Genome-Wide Analysis of the DNA Methylation Profile Identifies the Fragile Histidine Triad (FHIT) Gene as a New Promising Biomarker of Crohn’s Disease

 , and
, and 
Abstract
1. Introduction
2. Experimental Section
2.1. Patients and Clinical Samples
2.2. HumanMethylation450K BeadChip Array Analysis
2.3. Methylation Analyses; MSP and Semi-Quantitative MSP
2.4. Bisulfite Sequencing
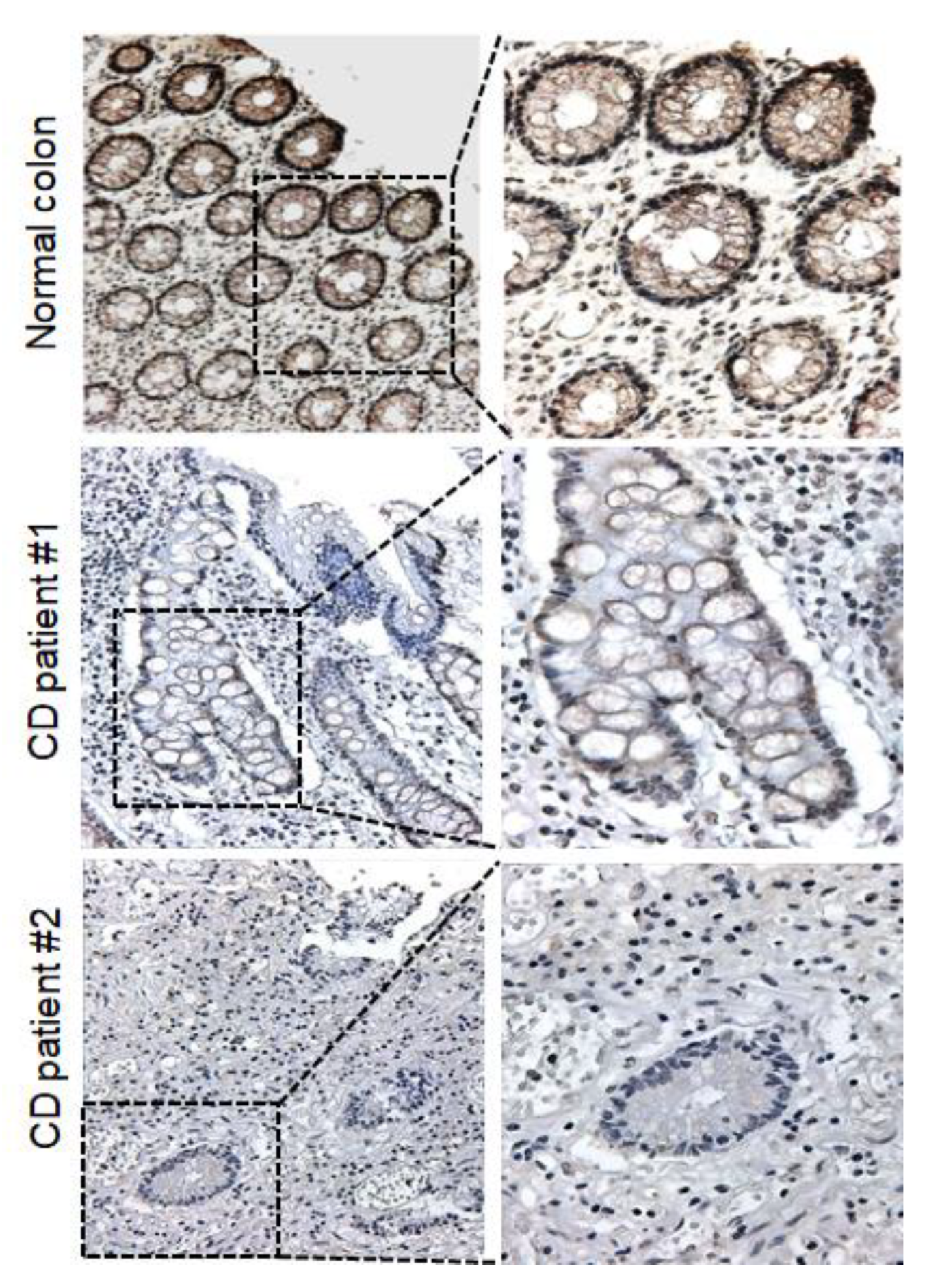
2.5. Immunohistochemistry
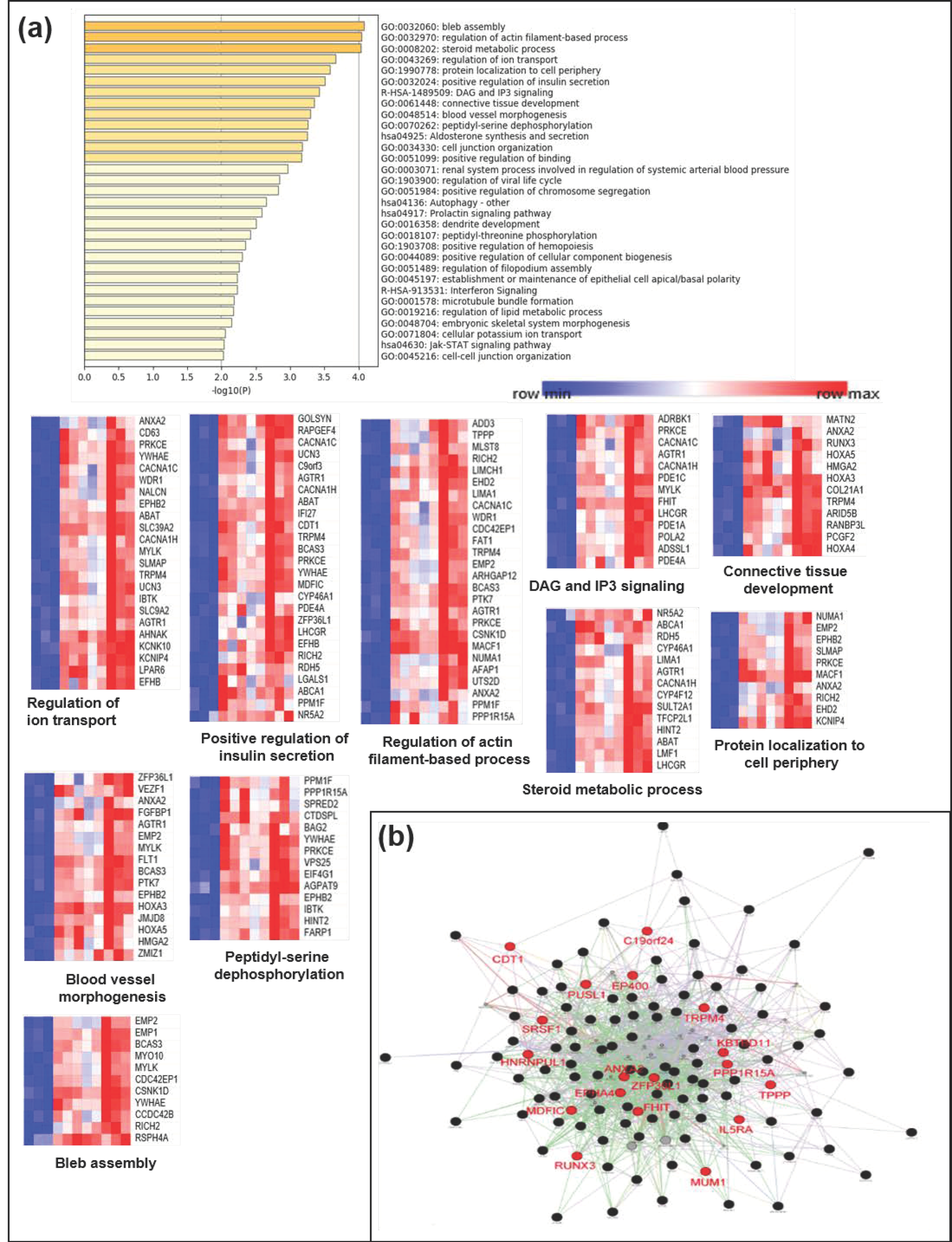
2.6. Gene Ontology and Network Analysis
2.7. Statistical Analysis
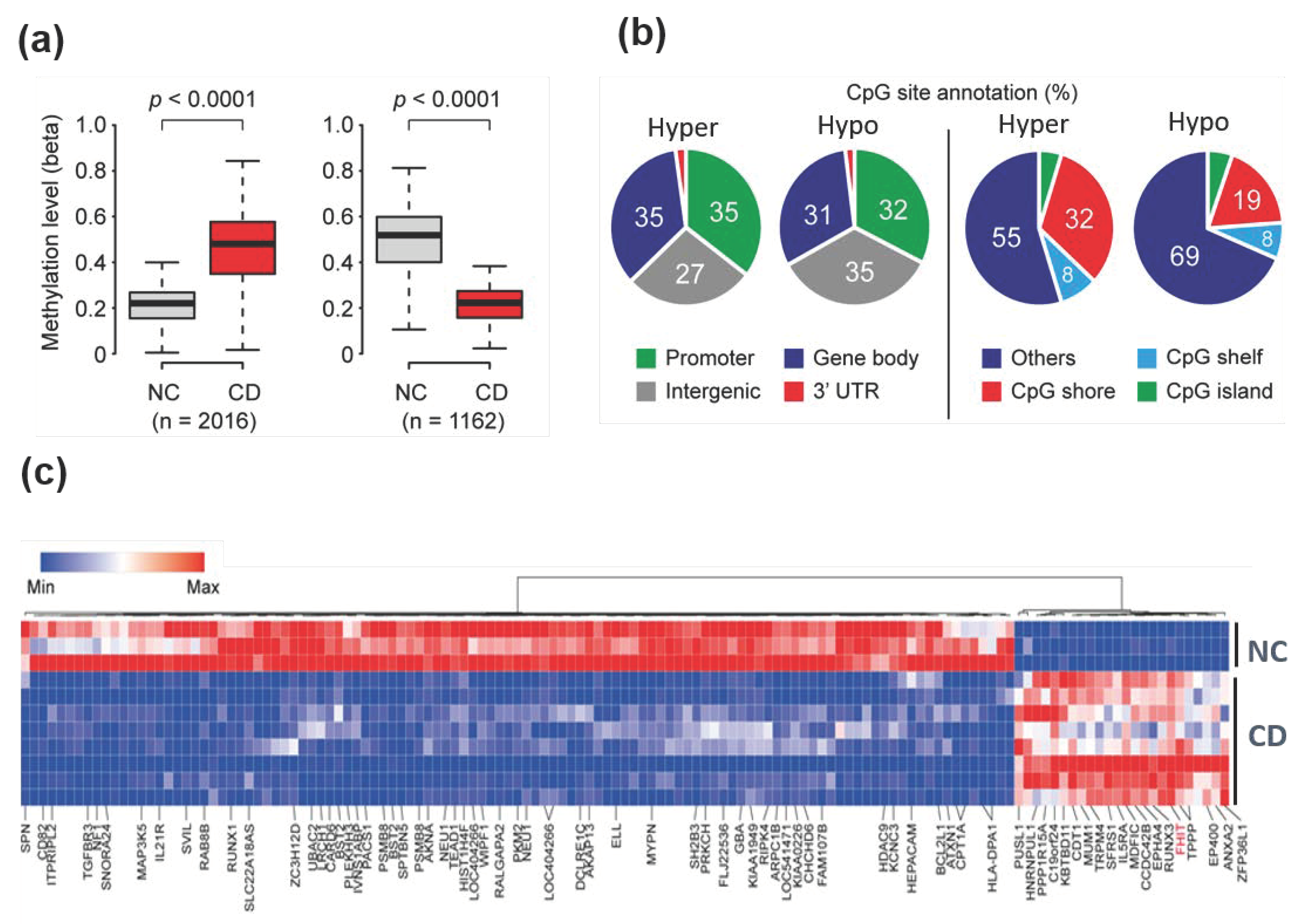
3. Results
3.1. Differential DNA Methylation Profiles in Patients with CD Compared to Normal Colon Tissues
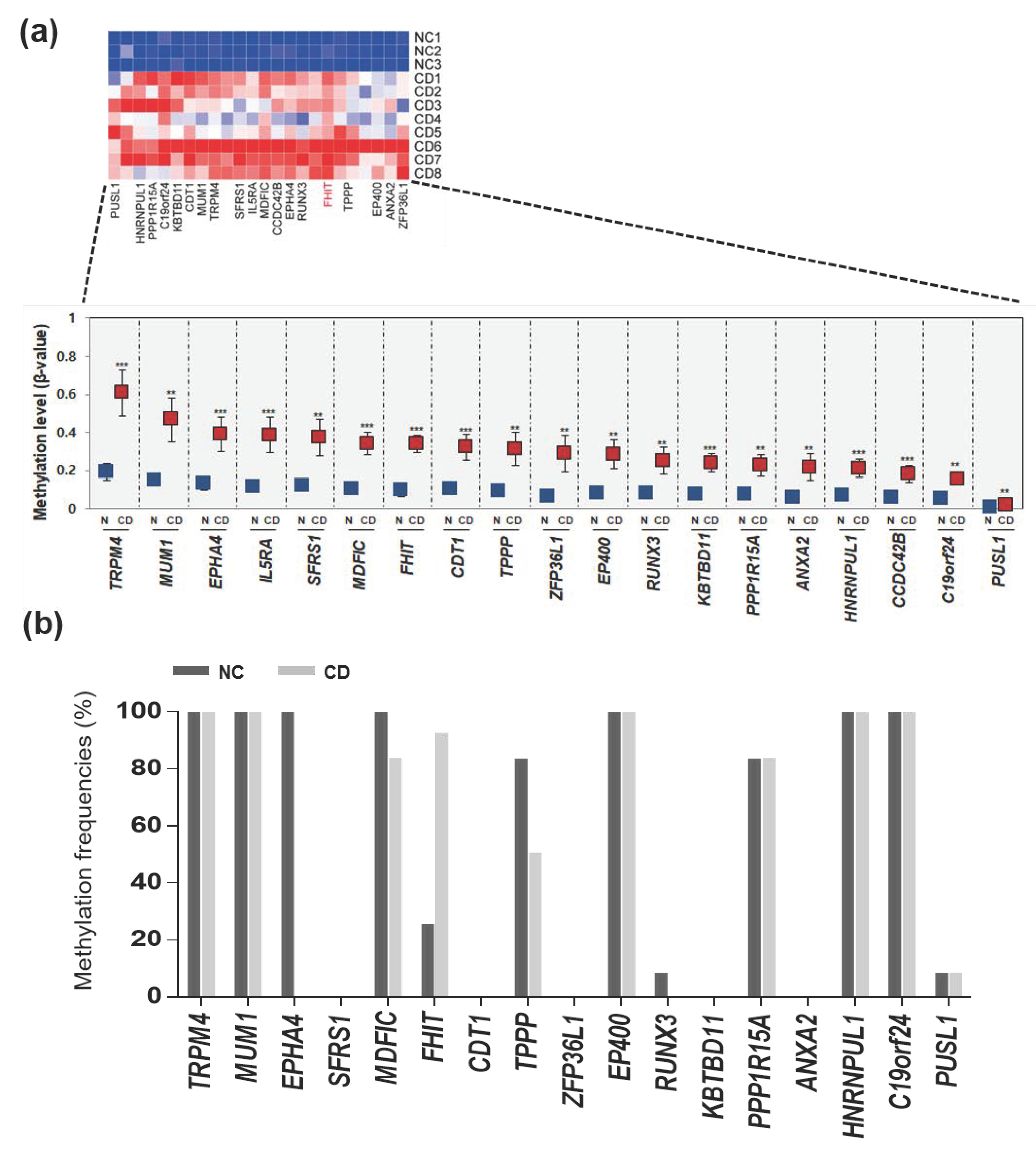
3.2. Selected Hypermethylated Candidate Genes in Patients with CD
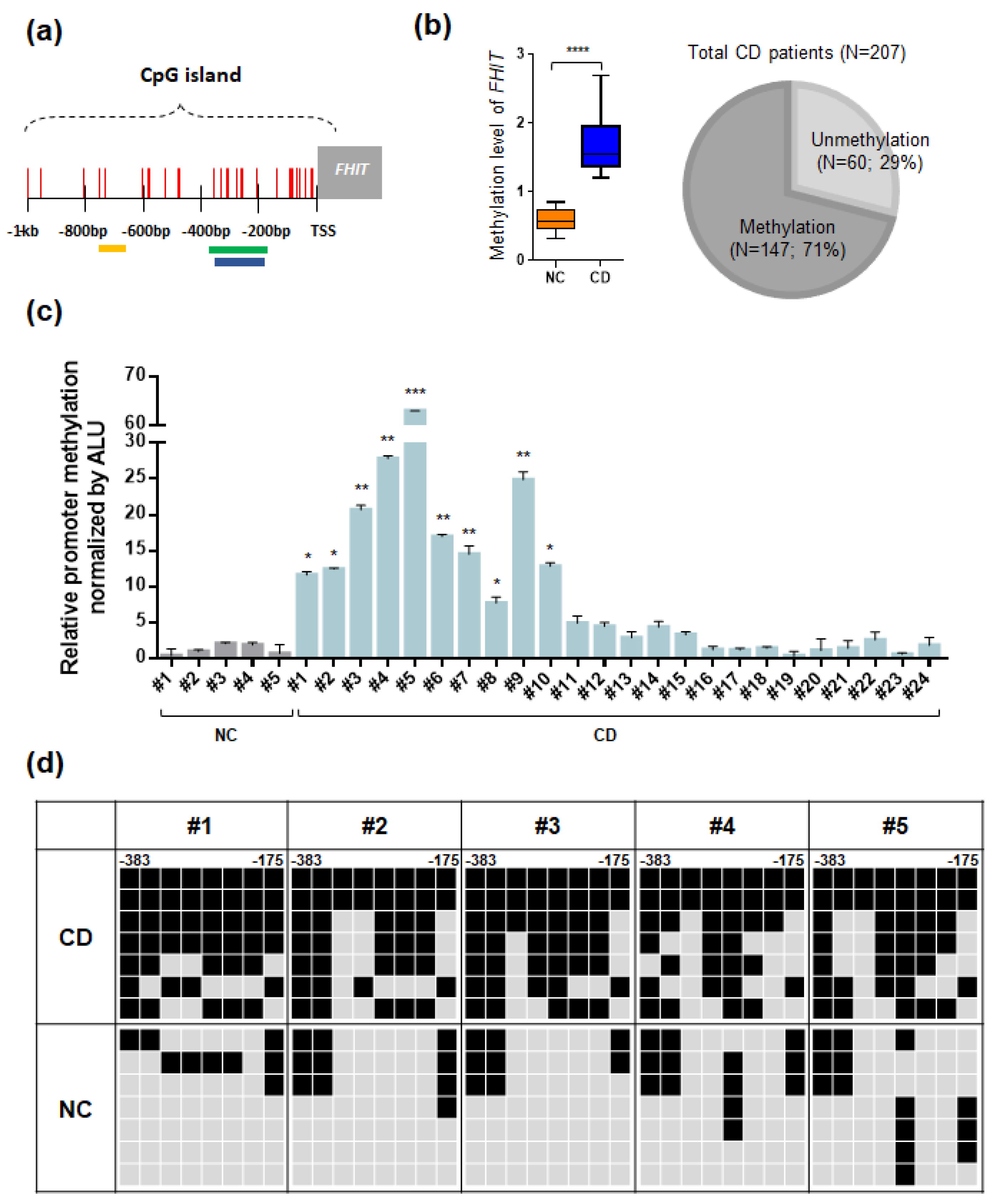
3.3. The FHIT Gene: A CD-Specific Hypermethylated Candidate Gene and its Biological Implications
3.4. Functional Implication of the Hypermethylated Genes in CD as Analyzed by a Gene Network
4. Discussion
5. Conclusions
6. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 2008, 8, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory Bowel Disease. Ann. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Van Limbergen, J.; Wilson, D.C.; Satsangi, J. The Genetics of Crohn’s Disease. Ann. Rev. Genom. Human Genet. 2009, 10, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Petronis, A.; Petroniene, R. Epigenetics of inflammatory bowel disease. Gut 2000, 47, 302. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Issa, J.P.; Ahuja, N.; Toyota, M.; Bronner, M.P.; Brentnall, T.A. Accelerated Age-related CpG Island Methylation in Ulcerative Colitis. Cancer Res. 2001, 61, 3573–3577. [Google Scholar]
- Maeda, O.; Ando, T.; Watanabe, O.; Ishiguro, K.; Ohmiya, N.; Niwa, Y.; Goto, H. DNA hypermethylation in colorectal neoplasms and inflammatory bowel disease: A mini review. Inflammopharmacology 2006, 14, 204–206. [Google Scholar] [CrossRef]
- Tahara, T.; Shibata, T.; Nakamura, M.; Yamashita, H.; Yoshioka, D.; Okubo, M.; Maruyama, N.; Kamano, T.; Kamiya, Y.; Nakagawa, Y.; et al. Effect of MDR1 gene promoter methylation in patients with ulcerative colitis. Int. J. Mol. Med. 2009, 23, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Kim, T.O. Epigenetic Alterations in Inflammatory Bowel Disease and Cancer. Intest. Res. 2015, 13, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Dhir, M.; Guzzetta, A.A.; Iacobuzio-Donahue, C.A.; Heo, K.; Yang, K.M.; Suzuki, H.; Toyota, M.; Kim, H.-M.; Ahuja, N. DNA methylation biomarker candidates for early detection of colon cancer. Tumor Biol. 2012, 33, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.; Zhang, H.; Greger, L.; Silva, A.-L.; Massey, D.; Dawson, C.; Metz, A.; Ibrahim, A.; Parkes, M. Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 2128–2137. [Google Scholar] [CrossRef]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Esteller, M.; Sanchez-Cespedes, M.; Rosell, R.; Sidransky, D.; Baylin, S.B.; Herman, J.G. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59, 67–70. [Google Scholar]
- DeVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grützmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating Methylated SEPT9 DNA in Plasma Is a Biomarker for Colorectal Cancer. Clin. Chem. 2009, 55, 1337–1346. [Google Scholar] [CrossRef]
- Bibikova, M. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef]
- Davis, S.; Du, P.; Bilke, S.; Triche, T.; Bootwalla, M. methylumi: Handle Illumina methylation data. R Package Version 2.24.1. Bioconductor. 2017. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Kang, K.; Bae, J.-H.; Han, K.; Kim, E.S.; Kim, T.-O.; Yi, J.M. A Genome-Wide Methylation Approach Identifies a New Hypermethylated Gene Panel in Ulcerative Colitis. Int. J. Mol. Sci. 2016, 17, 1291. [Google Scholar] [CrossRef]
- Tripathi, S.; Pohl, M.O.; Zhou, Y.; Rodriguez-Frandsen, A.; Wang, G.; Stein, D.A.; Moulton, H.M.; DeJesus, P.; Che, J.; Mulder, L.C.; et al. Meta- and Orthogonal Integration of Influenza “OMICS” data defines a role for UBR4 in virus budding. Cell Host Microbe 2015, 18, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Karatzas, P.S.; Gazouli, M.; Safioleas, M.; Mantzaris, G.J. DNA methylation changes in inflammatory bowel disease. Ann. Gastroent. 2014, 27, 125–132. [Google Scholar]
- Lin, Z.; Hegarty, J.P.; Yu, W.; Cappel, J.A.; Chen, X.; Faber, P.W.; Wang, Y.; Poritz, L.S.; Fan, J.-B.; Koltun, W.A. Identification of Disease-Associated DNA Methylation in B Cells from Crohn’s Disease and Ulcerative Colitis Patients. Dig. Dis. Sci. 2012, 57, 3145–3153. [Google Scholar] [CrossRef]
