Can Circulating Cardiac Biomarkers Be Helpful in the Assessment of LMNA Mutation Carriers?

 , , , ,
, , , ,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Definitions
2.2. Biomarkers’ Measurements
2.3. Mutation Screening
2.4. Statistical Analysis
3. Results
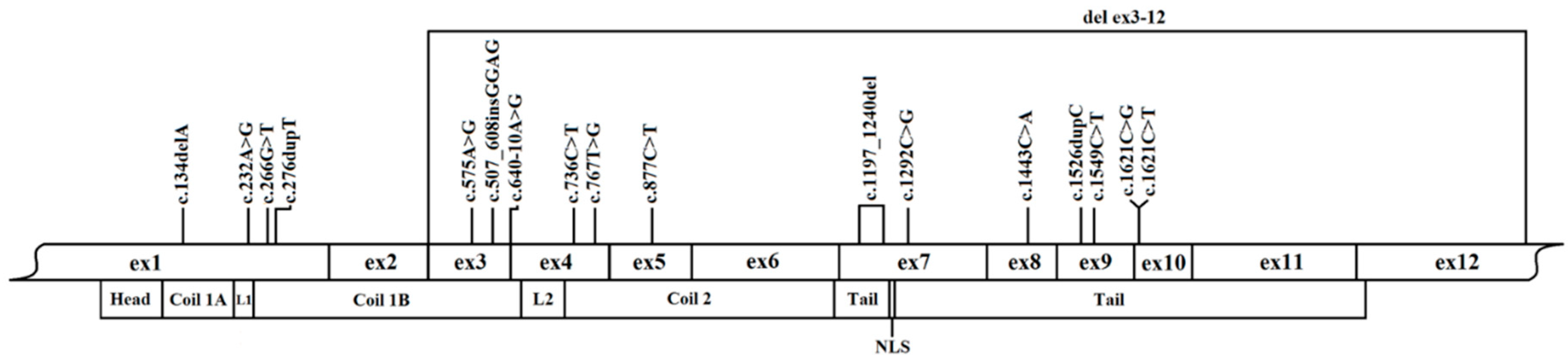
3.1. Molecular Findings in the Study Cohort
3.2. Study Population and Clinical Characteristics
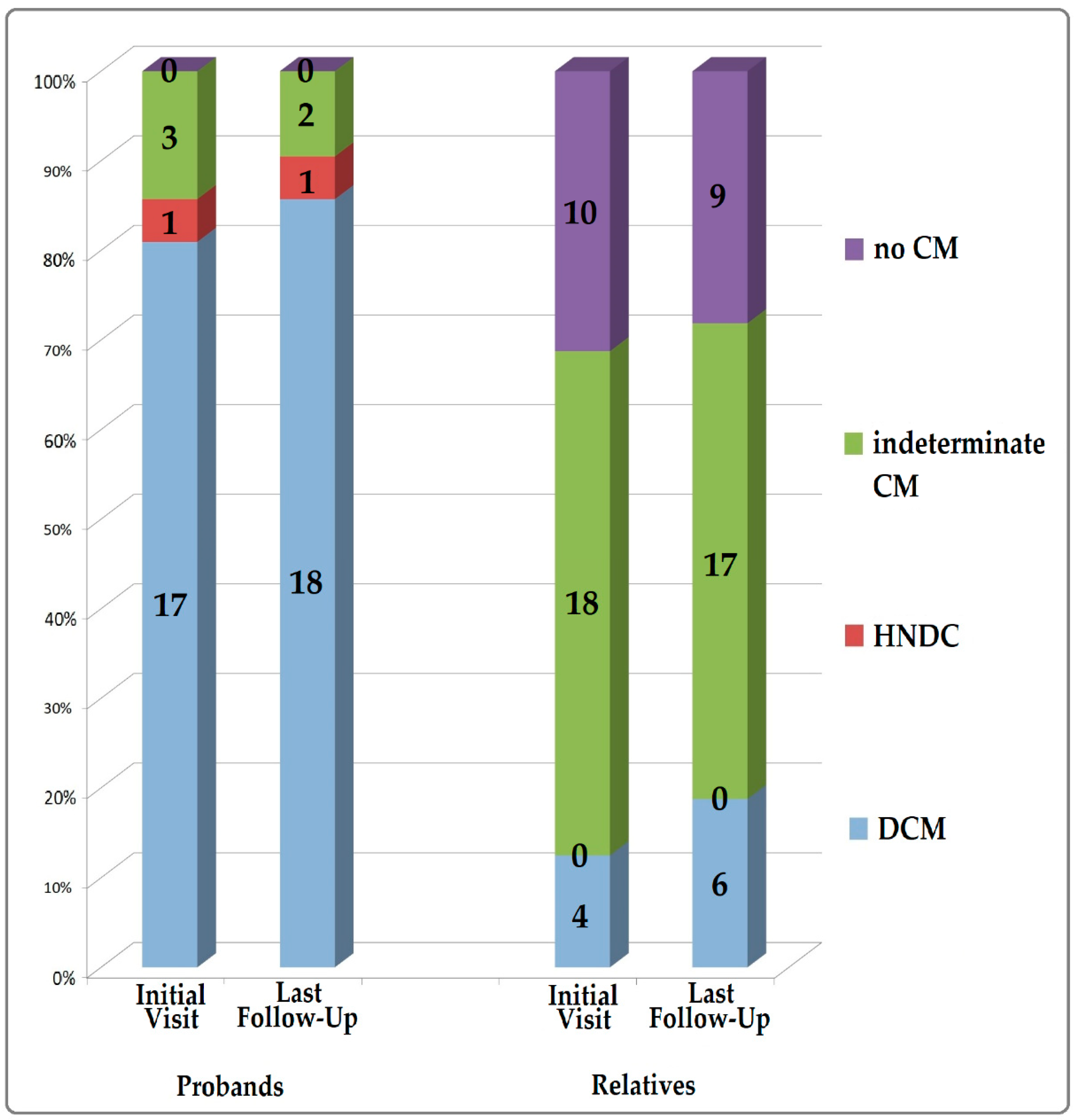
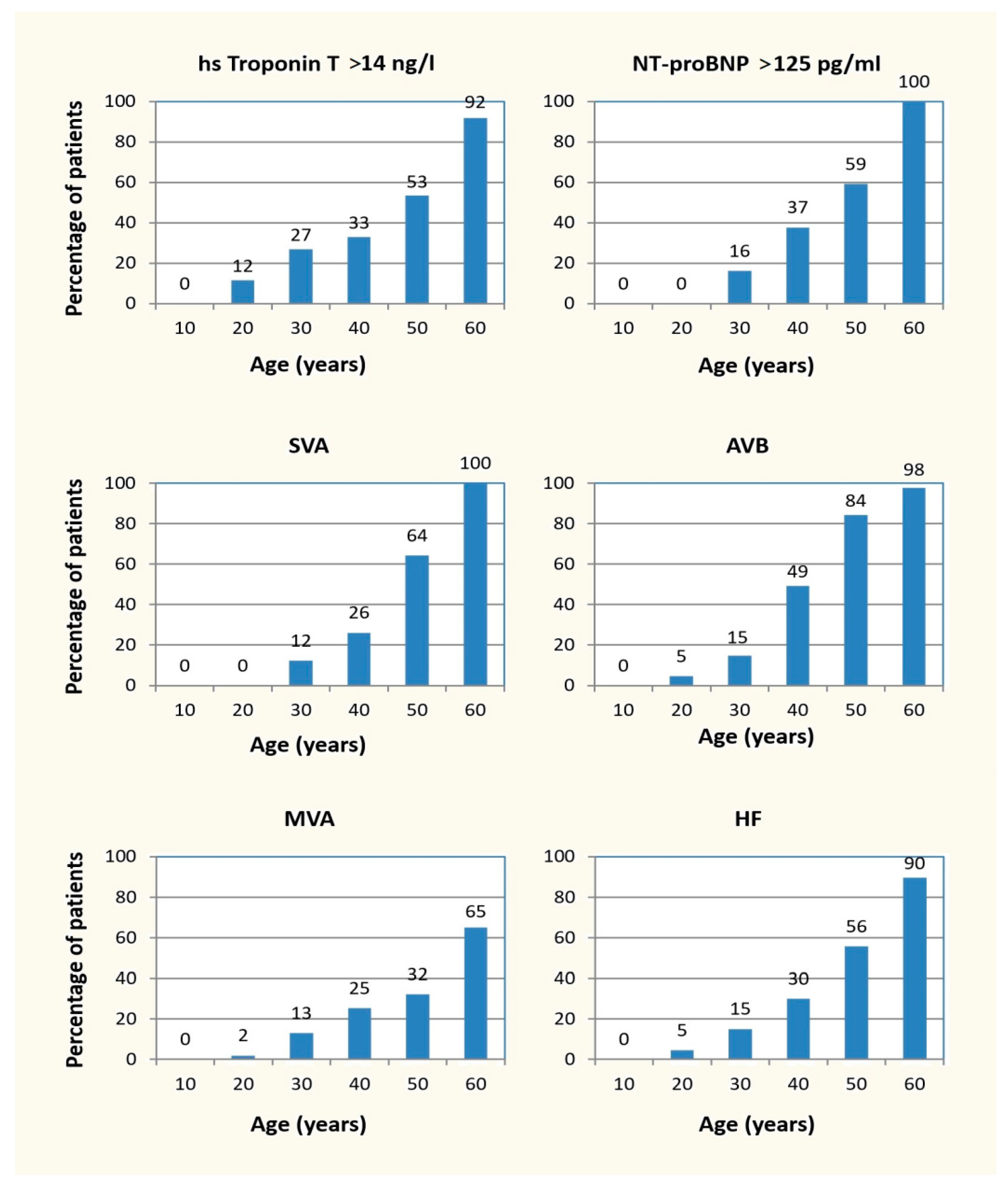
3.3. Penetrance of Cardiolaminopathy Indicators
3.4. Phenotypic Differences in Missense versus Non-Missense LMNA Variant Carriers
3.5. Follow-Up and Risk Stratification Including Biomarkers
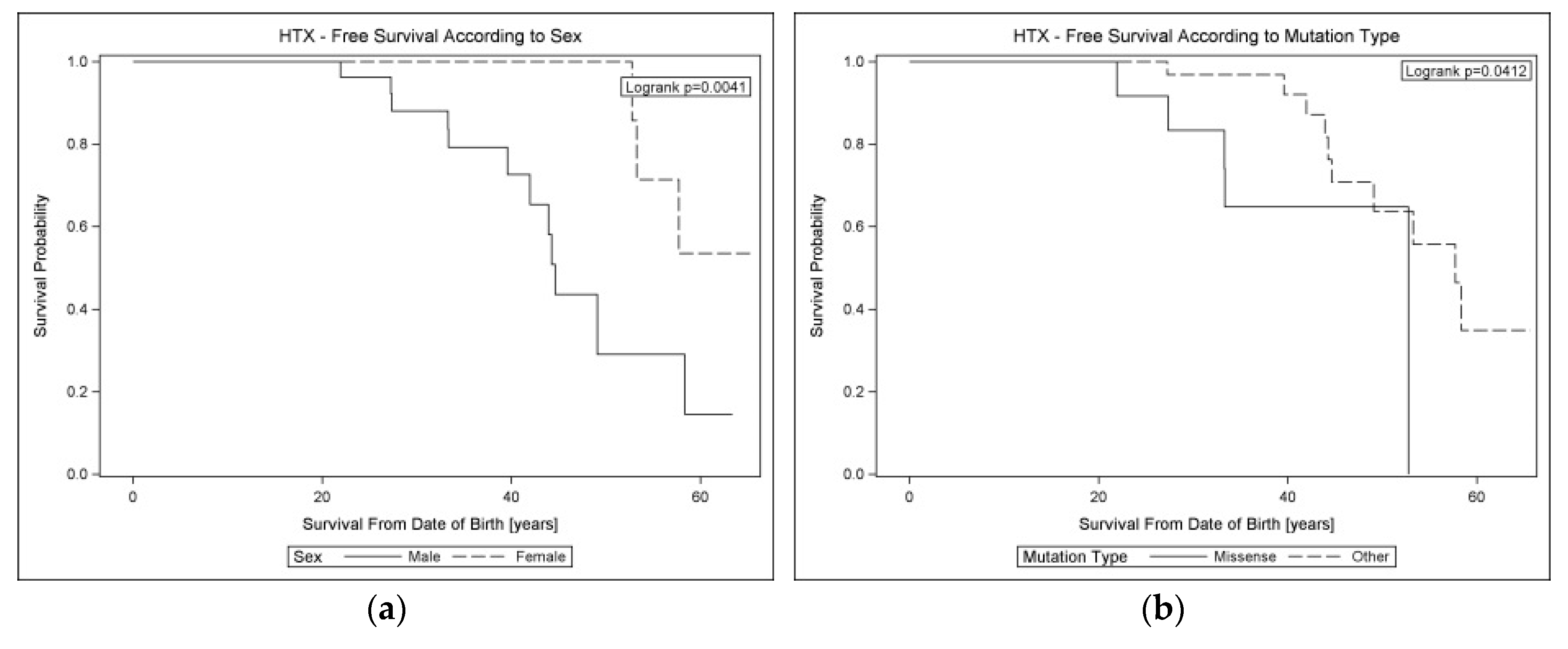
3.5.1. Arrhythmic Risk Stratification During the Follow-Up
3.5.2. Factors Affecting Lifelong Prognosis in Cardiolaminopathies
4. Discussion
4.1. Penetrance of Cardiolaminopathy Indicators
4.2. Phenotypic Differences in Missense versus Non-Missense LMNA Variants
4.3. Arrhythmic Risk Stratification Including Biomarkers
4.4. Molecular Findings in the Study Cohort
4.5. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hershberger, R.E.; Morales, A. Dilated Cardiomyopathy Overview; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews(R): Seattle, WA, USA, 1993. [Google Scholar]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.; Cuenca, S.; Ferro, M.D.; Zorio, E.; Aranda, R.S.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Augusto, J.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; A Treibel, T.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2019. [Google Scholar] [CrossRef]
- Van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Captur, G.; Bilińska, Z.; Arbustini, E. Lamin missense mutations-the spectrum of phenotype variability is increasing. Eur. J. Heart Fail. 2018, 20, 1413–1416. [Google Scholar] [CrossRef]
- Arbustini, E.; Pilotto, A.; Repetto, A.; Grasso, M.; Negri, A.; Diegoli, M.; Campana, C.; Scelsi, L.; Baldini, E.; Gavazzi, A.; et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J. Am. Coll. Cardiol. 2002, 39, 981–990. [Google Scholar] [CrossRef]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef]
- Ollila, L.; Nikus, K.; Holmström, M.; Jalanko, M.; Jurkko, R.; Kaartinen, M.; Koskenvuo, J.; Kuusisto, J.; Kärkkäinen, S.; Palojoki, E.; et al. Clinical disease presentation and ECG characteristics of LMNA mutation carriers. Open Heart 2017, 4, e000474. [Google Scholar] [CrossRef]
