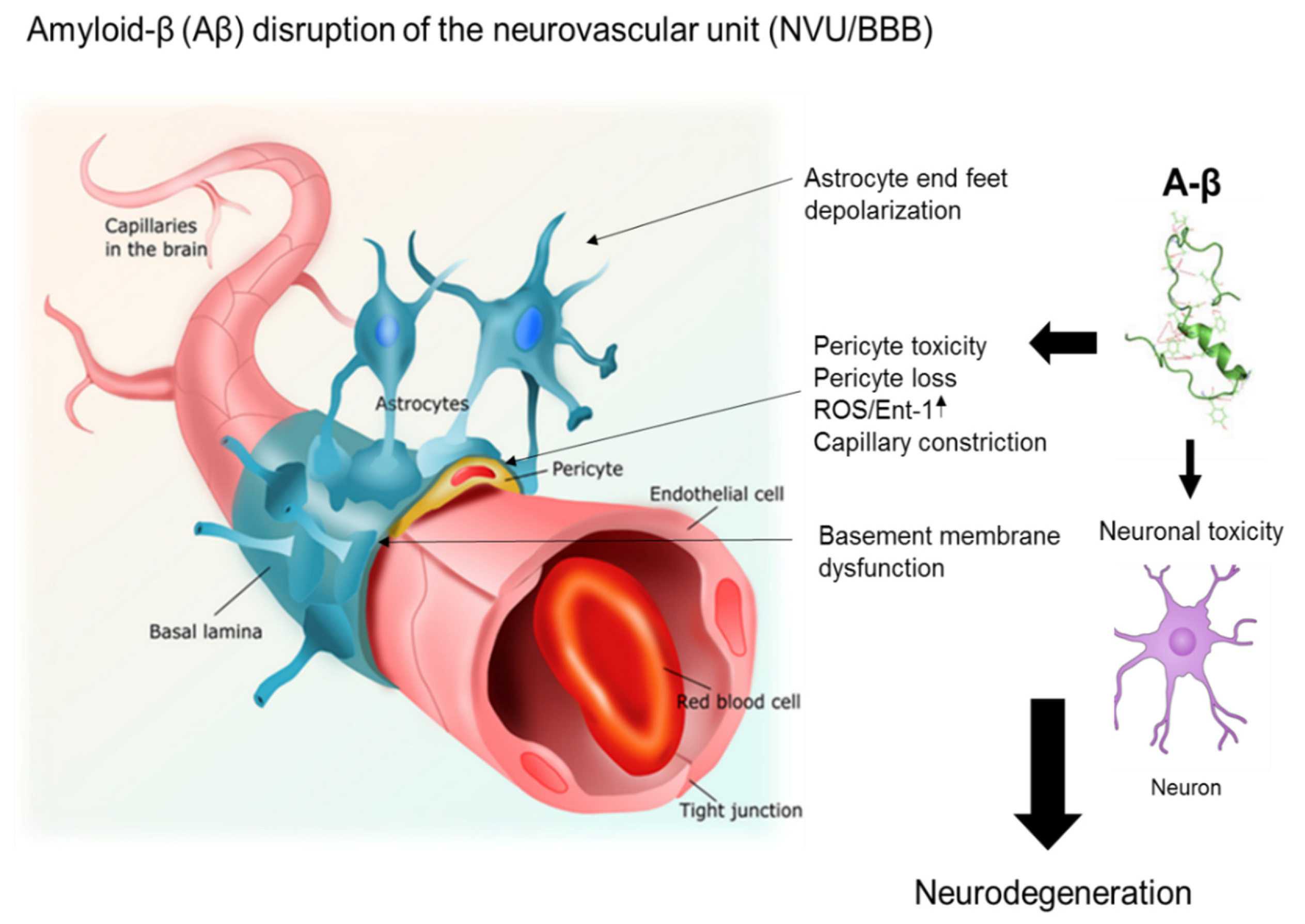{kind=link}
Abstract
1. Introduction
2. Aβ Proteins and Pericytes
3. The Interaction between Aβ Proteins and Brain Pericytes in AD Pathobiology
4. Aβ Proteins and Retinal Pericytes in the Ocular Compartment of AD Patients
5. Strategies to Replace Pericytes in the CNS or Protect Them from the Effects of Aβ
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Olichney, J.M.; Thal, L.J.; Mirra, S.S.; Morris, J.C.; Beekly, D.; Heyman, A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology 1996, 46, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Murray, M.E.; Frank, R.D.; DeTure, M.; Yamazaki, Y.; Tachibana, M.; Atagi, Y.; Davis, M.D.; Liu, C.C.; Zhao, N.; et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 2, 111–128. [Google Scholar]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 7, 419–434. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 1, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Zehendner, C.M.; White, R.; Hedrich, J.; Luhmann, H.J. A neurovascular blood-brain barrier in vitro model. Methods Mol. Biol. 2014, 1135, 403–413. [Google Scholar] [PubMed]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit-oncept review. Acta Physiol. (Oxf.) 2014, 4, 790–798. [Google Scholar] [CrossRef] [PubMed]
- ElAli, A.; Thériault, P.; Rivest, S. The role of pericytes in neurovascular unit remodeling in brain disorders. Int. J. Mol. Sci. 2014, 4, 6453–6474. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Hamilton, N.B.; Attwell, D.; Hall, C.N. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenerg. 2010, 2, 1–14. [Google Scholar] [CrossRef]
- Korn, C.; Augustin, H.G. Mechanisms of vessel pruning and regression. Dev. Cell 2015, 34, 5–17. [Google Scholar] [CrossRef]
- Alcendor, D.J.; Charest, A.M.; Zhu, W.Q.; Vigil, H.E.; Knobel, S.M. Infection and upregulation of proinflammatory cytokines in human brain vascular pericytes by human cytomegalovirus. J. Neuroinflamm. 2012, 9, 95. [Google Scholar] [CrossRef]
- Salmina, A.B.; Komleva, Y.K.; Lopatina, O.L.; Birbrair, A. Pericytes in Alzheimer’s Disease: Novel Clues to Cerebral Amyloid Angiopathy Pathogenesis. Adv. Exp. Med. Biol. 2019, 1147, 147–166. [Google Scholar]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 10, 1500–1513. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, S87–S100. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, 6450. [Google Scholar] [CrossRef] [PubMed]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L., Jr.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid β protein (A β) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A β 40 or A β 42(43). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 57. [Google Scholar] [CrossRef]
- Alcendor, D.J. Human Vascular Pericytes and Cytomegalovirus Pathobiology. Int. J. Mol. Sci. 2019, 6, 1456. [Google Scholar] [CrossRef]
- Baranello, R.J.; Bharani, K.L.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, A.M.; Sambamurti, K. Amyloid-β protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimer Res. 2015, 32–46. [Google Scholar] [CrossRef]