- Karatzas, P.S.; Mantzaris, G.J.; Safioleas, M.; Gazouli, M. DNA methylation profile of genes involved in inflammation and autoimmunity in inflammatory bowel disease. Medicine (Baltimore) 2014, 93, e309. [Google Scholar] [CrossRef]
- Nimmo, E.R.; Prendergast, J.G.; Aldhous, M.C.; Kennedy, N.A.; Henderson, P.; Drummond, H.E.; Ramsahoye, B.H.; Wilson, D.C.; Semple, C.A.; Satsangi, J. Genome-wide Methylation Profiling in Crohn’s Disease Identifies Altered Epigenetic Regulation of Key Host Defense Mechanisms Including the Th17 Pathway. Inflamm. Bowel Dis. 2011, 18, 889–899. [Google Scholar] [CrossRef]
- Bae, J.H.; Park, J.; Yang, K.M.; Kim, T.O.; Yi, J.M. Detection of DNA hypermethylation in sera of patients with Crohn’s disease. Mol. Med. Rep. 2014, 9, 725–729. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mathew, C.G. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat. Rev. Genet. 2008, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.A.; Tremelling, M.; Anderson, C.A.; Gwilliam, R.; Bumpstead, S.; Prescott, N.J.; Nimmo, E.R.; Massey, D.; Berzuini, C.; Johnson, C.; et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat. Genet. 2008, 40, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Granlund, A.v.B.; Flatberg, A.; Østvik, A.E.; Drozdov, I.; Gustafsson, B.I.; Kidd, M.; Beisvag, V.; Torp, S.H.; Waldum, H.L.; Martinsen, T.C.; et al. Whole Genome Gene Expression Meta-Analysis of Inflammatory Bowel Disease Colon Mucosa Demonstrates Lack of Major Differences between Crohn’s Disease and Ulcerative Colitis. PLoS ONE 2013, 8, e56818. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Nagy-Szakal, D.; Mir, S.A.V.; Frank, E.; Szigeti, R.; Kaplan, J.L.; Bronsky, J.; Opekun, A.; Ferry, G.D.; Winter, H.; et al. DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics 2014, 9, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.O.; Park, J.; Kang, M.J.; Lee, S.H.; Jee, S.R.; Rye, D.Y.; Yang, K.; Yi, J.M. DNA hypermethylation of a selective gene panel as a risk marker for colon cancer in patients with ulcerative colitis. Intern. J. Mol. Med. 2013, 31, 1255–1261. [Google Scholar] [CrossRef][Green Version]
- McDermott, E.; Ryan, E.J.; Tosetto, M.; Gibson, D.; Burrage, J.; Keegan, D.; Byrne, K.; Crowe, E.; Sexton, G.; Malone, K.; et al. DNA Methylation Profiling in Inflammatory Bowel Disease Provides New Insights into Disease Pathogenesis. J. Crohn’s Col. 2015, 10, 77–86. [Google Scholar] [CrossRef]
- Barnes, L.D.; Garrison, P.N.; Siprashvili, Z.; Guranowski, A.; Robinson, A.K.; Ingram, S.W.; Croce, C.M.; Ohta, M.; Huebner, K. Fhit, a Putative Tumor Suppressor in Humans, Is a Dinucleoside 5’,5’’’-P1,P3-Triphosphate Hydrolase. Biochemistry 1996, 35, 11529–11535. [Google Scholar] [CrossRef]
- Nishimura, A. The timing of cell division: Ap4A as a signal. Trends Biochem. Sci. 1998, 23, 157–159. [Google Scholar] [CrossRef]