- Fatkin, D.; Macrae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J.; Spudich, S.; De Girolami, U.; et al. Missense Mutations in the Rod Domain of the Lamin A/C Gene as Causes of Dilated Cardiomyopathy and Conduction-System Disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.G.; Fain, P.R.; Sinagra, G.; Robinson, M.L.; Robertson, A.D.; Carniel, E.; Di Lenarda, A.; Bohlmeyer, T.J.; A Ferguson, D.; Brodsky, G.L.; et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol. 2003, 41, 771–780. [Google Scholar] [CrossRef]
- Sylvius, N.; Bilinska, Z.T.; Veinot, J.P.; Fidzianska, A.; Bolongo, P.M.; Poon, S.; McKeown, P.; Davies, R.; Chan, K.-L.; Tang, A.S.L.; et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med Genet. 2005, 42, 639–647. [Google Scholar] [CrossRef]
- Saj, M.; Bilinska, Z.T.; Tarnowska, A.; Sioma, A.; Bolongo, P.; Sobieszczańska-Małek, M.; Michalak, E.; Golen, D.; Mazurkiewicz, L.; Małek, L.A.; et al. LMNA mutations in Polish patients with dilated cardiomyopathy: Prevalence, clinical characteristics, and in vitro studies. BMC Med Genet. 2013, 14, 55. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Bilinska, Z.T.; Sylvius, N.; Boudreau, E.; Veinot, J.P.; Labib, S.; Bolongo, P.M.; Hamza, A.; Jackson, T.; Płoski, R.; et al. Genetic and ultrastructural studies in dilated cardiomyopathy patients: A large deletion in the lamin A/C gene is associated with cardiomyocyte nuclear envelope disruption. Basic Res. Cardiol. 2010, 105, 365–377. [Google Scholar] [CrossRef]
- Płoski, R.; Pollak, A.; Müller, S.; Franaszczyk, M.; Michalak, E.; Kosińska, J.; Stawinski, P.; Spiewak, M.; Seggewiss, H.; Bilinska, Z.T. Does p.Q247X in TRIM63 cause human hypertrophic cardiomyopathy? Circ. Res. 2014, 114, 2–5. [Google Scholar] [CrossRef]
- Kourgiannidis, G.; Anastasakis, A.; Lampropoulos, K.; Iliopoulos, T. A patient with ventricular tachycardia due to a novel mutation of the lamin A/C gene: Case presentation and mini review. Hellenic. J. Cardiol. 2013, 54, 326–330. [Google Scholar]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.I.; Favalli, V.; Grasso, M.; et al. Long-Term Outcome and Risk Stratification in Dilated Cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef]
- Małek, Ł.A.; Labib, S.; Mazurkiewicz, Ł.; Saj, M.; Płoski, R.; Tesson, F.; Bilinska, Z.T. A new c.1621 C>G, p.R541G lamin A/C mutation in a family with DCM and regional wall motion abnormalities (akinesis/dyskinesis): Genotype–phenotype correlation. J. Hum. Genet. 2010, 56, 83–86. [Google Scholar] [CrossRef]
- Stallmeyer, B.; Koopmann, M.; Schulze-Bahr, E. Identification of Novel Mutations in LMNA Associated with Familial Forms of Dilated Cardiomyopathy. Genet. Test. Mol. Biomarkers 2012, 16, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Forissier, J.F.; Bonne, G.; Bouchier, C.; Duboscq-Bidot, L.; Richard, P.; Wisnewski, C.; Briault, S.; Moraine, C.; Dubourg, O.; Schwartz, K.; et al. Apical left ventricular aneurysm without atrio-ventricular block due to a lamin A/C gene mutation. Eur. J. Heart Fail. J. Work. Group Heart Fail. Eur. Soc. Cardiol. 2003, 5, 821–825. [Google Scholar]
