Inhibition of Protein-Tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-Challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress


 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell and Culture Conditions
2.2. FFA/BSA Complex Preparation and Related Cell Treatments
2.3. Cytotoxicity Assay
2.4. Oil Red O Staining
2.5. Fluorescent Detection of Intracellular Lipid Droplets
2.6. Flow Cytometric Analysis of Early Apoptosis
2.7. Quantification of Multicaspase Activity
2.8. Mitochondrial Transmembrane Potential
2.9. Measurement of Intracellular ROS
2.10. Protein Carbonylation Assay
2.11. Gene Expression Analysis
2.12. Metabolomic Profiling of HepG2 FAAs
2.12.1. Cell lysis and Metabolite Extraction
2.12.2. Preparation of HepG2 Fatty Acid Methyl Esters (FAMEs)
2.12.3. FAME Qualitative and Quantitative Analysis
2.13. Statistics
3. Results
3.1. Effects of FFA and PTP Inhibitor Treatments on HepG2 Cell Viability
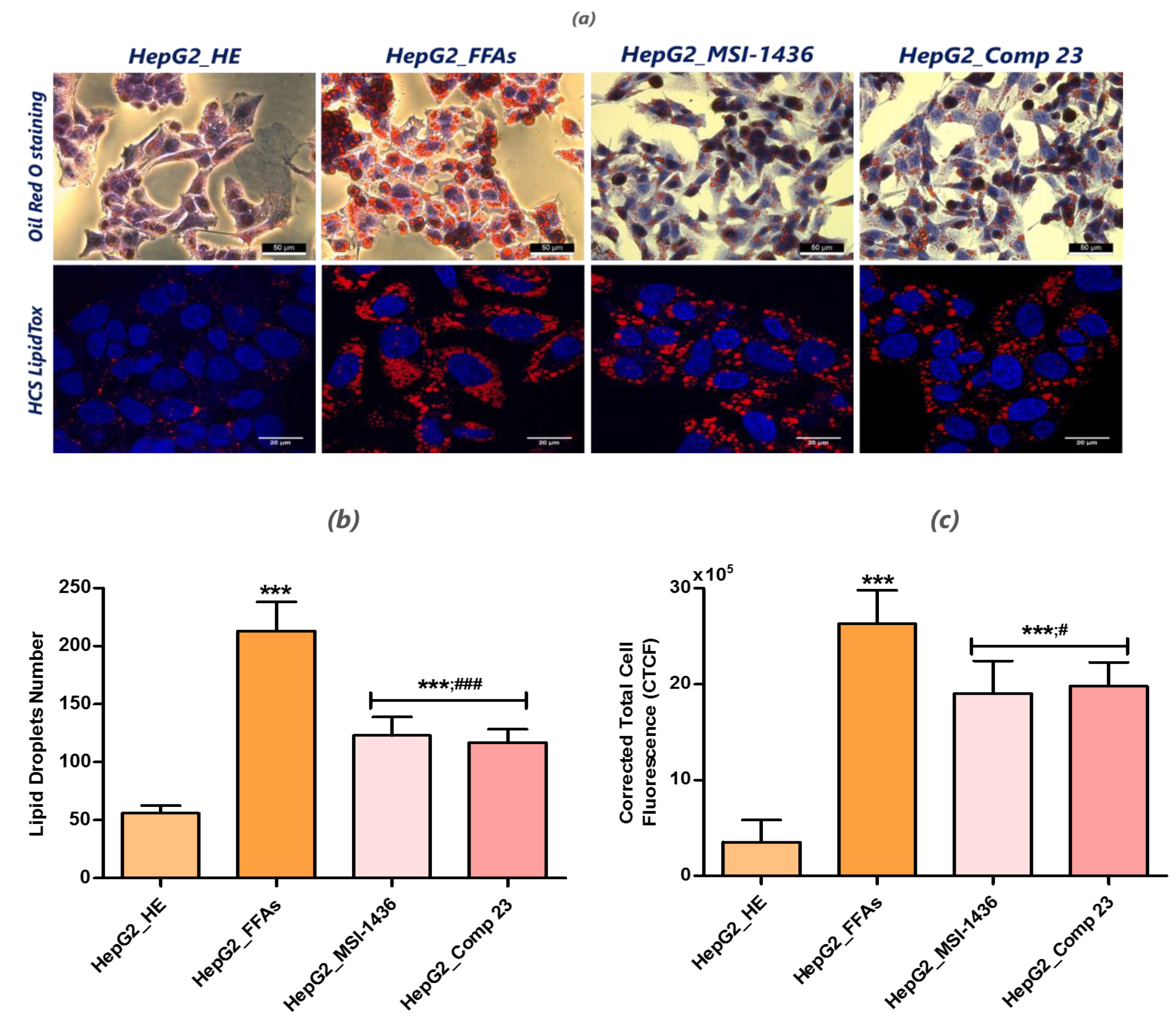
3.2. Analysis of Lipid Accumulation in the FFA-Overloaded and Treated Cells
3.3. MSI-1436 and Compound 23 Inhibit FFA-Induced Lipo-Apoptosis in HepG2 Cells
3.4. MSI-1436 and Compound 23 Improve Mitochondrial Dynamics in HepG2 FFA-Treated Cells
3.5. MSI-1436 and Compound 23 Reduce the Oxidative Stress Generated in HepG2 FFA-Treated Cells
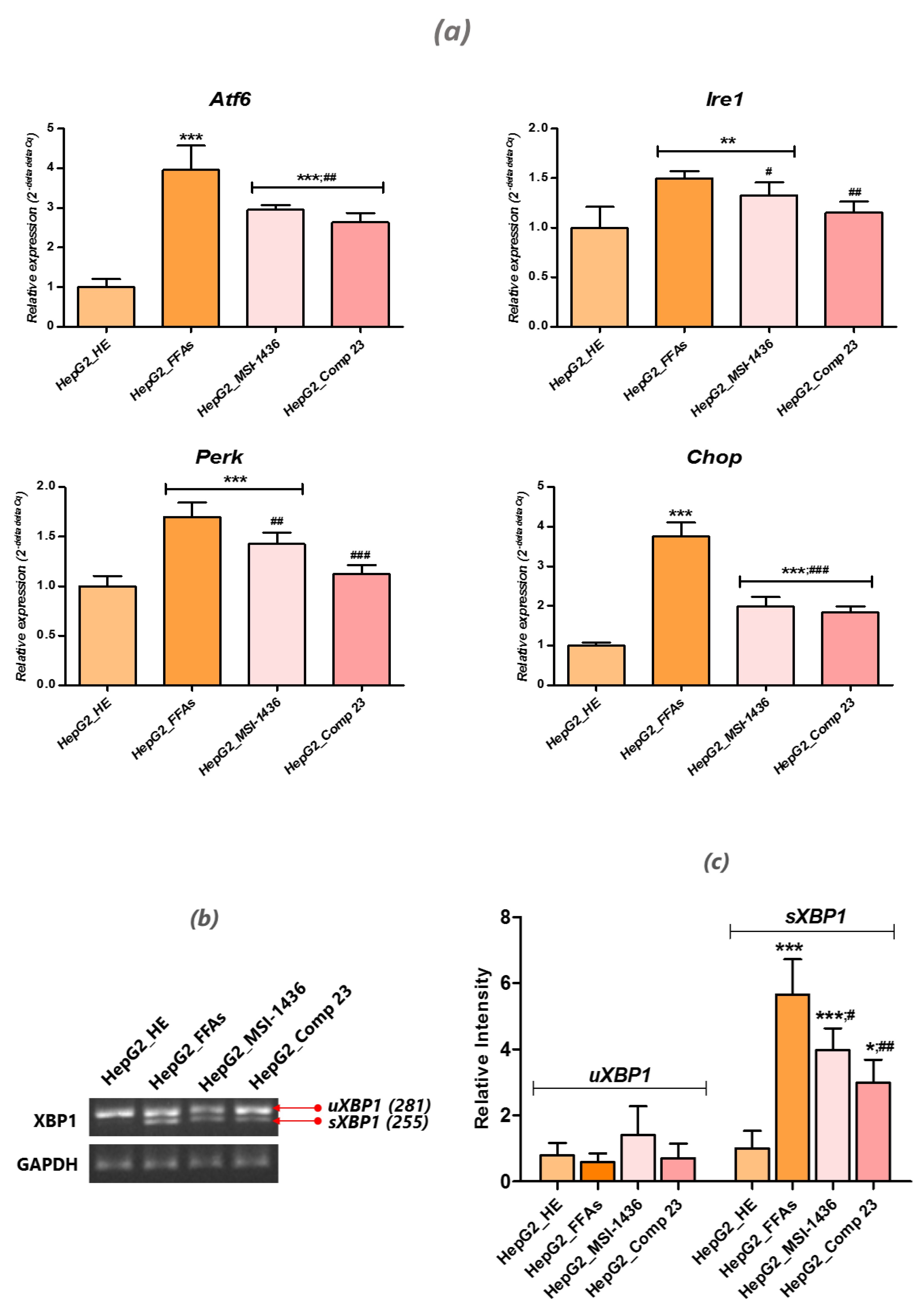
3.6. PTP Inhibitors Attenuate FFA-Induced ER Stress in HepG2 Cells
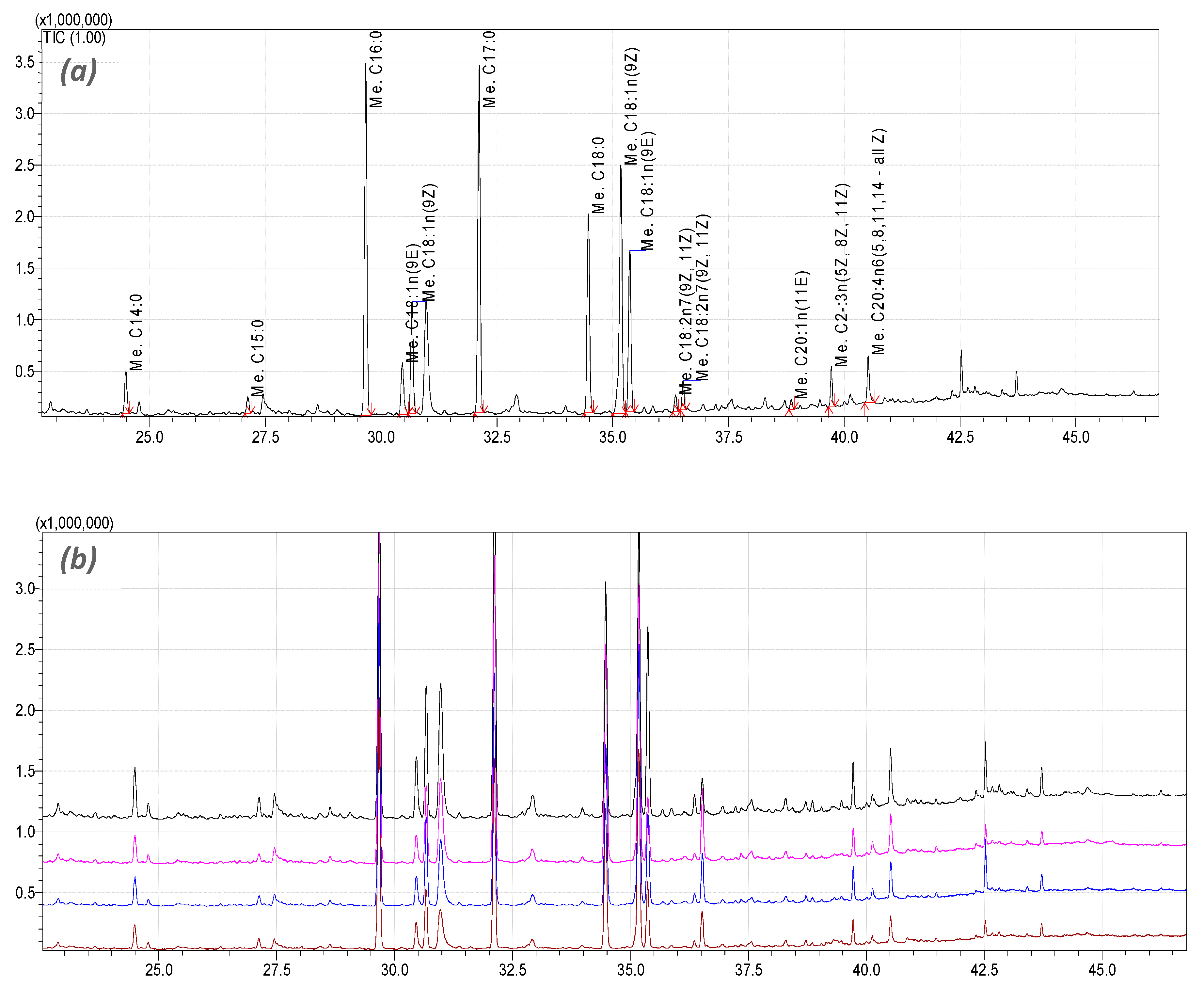
3.7. PTP Inhibitors Ameliorate the Free Fatty Acid Composition Changes in HepG2 Cells Challenged with the PA/OA Mixture
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 7-AAD | cid Phosphatase 1 |
| AM | Acetoxymethyl |
| DAG | Diacylglycerol |
| DNPH | 2,4-Dinitrophenyl Hydrazine |
| EFAs | Essential Fatty Acids |
| FAMEs | Fatty Acid Methyl Esters |
| FFAs | Free Fatty Acids |
| GC-MS | Gas chromatography–mass spectrometry |