- Huebner, K.; Garrison, P.N.; Barnes, L.D.; Croce, C.M. The role of the fhit/fra3b locus in cancer. Ann. Rev. Genet. 1998, 32, 7–31. [Google Scholar] [CrossRef]
- Wang, H.-L.; Zhou, P.-Y.; Liu, P.; Zhang, Y. Abnormal FHIT protein expression may be correlated with poor prognosis in gastric cancer: A meta-analysis. Tumor Biol. 2014, 35, 6815–6821. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.Q.; Wang, S.J.; Zhang, L.W.; Wang, X.L.; Zhang, J.H.; Guo, W. DNA methylation and loss of protein expression in esophageal squamous cell carcinogenesis of high-risk area. J. Exp. Clin. Cancer Res. 2007, 26, 587–594. [Google Scholar] [PubMed]
- Younes, S.F.; Aiad, H.A.; Asaad, N.Y.; Kandil, M.A.; Natkunam, Y.; Mokhtar, N.M. FHIT, EGFR, and MSH2: Possible Etiopathologic, Prognostic, and Predictive Role in Non–Small Cell Lung Carcinoma in Egyptian Patients. Appl. Immunohistochem. Mol. Morph. 2014, 22. [Google Scholar] [CrossRef] [PubMed]
- Kapitanović, S.; Čačev, T.; Lončar, B.; Catela Ivković, T.; Križanac, Š.; Pavelić, K. Reduced FHIT expression is associated with tumor progression in sporadic colon adenocarcinoma. Exp. Mol. Pathol. 2014, 96, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.Y.Y.; Fidanza, V.; Zanesi, N.; Lock, L.F.; Siracusa, L.D.; Mancini, R.; Siprashvili, Z.; Ottey, M.; Martin, S.E.; Druck, T.; et al. Muir–Torre-like syndrome in Fhit-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4742–4747. [Google Scholar] [CrossRef]
- Zanesi, N.; Fidanza, V.; Fong, L.Y.; Mancini, R.; Druck, T.; Valtieri, M.; Rüdiger, T.; McCue, P.A.; Croce, C.M.; Huebner, K. The tumor spectrum in FHIT-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10250–10255. [Google Scholar] [CrossRef]