- Hookana, E.; Junttila, M.J.; Särkioja, T.; Sormunen, R.; Niemelä, M.; Raatikainen, M.P.; Uusimaa, P.; Lizotte, E.; Peuhkurinen, K.; Brugada, R.; et al. Cardiac Arrest and Left Ventricular Fibrosis in a Finnish Family with the Lamin A/C Mutation. J. Cardiovasc. Electrophysiol. 2008, 19, 743–747. [Google Scholar] [CrossRef]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.D.; Seidman, M.; Baxter, S.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef]
- Ito, K.; Patel, P.N.; Gorham, J.M.; McDonough, B.; DePalma, S.R.; Adler, E.E.; Lam, L.; MacRae, C.A.; Mohiuddin, S.M.; Fatkin, D.; et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, 7689–7694. [Google Scholar] [CrossRef]
- Fidzianska, A.; Bilinska, Z.T.; Tesson, F.; Wagner, T.; Walski, M.; Grzybowski, J.; Ruzyłło, W.; Hausmanowa-Petrusewicz, I. Obliteration of cardiomyocyte nuclear architecture in a patient with LMNA gene mutation. J. Neurol. Sci. 2008, 271, 91–96. [Google Scholar] [CrossRef]
- Otomo, J.; Kure, S.; Shiba, T.; Karibe, A.; Shinozaki, T.; Yagi, T.; Naganuma, H.; Tezuka, F.; Miura, M.; Ito, M.; et al. Electrophysiological and Histopathological Characteristics of Progressive Atrioventricular Block Accompanied by Familial Dilated Cardiomyopathy Caused by a Novel Mutation of Lamin A/C Gene. J. Cardiovasc. Electrophysiol. 2005, 16, 137–145. [Google Scholar] [CrossRef]
- Nakajima, K.; Aiba, T.; Makiyama, T.; Nishiuchi, S.; Ohno, S.; Kato, K.; Yamamoto, Y.; Doi, T.; Shizuta, S.; Onoue, K.; et al. Clinical Manifestations and Long-Term Mortality in Lamin A/C Mutation Carriers From a Japanese Multicenter Registry. Circ. J. 2018, 82, 2707–2714. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Blanes, J.G.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2012, 34, 1448–1458. [Google Scholar] [CrossRef]
- McKie, P.M.; AbouEzzeddine, O.F.; Scott, C.G.; Mehta, R.; Rodeheffer, R.J.; Redfield, M.M.; Burnett, J.C.; Jaffe, A.S. High-Sensitivity Troponin I and Amino-Terminal Pro–B-Type Natriuretic Peptide Predict Heart Failure and Mortality in the General Population. Clin. Chem. 2014, 60, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Grzybowski, J.; Bilinska, Z.T.; Janas, J.; Michalak, E.; Ruzyllo, W. Plasma concentrations of N-terminal atrial natriuretic peptide are raised in asymptomatic relatives of dilated cardiomyopathy patients with left ventricular enlargement. Heart 2002, 88, 191–192. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wess, G.; Domenech, O.; Dukes-McEwan, J.; Häggström, J.; Gordon, S. European Society of Veterinary Cardiology screening guidelines for dilated cardiomyopathy in Doberman Pinschers. J. Veter. Cardiol. 2017, 19, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Charron, P.; Elliott, P.; Gimeno, J.R.; Caforio, A.L.P.; Kaski, J.P.; Tavazzi, L.; Tendera, M.; Maupain, C.; Laroche, C.; Rubiś, P.; et al. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: Baseline data and contemporary management of adult patients with cardiomyopathies. Eur. Heart J. 2018, 39, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Nishiuchi, S.; Makiyama, T.; Aiba, T.; Nakajima, K.; Hirose, S.; Kohjitani, H.; Yamamoto, Y.; Harita, T.; Hayano, M.; Wuriyanghai, Y.; et al. Gene-Based Risk Stratification for Cardiac Disorders in LMNA Mutation Carriers. Circ. Cardiovasc. Genet. 2017, 10, e001603. [Google Scholar] [CrossRef]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, M.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2016, 17, 2793–2867. [Google Scholar]
- Captur, G.; Arbustini, E.; Syrris, P.; Radenkovic, D.; O’Brien, B.; McKenna, W.J.; Moon, J.C. Lamin mutation location predicts cardiac phenotype severity: Combined analysis of the published literature. Open Heart 2018, 5, e000915. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Özmen, Ç.; Deniz, A.; Deveci, O.S.; Cagliyan, C.E.; Celik, A.I.; Yildiz, I.; Yildiz, P.Ö.; Demir, M.; Kanadaşı, M. Association among tenascin-C and NT-proBNP levels and arrhythmia prevalence in heart failure. Clin. Investig. Med. 2017, 40, 219. [Google Scholar] [CrossRef]
- Medina, A.; Moss, A.J.; McNitt, S.; Zareba, W.; Wang, P.J.; Goldenberg, I. Brain natriuretic peptide and the risk of ventricular tachyarrhythmias in mildly symptomatic heart failure patients enrolled in MADIT-CRT. Heart Rhythm. 2016, 13, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Aimo, A.; Januzzi, J.L.; Vergaro, G.; Ripoli, A.; Latini, R.; Masson, S.; Magnoli, M.; Anand, I.S.; Cohn, J.N.; Tavazzi, L.; et al. Prognostic Value of High-Sensitivity Troponin T in Chronic Heart Failure. Circulation 2018, 137, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Alawieh, H.; Chemaly, T.; Alam, S.; Khraiche, M. Towards Point-of-Care Heart Failure Diagnostic Platforms: BNP and NT-proBNP Biosensors. Sensors 2019, 19, 5003. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LMNA Gene Mutation | Protein Mutation | Type | Exon (NLS Class) | ACMG Classification | No. of Probands/Relatives | Proband Phenotype | References |
|---|---|---|---|---|---|---|---|