- Ruzali, W.A.; Kehoe, P.G.; Love, S. Influence of LRP-1 and apolipoprotein E on amyloid-β uptake and toxicity to cerebrovascular smooth muscle cells. J. Alzheimers Dis. 2013, 1, 95–110. [Google Scholar] [CrossRef]
- Sagare, A.P.; Deane, R.; Zetterberg, H.; Wallin, A.; Blennow, K.; Zlokovic, B.V. Impaired lipoprotein receptor-mediated peripheral binding of plasma amyloid-β is an early biomarker for mild cognitive impairment preceding Alzheimer’s disease. J. Alzheimers. Dis. 2011, 24, 25–34. [Google Scholar] [CrossRef]
- Shinohara, M.; Fujioka, S.; Murray, M.E.; Wojtas, A.; Baker, M.; Rovelet-Lecrux, A.; Rademakers, R.; Das, P.; Parisi, J.E.; Graff-Radford, N.R.; et al. Regional distribution of synaptic markers and APP correlate with distinct clinicopathological features in sporadic and familial Alzheimer’s disease. Brain 2014, 137, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Zur cellulose. Arch. Pathol. 1854, 6, 416–426. [Google Scholar] [CrossRef]
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Friedreich, N.; Kekulé, A. Zur amyloidfrage. Virchows Arch. 1859, 16, 50–65. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.-I.; Masters, C.L.; Merlini, G.; Saraiva, M.J.; Sipe, J.D. Amyloid: Toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2005, 12, 1–4. [Google Scholar] [CrossRef]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 12, 755–773. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillary oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef]
- Kayed, R.; Pensalfini, A.; Margol, L.; Sokolov, Y.; Sarsoza, F.; Head, E.; Hall, J.; Glabe, C. Annular protofibrils are a structurally and functionally distinct typeof amyloid oligomer. J. Biol. Chem. 2009, 284, 4230–4237. [Google Scholar] [CrossRef]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef]
- Ma, B.; Nussinov, R. Polymorphic C-terminal beta-sheet interactions determine the formation of fibril or amyloid beta-derived diffusible ligand-like globulomer for the alzheimer A{beta}42 dodecamer. J. Biol. Chem. 2010, 285, 37102–37110. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Kurouski, D.; Rasool, S.; Milton, S.; Wu, J.W.; Uversky, V.N.; Lednev, I.K.; Glabe, C.G. Structural differences between amyloid beta oligomers. Biochem. Biophys. Res. Commun. 2016, 4, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. Yearly Growth of Total Structures. 2018. Available online: http://www.rcsb.org/pdb/statistics/contentGrowthChart.do?content=total (accessed on 20 September 2018).
- Alzheimer, A. Über einen eigenartigen schweren Erkrankungsprozeβ der Hirnrincle. Neurol. Cent. 1906, 25, 1134. [Google Scholar]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. Psych. Gerichtl. Med. 1907, 641, 46–48. [Google Scholar]
- Lott, I.T.; Head, E. Alzheimer disease and Down syndrome: Factors in pathogenesis. Neurobiol. Aging 2005, 26, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid beta peptides. J. Physiol. 2006, 575, 5–10. [Google Scholar] [CrossRef]