- Joannes, A.; Grelet, S.; Duca, L.; Gilles, C.; Kileztky, C.; Dalstein, V.; Birembaut, P.; Polette, M.; Nawrocki-Raby, B. Fhit Regulates EMT Targets through an EGFR/Src/ERK/Slug Signaling Axis in Human Bronchial Cells. Mol. Cancer Res. 2014, 12, 775. [Google Scholar] [CrossRef]
- Joannes, A.; Bonnomet, A.; Bindels, S.; Polette, M.; Gilles, C.; Burlet, H.; Cutrona, J.; Zahm, J.M.; Birembaut, P.; Nawrocki-Raby, B. Fhit regulates invasion of lung tumor cells. Oncogene 2009, 29, 1203–1213. [Google Scholar] [CrossRef][Green Version]
- Liu, L.; Sun, L.; Li, C.; Li, X.; Zhang, Y.; Yu, Y.; Xia, W. Quantitative detection of methylation of FHIT and BRCA1 promoters in the serum of ductal breast cancer patients. Bio-med. Mat. Eng. 2015, 26 (Suppl. 1), S2217–S2222. [Google Scholar] [CrossRef]
- Geng, X.; Pu, W.; Tan, Y.; Lu, Z.; Wang, A.; Tan, L.; Chen, S.; Guo, S.; Wang, J.; Chen, X. Quantitative assessment of the diagnostic role of FHIT promoter methylation in non-small cell lung cancer. Oncotarget 2017, 8, 6845–6856. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | n = 207 |
|---|---|
| Age (years) | |
| Median | 33 |
| Gender | |
| Male | 152 (73.6%) |
| Female | 55 (26.4%) |
| Duration from diagnosis to sampling | |
| ≤1 year | 26 (12.5%) |
| 1–5 year | 76 (37.0%) |
| ≥5 year | 105 (50.5%) |
| Months (median) | 63 |
| Disease location at diagnosis | |
| Small bowel alone | 58 (27.9%) |