| c.134delA | p.Tyr45Ser fsTer51 | truncation | 1 (1) | pathogenic | 1/1 | HNDC, AVB, AF, nsVT, HTX | novel |
| c.232A>G | p.Lys78Glu | missense | 1 (1) | likely pathogenic | 1/1 | DCM, SCA, AVB, HTX | Kourgiannidis et al. [20] |
| c.266G>T | p.Arg89Leu | missense | 1 (1) | likely pathogenic | 1/0 | DCM, AVB, AF, HTX | Pasotti et al. [21], Taylor et al. [13], Saj et al. [15] |
| c.276dupT | p.Asp93Ter | truncation | 1 (1) | pathogenic | 1/3 | DCM, AF, nsVT | novel |
| del_ex3-12 | n/a | truncation | 2/3 (1) | pathogenic | 1/0 | DCM, AVB, AF, nsVT, ICD shocks, HTX | Gupta et al. [18], Saj et al. [15] |
| c.575A>G | p.Asp192Gly | missense | 3 (1) | likely pathogenic | 2/1 | DCM, AVB, AF, nsVT, HTX/HF death | Sylvius et al. [14], Saj et al. [15], Fidziańska et al. [28] |
| c.607_608 insGGAG | p.Glu203GlyfsTer12 | truncation | 3 (1) | pathogenic | 1/4 | DCM, AVB, sVT, HTX | novel |
| c.640-10A>G | n/a | inframe insertion | 3/4 (1) | likely pathogenic | 1/2 | DCM, sVT | Otomo et al. [29], Ito et al. [27] |
| c.736C>T | p.Gln246Ter | truncation | 4 (1) | pathogenic | 1/0 | DCM, AVB, AF, nsVT, HF death | Pasotti et al. [21], Saj et al. [15] |
| c.767T>G | p.Val256Gly | missense | 4 (1) | likely pathogenic | 1/0 | DCM, AF, AVB, nsVT, HF death | Saj et al. [15] |
| c.877C>T | p.Gln293Ter | truncation | 5 (1) | pathogenic | 1/0 | LVE, AVB, AF, nsVT | novel |
| c.1197_1240del | p.Gly400Arg fsTer11 | truncation | 7 (1) | pathogenic | 1/2 | DCM, SCA, ICD shocks, AVB, AF | Saj et al. [15] |
| c.1292C>G | p.Ser431Ter | truncation | 7 (2) | pathogenic | 1/5 | DCM, AVB, nsVT, HTX | Saj et al. [15] |
| c.1443C>A | p.Tyr481Ter | truncation | 8 (2) | pathogenic | 1/3 | DCM, AVB, AF, nsVT, HTX | Sylvius et al. [14] |
| c.1526dupC | p.Thr510Tyr fsTer42 | truncation | 9 (2) | pathogenic | 1/4 | DCM, AVB, AF, nsVT, HTX | Saj et al. [15], Pugh et al. [26] |
| c.1549C>T | p.Gln517Ter | truncation | 9 (2) | pathogenic | 2/3 | DCM, SCA, AVB, AF, nsVT | Stallmeyer et al. [23] |
| c.1621C>G | p.Arg541Gly | missense | 10 (2) | likely pathogenic | 1/2 | DCM, nsVT | Malek et al. [22], Saj et al. [15] |
| c.1621C>T | p.Arg541Cys | missense | 10 (2) | likely pathogenic | 2/1 | DCM, AVB, SCA, HTX | Forissier et al. [24], Hookana et al. [25], Saj et al. [15], Pugh et al. [26] |
| Total n = 53 | Probands n = 21 (39.6%) | Relatives n = 32 (60.4%) | p | |
|---|---|---|---|---|
| Age (years) | 33.2 ± 12.4 | 39.6 ± 10.0 | 29.0 ± 12.2 | 0.002 |
| Men, n (%) | 31 (58.5%) | 14 (66.7%) | 17 (53.1%) | 0.328 |
| LMNA missense variants, n (%) | 13 (24.5%) | 8 (38.1%) | 5 (15.6%) | 0.063 |
| Symptoms | ||||
| Syncope, n (%) (n = 49) | 12 (24.5%) | 8 (42.1%) | 4 (13.3%) | 0.039 |