- Du, W.J.; Guo, J.-J.; Gao, M.-T.; Hu, S.-Q.; Dong, X.-Y.; Han, Y.-F.; Liu, F.-F.; Jiang, S.; Sun, Y. Brazilin inhibits amyloid beta-protein fibrillogenesis, remodels amyloid fibrils and reduces amyloid cytotoxicity. Sci. Rep. 2015, 5, 7992. [Google Scholar] [CrossRef]
- Eberth, C.J. Handbuch der Lehre von der Gewegen des Menschen und der Tiere; Engelmann: Leipzig, Germany, 1871; Volume 1. [Google Scholar]
- Rouget, C. Me’moire sur le de’veloppement, la structure et les propriete’s physiologiques des capillaires sanguins et lymphatiques. Arch. Physiol. Norm. Path. 1873, 5, 603–663. [Google Scholar]
- Zimmermann, K.W. Der feinere Bau der Blutkapillaren. Z. Anat. Entwicklungsgesch. 1923, 68, 29–109. [Google Scholar] [CrossRef]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? J. Cereb. Blood Flow Metab. 2016, 2, 451–455. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Rustenhoven, J.; Scotter, E.L.; Schweder, P.; Faull, R.L.M.; Park, T.I.H.; Dragunow, M. Markers for human brain pericytes and smooth muscle cells. J. Chem. Neuroanat. 2018, 92, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N.; Turczyn, T.J.; Das, A. Pericyte coverage of retinal and cerebral capillaries. Investig. Ophthalmol. Vis. Sci. 1990, 31, 999–1007. [Google Scholar]
- Frank, R.N.; Dutta, S.; Mancini, M.A. Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Investig. Ophthalmol. Vis. Sci. 1987, 28, 1086–1091. [Google Scholar]
- Gaengel, K.; Genove, G.; Armulik, A.; Betsholtz, C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-β in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 2, 173–185. [Google Scholar] [CrossRef]
- Olson, L.E.; Soriano, P. PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev. Cell 2011, 6, 815–826. [Google Scholar] [CrossRef]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001, 3, 543–553. [Google Scholar] [CrossRef]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B., Jr.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Miners, J.S.; Schulz, I.; Love, S. Differing associations between Aβ accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2018, 1, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, M.M.; Otte-Holler, I.; van Triel, J.J.; Veerhuis, R.; Maat-Schieman, M.L.; Bu, G.; Verbeek, M.M. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am. J. Pathol. 2007, 171, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Nielsen, H.M.; Minthon, L.; Wennstrom, M. Involvement of matrix metalloproteinase-9 in amyloid-beta 1-42- induced shedding of the pericyte proteoglycan NG2. J. Neuropathol. Exp. Neurol. 2014, 73, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.F.; Mackenzie, I.R.; Feldman, H.H. Clinical phenotypes of cerebral amyloid angiopathy. J. Neurol. Sci. 2007, 257, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; Van Nostrand, W.E.; Otte-Holler, I.; Wesseling, P.; De Waal, R.M. Amyloid-beta-induced degeneration of human brain pericytes is dependent on the apolipoprotein E genotype. Ann. N. Y. Acad. Sci. 2000, 903, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; de Waal, R.M.; Schipper, J.J.; Van Nostrand, W.E. Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J. Neurochem. 1997, 68, 1135–1141. [Google Scholar] [CrossRef]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Netherlands Brain Bank; Olofsson, A.; Wennström, M. Amyloid-beta 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 3, e12728. [Google Scholar] [CrossRef]