| Colon alone | 23 (11.1%) |
| Small bowel and colon | 126 (61.1%) |
| Disease behavior at diagnosis | |
| Inflammatory (B1) | 130 (62.5%) |
| Stricturing (B2) | 32 (15.9%) |
| Penetrating (B3) | 45 (21.6%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.-O.; Park, D.-I.; Han, Y.K.; Kang, K.; Park, S.-G.; Park, H.R.; Yi, J.M. Genome-Wide Analysis of the DNA Methylation Profile Identifies the Fragile Histidine Triad (FHIT) Gene as a New Promising Biomarker of Crohn’s Disease. J. Clin. Med. 2020, 9, 1338. https://doi.org/10.3390/jcm9051338
Kim T-O, Park D-I, Han YK, Kang K, Park S-G, Park HR, Yi JM. Genome-Wide Analysis of the DNA Methylation Profile Identifies the Fragile Histidine Triad (FHIT) Gene as a New Promising Biomarker of Crohn’s Disease. Journal of Clinical Medicine. 2020; 9(5):1338. https://doi.org/10.3390/jcm9051338
Chicago/Turabian StyleKim, Tae-Oh, Dong-Il Park, Yu Kyeong Han, Keunsoo Kang, Sae-Gwang Park, Hae Ryoun Park, and Joo Mi Yi. 2020. "Genome-Wide Analysis of the DNA Methylation Profile Identifies the Fragile Histidine Triad (FHIT) Gene as a New Promising Biomarker of Crohn’s Disease" Journal of Clinical Medicine 9, no. 5: 1338. https://doi.org/10.3390/jcm9051338
APA StyleKim, T.-O., Park, D.-I., Han, Y. K., Kang, K., Park, S.-G., Park, H. R., & Yi, J. M. (2020). Genome-Wide Analysis of the DNA Methylation Profile Identifies the Fragile Histidine Triad (FHIT) Gene as a New Promising Biomarker of Crohn’s Disease. Journal of Clinical Medicine, 9(5), 1338. https://doi.org/10.3390/jcm9051338