| Family history of SCD <60 years, n (%) (n = 50) | 25 (50%) | 8 (44.4%) | 17 (53.1%) | 0.556 |
| Heart failure, n (%) | 19 (35.9%) | 15 (71.4%) | 4 (12.5%) | <0.0001 |
| NYHA class ≥ 3, n (%) | 7 (13.2%) | 6 (28.6%) | 1 (3.1%) | 0.012 |
| Arrhythmias | ||||
| Atrial arrhythmias, n (%) (n = 52) | 19 (36.5%) | 13 (61.9%) | 6 (19.4%) | 0.002 |
| nsVT, n (%) (n = 50) | 30 (60%) | 19 (100%) | 11 (35.5%) | <0.0001 |
| SCA/sVT, n (%) (n = 50) | 9 (18.0%) | 7 (36.8%) | 2 (6.4%) | 0.018 |
| CCD | ||||
| LBBB, n (%) (n = 47) | 8 (17.0%) | 8 (50.0%) | 0 (0%) | <0.0001 |
| AV block (≥1), n (%) (n = 52) | 31 (59.6%) | 17 (85.0%) | 14 (43.7%) | 0.003 |
| Cardiomyopathies | ||||
| LVEF < 50%, n (%) | 19 (35.8%) | 15 (71.4%) | 4 (12.5%) | <0.0001 |
| LVEF (%) | 50.5 ± 16.2 | 36.9 ± 15.7 | 59.4 ± 8.9 | <0.0001 |
| LVE > 112%, n (%) | 28 (52.8%) | 18 (85.7%) | 10 (31.3%) | 0.0001 |
| LVEDD (mm) | 53.7 ± 8.7 | 59.1 ± 8.2 | 50.2 ± 7.2 | 0.0001 |
| Biomarkers | ||||
| CK (IU/l) (n = 46) | 162.5 (93–291) | 121 (83–253) | 178 (93–458) | 0.157 |
| elevated CK, n (%) (n = 46) | 13 (28.3%) | 2 (10.5%) | 11 (40.7%) | 0.025 |
| hs Troponin T (ng/L) (n = 42) | 13.6 (7.0–23.9) | 19.2 (13.4–28.9) | 11.9 (5.7–19.9) | 0.018 |
| elevated hs Troponin T, n (%) (n = 42) | 20 (47.6%) | 10 (66.7%) | 10 (37.0%) | 0.065 |
| NT-proBNP (pg/mL) (n = 42) | 161.0 (72.7–683.7) | 683.7 (224–1211) | 84.9 (52.4–183.0) | <0.001 |
| elevated NT-proBNP, n (%) (n = 42) | 23 (54.8%) | 14 (82.3%) | 9 (36.0%) | 0.003 |
| Comorbidities | ||||
| Coronary artery disease, n (%) | 2 (3.8%) | 1 (4.8%) | 1 (3.1%) | 1.000 |
| Hypertension, n (%) | 6 (11.3) | 1 (4.8%) | 5 (15.6%) | 0.384 |
| Implantable devices | ||||
| ICD in primary PPX, n (%) | 8 (15.1%) | 5 (23.8%) | 3 (9.4%) | 0.204 |
| ICD in secondary PPX, n (%) | 8 (15.1%) | 6 (28.6%) | 2 (6.3%) | 0.047 |
| ICD/CRT-D implantation, n (%) | 16 (30.2%) | 11 (52.4%) | 5 (15.6%) | 0.004 |
| Medication | ||||
| β-Blocker, n (%) | 25 (47.2%) | 16 (76.2%) | 9 (28.1%) | <0.001 |
| ACE-I or ARB, n (%) | 21 (39.6%) | 17 (81.0%) | 4 (12.5%) | <0.0001 |
| MRA, n (%) | 6 (11.3%) | 6 (28.6%) | 0 (0%) | 0.002 |
| Biomarker | Baseline | Last Measurement | Relative Change | p |
|---|---|---|---|---|
| hs Troponin T (ng/L) n = 32 | 15.1 (7.6–24.7) | 17.4 (9.6–30.6) | +15.2% | 0.002 |
| NT-proBNP (pg/mL) n = 27 | 223.8 (72.7–683.7) | 478.3 (86.3–1353) | +114% | 0.0003 |
| Total n = 53 | Non-Missense n = 40 | Missense n = 13 | p | |
|---|---|---|---|---|
| Age (years) | 38.6 ± 12.5 | 39.6 ± 13.3 | 35.5 ± 9.6 | 0.318 |
| Men, n (%) | 31 (58.5%) | 24 (60.0%) | 7 (53.8%) | 0.696 |
| Probands, n (%) | 21 (39.6%) | 13 (32.5%) | 8 (61.5%) | 0.063 |