- Roberts, K.F.; Elbert, D.L.; Kasten, T.P.; Patterson, B.W.; Sigurdson, W.C.; Connors, R.E.; Ovod, V.; Munsell, L.Y.; Mawuenyega, K.G.; Miller-Thomas, M.M.; et al. Amyloidbeta efflux from the central nervous system into the plasma. Ann. Neurol. 2014, 76, 837–844. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Abeta in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Sagare, A.P.; Friedman, A.E.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport pathways for clearance of human Alzheimer’s amyloid betapeptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 2007, 27, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss (1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef]
- Casey, C.S.; Atagi, Y.; Yamazaki, Y.; Shinohara, M.; Tachibana, M.; Fu, Y.; Bu, G.; Kanekiyo, T. Apolipoprotein E Inhibits Cerebrovascular Pericyte Mobility through a RhoA Protein-mediated Pathway. J. Biol. Chem. 2015, 22, 14208–14217. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Guillamon, M.; Mawhirt, S.; Blais, S.; Montaner, J.; Neubert, T.A.; Rostagno, A.; Ghiso, J. Sequential Amyloid-β Degradation by the Matrix Metalloproteases MMP-2 and MMP-9. J. Biol. Chem. 2015, 24, 15078–15091. [Google Scholar] [CrossRef]
- Olsson, F.; Schmidt, S.; Althoff, V.; Munter, L.M.; Jin, S.; Rosqvist, S.; Lendahl, U.; Multhaup, G.; Lundkvist, J. Characterization of intermediate steps in amyloid beta (Aβ) production under near-native conditions. J. Biol. Chem. 2014, 3, 1540–1550. [Google Scholar] [CrossRef]
- Fluhrer, R.; Multhaup, G.; Schlicksupp, A.; Okochi, M.; Takeda, M.; Lammich, S.; Willem, M.; Westmeyer, G.; Bode, W.; Walter, J.; et al. Identification of a beta-secretase activity, which truncates amyloid beta-peptide after its presenilin-dependent generation. J. Biol. Chem. 2003, 8, 5531–5538. [Google Scholar] [CrossRef]
- Kirabali, T.; Rigotti, S.; Siccoli, A.; Liebsch, F.; Shobo, A.; Hock, C.; Nitsch, R.M.; Multhaup, G.; Kulic, L. The amyloid-β degradation intermediate Aβ34 is pericyte-associated and reduced in brain capillaries of patients with Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 1, 194. [Google Scholar] [CrossRef]
- Díaz-Coránguez, M.; Ramos, C.; Antonetti, D.A. The inner blood-retinal barrier: Cellular basis and development. Vis. Res. 2017, 139, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. A New Human Blood-Retinal Barrier Model Based on Endothelial Cells, Pericytes, and Astrocytes. Int. J. Mol. Sci. 2020, 21, 1636. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, K.; Tachikawa, M. The inner blood-retinal barrier: Molecular structure and transport biology. Adv. Exp. Med. Biol. 2012, 763, 85–104. [Google Scholar] [PubMed]