| Symptoms | ||||
| Heart failure, n (%) | 27 (50.9%) | 18 (45.0%) | 9 (69.2%) | 0.129 |
| NYHA class ≥ 3, n (%) | 17 (32.1%) | 11 (27.5%) | 6 (46.2%) | 0.306 |
| Arrhythmias | ||||
| Atrial arrhythmias, n (%) (n = 52) | 24 (46.2%) | 20 (51.3%) | 4 (30.8%) | 0.199 |
| nsVT, n (%) (n = 51) | 35 (68.6%) | 27 (69.2%) | 8 (66.7%) | 1.000 |
| CCD | ||||
| LBBB, n (%) (n = 39) | 10 (25.6%) | 4 (14.3%) | 6 (54.5%) | 0.017 |
| AV block (≥1), n (%) | 36 (67.9%) | 29 (72.5%) | 7 (53.8%) | 0.306 |
| Cardiomyopathies | ||||
| LVEF < 50%, n (%) | 22 (41.5%) | 13 (32.5%) | 9 (69.2%) | 0.019 |
| LVEF (%) | 46.9 ± 17.4 | 51.9 ± 15.3 | 38.0 ± 17.9 | 0.012 |
| LVE > 112%, n (%) | 29 (54.7%) | 19 (47.5%) | 10 (76.9%) | 0.064 |
| LVEDD (mm) | 54.8 ± 9.1 | 51.0 ± 10.5 | 60.1 ± 12.2 | 0.016 |
| Biomarkers | ||||
| hs Troponin T (ng/L) (n = 42) | 16.1 (9.9–29.8) | 16.2 (9.9–31.5) | 13.9 (10.4–17.1) | 0.520 |
| elevated hs Troponin T, n (%) (n = 42) | 24 (57.1%) | 20 (60.6%) | 4 (44.4%) | 0.462 |
| NT-proBNP (pg/mL) (n = 41) | 397.2 (85–1037) | 359.3 (84–1012) | 722.0 (86–2245) | 0.324 |
| elevated NT-proBNP, n (%) (n = 41) | 26 (63.4%) | 18 (60%) | 8 (72.7%) | 0.716 |
| Implantable devices | ||||
| ICD in primary PPX, n (%) | 26 (49.1%) | 20 (50.0%) | 6 (46.2%) | 0.810 |
| ICD in secondary PPX, n (%) | 8 (15.1%) | 5 (12.5%) | 3 (23.1%) | 0.389 |
| ICD/CRT-D implantation, n (%) | 34 (64.2%) | 25 (62.5%) | 9 (69.2%) | 0.749 |
| Events during follow-up | ||||
| Malignant ventricular arrhythmia, n (%) | 13 (24.5%) | 9 (22.5%) | 4 (30.8%) | 0.712 |
| Appropriate ICD shock, n (%) | 11 (31.4%) | 7 (26.9%) | 4 (44.4%) | 0.416 |
| RF ablation for VA, n (%) | 7 (13.2%) | 4 (10%) | 3 (23.1%) | 0.343 |
| Cardiopulmonary resuscitation, n (%) | 1 (1.9%) | 1 (2.5%) | 0 (0%) | 1.000 |
| Sudden cardiac death, n (%) | 1 (1.9%) | 1 (2.5%) | 0 (0%) | 1.000 |
| End-stage heart failure, n (%) | 14 (26.4%) | 9 (22.5%) | 5 (38.5%) | 0.292 |
| Heart transplantation, n (%) | 11 (20.8%) | 8 (20.0%) | 3 (23.1%) | 1.000 |
| HF death, n (%) | 3 (5.7%) | 1 (2.5%) | 2 (15.4%) | 0.145 |
| Cumulate Incidence | p-Value Log-Rank | Univariable | Mutivariable | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | p-Value Wald | HR (95% CI) | p-Value Wald | |||
| MVA, from date of first visit | at 8 years of follow-up (n = 44) | |||||
| Sex: Male vs. Female | 24 vs. 20 | 0.929 | 1.07 (0.24–4.79) | 0.929 | - | |
| Mutation type: Missense vs. Other | 25 vs. 22 | 0.727 | 0.69 (0.08–5.73) | 0.729 | - | |
| AV block: yes vs. no | 42 vs. 0 | 0.007 | NA | - | ||
| nsVT: yes vs. no | 37 vs. 9 | 0.031 | 7.38 (0.88–61.7) | 0.064 | - | |
| LVEF: <45% vs. >45% | 48 vs. 16 | 0.100 | 3.32 (073–15.04) | 0.120 | ||