- Wilkerson, I.; Laban, J.; Mitchell, J.M.; Sheibani, N.; Alcendor, D.J. Retinal pericytes and cytomegalovirus infectivity: Implications for HCMV-induced retinopathy and congenital ocular disease. J. Neuroinflamm. 2015, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 9, 1591–1598. [Google Scholar]
- Enge, M.; Bjarnegård, M.; Gerhardt, H.; Gustafsson, E.; Kalén, M.; Asker, N.; Hammes, H.P.; Shani, M.; Fässler, R.; Betsholtz, C. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002, 16, 4307–4316. [Google Scholar] [CrossRef]
- Ong, S.S.; Proia, A.D.; Whitson, H.E.; Farsiu, S.; Doraiswamy, P.M.; Lad, E.M. Ocular amyloid imaging at the crossroad of Alzheimer’s disease and age-related macular degeneration: Implications for diagnosis and therapy. J. Neurol. 2019, 7, 1566–1577. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J. Alzheimers Dis. 2011, 24, 615–631. [Google Scholar] [CrossRef]
- Muraleva, N.A.; Kozhevnikova, O.S.; Fursova, A.Z.; Kolosova, N.G. Suppression of AMD-Like Pathology by Mitochondria-Targeted Antioxidant SkQ1 Is Associated with a Decrease in the Accumulation of Amyloid β and in mTOR Activity. Antioxidants 2019, 6, 177. [Google Scholar] [CrossRef]
- Kolosova, N.G.; Tyumentsev, M.A.; Muraleva, N.A.; Kiseleva, E.; Vitovtov, A.O.; Stefanova, N.A. Antioxidant SkQ1 Alleviates Signs of Alzheimer’s Disease-like Pathology in Old OXYS Rats by Reversing Mitochondrial Deterioration. Curr. Alzheimer Res. 2017, 12, 1283–1292. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Muraleva, N.A.; Maksimova, K.Y.; Rudnitskaya, E.A.; Kiseleva, E.; Telegina, D.V.; Kolosova, N.G. An antioxidant specifically targeting mitochondria delays progression of Alzheimer’s disease-like pathology. Aging (Albany N. Y.) 2016, 8, 2713–2733. [Google Scholar] [CrossRef] [PubMed]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ljubimov, A.V.; Miller, C.A.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 2011, 54 (Suppl. 1), S204–S217. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Byman, E. Netherlands Brain Bank, Wennström M1. Levels of Retinal Amyloid-β Correlate with Levels of Retinal IAPP and Hippocampal Amyloid-β in Neuropathologically Evaluated Individuals. J. Alzheimers. Dis. 2020, 3, 1201–1209. [Google Scholar]
- Salobrar-García, E.; de Hoz, R.; Ramírez, A.I.; López-Cuenca, I.; Rojas, P.; Vazirani, R.; Amarante, C.; Yubero, R.; Gil, P.; Pinazo-Durán, M.D.; et al. Changes in visual function and retinal structure in the progression of Alzheimer’s disease. PLoS ONE 2019, 8, e0220535. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Thirgood, R.A.; Oliphant, H.; Frizzati, A.; Littlewood, E.; Votruba, M.; Good, M.A.; Williams, J.; Morgan, J.E. Retinal ganglion cell dendritic degeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 7, 1799–1806. [Google Scholar] [CrossRef]
- Liu, B.; Rasool, S.; Yang, Z.; Glabe, C.G.; Schreiber, S.S.; Ge, J.; Tan, Z. Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer’s transgenic mice. Am. J. Pathol. 2009, 5, 2099–2110. [Google Scholar] [CrossRef]