| LVEF: <55% vs. >55% | 42 vs. 11 | 0.026 | 5.54 (1.02–27.78) | 0.047 | - | |
| NT-proBNP: >150 vs. <150 pg/mL | 67 vs. 6 | 0.002 | 13.40 (1.6–112.7) | 0.017 | 10.4 (1.21–89.79) | 0.010 |
| hsTn T: >20 vs. <20 ng/L | 66 vs. 6 | 0.003 | 13.16 (1.49–115.8) | 0.020 | - | |
| MVA, from date of birth | at 60 years (n = 53) | |||||
| Status: Proband vs. Relative | 65 vs. 62 | 0.746 | 1.18 (0.43; 3.29) | 0.746 | - | |
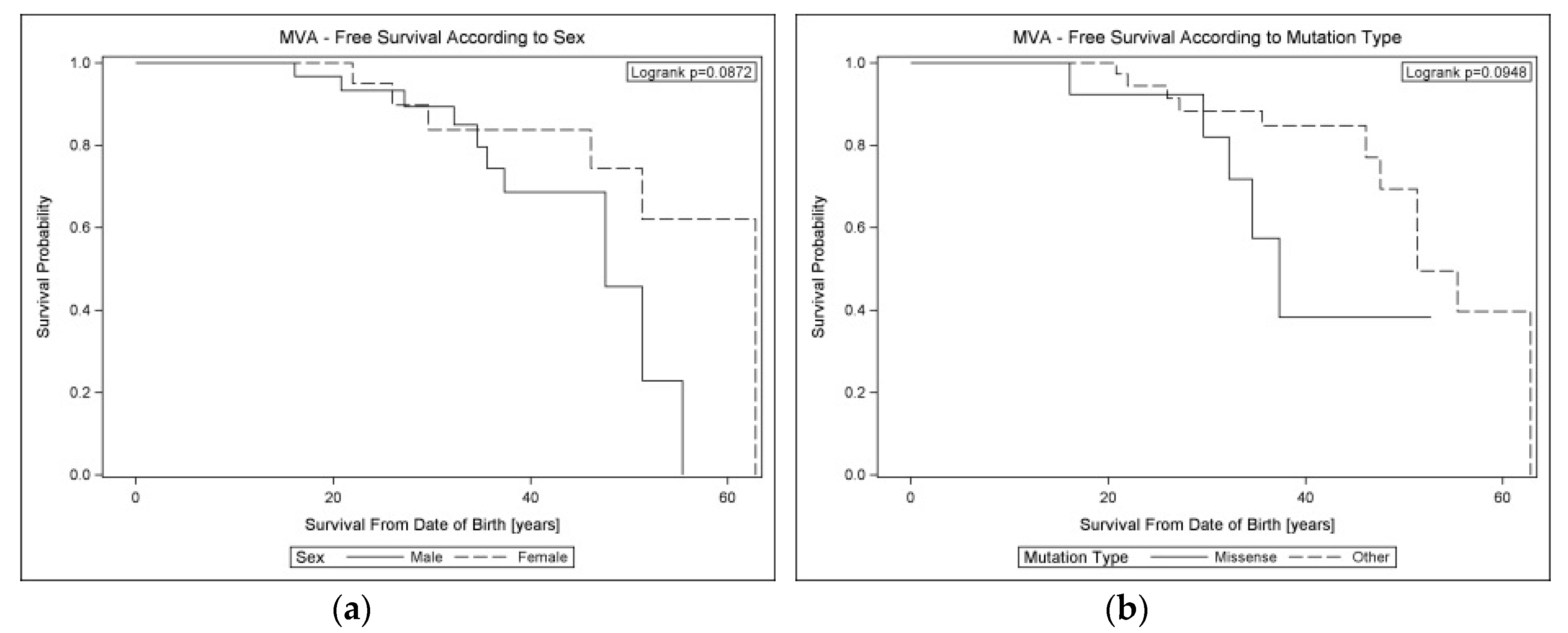
| Sex: Male vs. Female | 100 vs. 41 | 0.087 | 2.61 (0.84; 8.05) | 0.096 | - | |
| Mutation type: Missense vs. Other | 58 vs. 64 | 0.095 | 2.52 (0.82–7.72] | 0.106 | - | |
| End-stage HF, from date of birth | at 60 years (n = 53) | |||||
| Status: Proband vs. Relative | 83 vs. 41 | 0.034 | 3.71 (1.02; 13.47) | 0.046 | - | |
| Sex: Male vs. Female | 84 vs. 50 | 0.004 | 5.55 (1.52; 20.27) | 0.009 | 6.18 (1.66–23.0) | 0.007 |
| Mutation type: Missense vs. other | 100 vs. 63 | 0.041 | 3.18 (0.99; 10.25) | 0.052 | 3.83 (1.14–12.85) | 0.029 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmielewski, P.; Michalak, E.; Kowalik, I.; Franaszczyk, M.; Sobieszczanska-Malek, M.; Truszkowska, G.; Stepien-Wojno, M.; Biernacka, E.K.; Foss-Nieradko, B.; Lewandowski, M.; et al. Can Circulating Cardiac Biomarkers Be Helpful in the Assessment of LMNA Mutation Carriers? J. Clin. Med. 2020, 9, 1443. https://doi.org/10.3390/jcm9051443
Chmielewski P, Michalak E, Kowalik I, Franaszczyk M, Sobieszczanska-Malek M, Truszkowska G, Stepien-Wojno M, Biernacka EK, Foss-Nieradko B, Lewandowski M, et al. Can Circulating Cardiac Biomarkers Be Helpful in the Assessment of LMNA Mutation Carriers? Journal of Clinical Medicine. 2020; 9(5):1443. https://doi.org/10.3390/jcm9051443
Chicago/Turabian StyleChmielewski, Przemyslaw, Ewa Michalak, Ilona Kowalik, Maria Franaszczyk, Malgorzata Sobieszczanska-Malek, Grazyna Truszkowska, Malgorzata Stepien-Wojno, Elzbieta Katarzyna Biernacka, Bogna Foss-Nieradko, Michal Lewandowski, and et al. 2020. "Can Circulating Cardiac Biomarkers Be Helpful in the Assessment of LMNA Mutation Carriers?" Journal of Clinical Medicine 9, no. 5: 1443. https://doi.org/10.3390/jcm9051443
APA StyleChmielewski, P., Michalak, E., Kowalik, I., Franaszczyk, M., Sobieszczanska-Malek, M., Truszkowska, G., Stepien-Wojno, M., Biernacka, E. K., Foss-Nieradko, B., Lewandowski, M., Oreziak, A., Bilinska, M., Kusmierczyk, M., Tesson, F., Grzybowski, J., Zielinski, T., Ploski, R., & Bilinska, Z. T. (2020). Can Circulating Cardiac Biomarkers Be Helpful in the Assessment of LMNA Mutation Carriers? Journal of Clinical Medicine, 9(5), 1443. https://doi.org/10.3390/jcm9051443