- Hale, G.; Good, M. Impaired visuospatial recognition memory but normal object novelty detection and relative familiarity judgments in adult mice expressing the APPswe Alzheimer’s disease mutation. Behav. Neurosci. 2005, 4, 884–891. [Google Scholar] [CrossRef]
- Perez, S.E.; Lumayag, S.; Kovacs, B.; Mufson, E.J.; Xu, S. Betaamyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer’s disease. Investig. Ophthalmol. Vis. Sci. 2009, 2, 793–800. [Google Scholar] [CrossRef]
- Ning, A.; Cui, J.; To, E.; Ashe, K.H.; Matsubara, J. Amyloidbeta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Investig. Ophthalmol. Vis. Sci. 2008, 11, 5136–5143. [Google Scholar] [CrossRef]
- Edwards, M.M.; Rodriguez, J.J.; Gutierrez-Lanza, R.; Yates, J.; Verkhratsky, A.; Lutty, G.A. Retinal macroglia changes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Eye Res. 2014, 127, 252–260. [Google Scholar] [CrossRef]
- Den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 1, 147. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, L.; Huang, S.C.; Magnani, G.; Ambrosi, A.; Comi, G.; Leocani, L. Optical Coherence Tomography Reveals Retinal Neuroaxonal Thinning in Frontotemporal Dementia as in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 3, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Ascaso, F.J.; Cruz, N.; Modrego, P.J.; Lopez-Anton, R.; Santabarbara, J.; Pascual, L.F. Retinal alterations in mild cognitive impairment and Alzheimer’s disease: An optical coherence tomography study. J. Neurol. 2014, 261, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- McGrory, S.; Cameron, J.R.; Pellegrini, E.; Warren, C.; Doubal, F.N.; Deary, I.J.; Dhillon, B.; Wardlaw, J.M.; Trucco, E.; MacGillivray, T.J. The application of retinal fundus camera imaging in dementia: A systematic review. Alzheimers Dement. (Amst.) 2016, 6, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Szegedi, S.; Dal-Bianco, P.; Stögmann, E.; Traub-Weidinger, T.; Rainer, M.; Masching, A.; Schmidl, D.; Werkmeister, R.M.; Chua, J.; Schmetterer, L.; et al. Anatomical and functional changes in the retina in patients with Alzheimer’s disease and mild cognitive impairment. Acta Ophthalmol. 2020. [CrossRef] [PubMed]
- Busche, M.A. In Vivo Two-Photon Calcium Imaging of Hippocampal Neurons in Alzheimer Mouse Models. Methods Mol. Biol. 2018, 1750, 341–351. [Google Scholar]
- Tachibana, M.; Yamazaki, Y.; Liu, C.C.; Bu, G.; Kanekiyo, T. Pericyte, implantation in the brain enhances cerebral blood flow and reduces amyloid-β pathology in amyloid model mice. Exp. Neurol. 2018, 300, 13–21. [Google Scholar] [CrossRef]
- Ferland-McCollough, D.; Slater, S.; Richard, J.; Reni, C.; Mangialardi, G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol. Ther. 2017, 171, 30–42. [Google Scholar] [CrossRef]
- Caplan, A.I. Adult mesenchymal stem cells: When, where, and how. Stem Cells Int. 2015, 2015, 628767. [Google Scholar] [CrossRef]
- Siqueira, R.C.; Messias, A.; Messias, K.; Arcieri, R.S.; Ruiz, M.A.; Souza, N.F.; Jorge, R. Quality of life in patients with retinitis pigmentosa submitted to intravitreal use of bone marrow-derived stem cells (Reticell-clinical trial). Stem Cell Res. Ther. 2015, 6, 29. [Google Scholar] [CrossRef]
- Yamagishi, S.; Inagaki, Y.; Amano, S.; Okamoto, T.; Takeuchi, M.; Makita, Z. Pigment epithelium-derived factor protects cultured retinal pericytes from advanced glycation end product-induced injury through its antioxidative properties. Biochem. Biophys. Res. Commun. 2002, 296, 877–882. [Google Scholar] [CrossRef]
- Zhang, S.X.; Wang, J.J.; Gao, G.; Parke, K.; Ma, J.X. Pigment epithelium-derived factor downregulates vascular endothelial growth factor (VEGF) expression and inhibits VEGF-VEGF receptor 2 binding in diabetic retinopathy. J. Mol. Endocrinol. 2006, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Leo, L.F.; McGregor, C.; Grivitishvili, A.; Barnstable, C.J.; Tombran-Tink, J. Pigment epithelium-derived factor (PEDF) peptide eye drops reduce inflammation, cell death and vascular leakage in diabetic retinopathy in Ins2(Akita) mice. Mol. Med. 2012, 18, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, K.; Wu, D.M.; Puro, D.G. Physiology of rat retinal pericytes: Modulation of ion channel activity by serum-derived molecules. J. Physiol. 1999, 3, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Franks, M.E.; Macpherson, G.R.; Figg, W.D. Thalidomide. Lancet 2004, 363, 1802–1811. [Google Scholar] [CrossRef]
- Lebrin, F.; Srun, S.; Raymond, K.; Martin, S.; van den Brink, S.; Freitas, C.; Breant, C.; Mathivet, T.; Larrivee, B.; Thomas, J.L.; et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat. Med. 2010, 16, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-alpha in an animal model of inflamed Alzheimer’s disease brain. Neurobiol. Dis. 2008, 29, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Soussain, C.; Thalgott, J. 2015. Available online: https://patents.google.com/patent/WO2015107196A1/en (accessed on 6 August 2018).
- Harrell, R.; Simovic, B.; Fellabaum, M.; Arsenijevic, A.; Djonov, V.; Volarevic, V. Molecular Mechanisms Underlying Therapeutic Potential of Pericytes. J. Biomed. Sci. 2018, 1, 21. [Google Scholar] [CrossRef]
- Avolio, E.; Alvino, V.V.; Ghorbel, M.T.; Campagnolo, P. Perivascular cells and tissue engineering: Current applications and untapped potential. Pharmacol. Ther. 2017, 171, 83–92. [Google Scholar] [CrossRef]



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
J. Alcendor, D. Interactions between Amyloid-Β Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 1490. https://doi.org/10.3390/jcm9051490
J. Alcendor D. Interactions between Amyloid-Β Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease. Journal of Clinical Medicine. 2020; 9(5):1490. https://doi.org/10.3390/jcm9051490
Chicago/Turabian StyleJ. Alcendor, Donald. 2020. "Interactions between Amyloid-Β Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease" Journal of Clinical Medicine 9, no. 5: 1490. https://doi.org/10.3390/jcm9051490
APA StyleJ. Alcendor, D. (2020). Interactions between Amyloid-Β Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease. Journal of Clinical Medicine, 9(5), 1490. https://doi.org/10.3390/jcm9051490