| HBSS | Hanks’ Balanced Salt solution |
| HCC | Hepatocellular Carcinoma |
| HepG2 | Human hepatocarcinoma |
| HER | Human Epidermal Growth Factor Receptor |
| HFD | High-Fat diet |
| HPCs | Equine Hepatic Progenitor-like Cells |
| HTN | Systemic Hypertension |
| INRS | Insulin Receptor |
| JAK2 | Janus Kinase 2 |
| KO | Knockout |
| LMPTP | Low Molecular Weight Protein Tyrosine Phosphatase |
| MetS | Metabolic Syndrome |
| MSI-1436 | Trodusquemine |
| MUFAs | Monounsaturated Fatty Acids |
| NAFLD | Non-alcoholic Fatty Liver Disease |
| NASH | Nonalcoholic Steatohepatitis |
| OA | oleate |
| PA | palmitate |
| PGC-1 | Peroxisome Proliferator-activated Receptor γ Coactivator 1 |
| PI | Propidium Iodide |
| Prkn | Parkin RBR E3 Ubiquitin Protein Ligase |
| PTP1B | Protein-tyrosine phosphatase 1B |
| PTPN1 | Protein Tyrosine Phosphatase Non-receptor type 1 |
| PTPs | Protein Tyrosine Phosphatases |
| PUFA | Polyunsaturated Fatty Acids |
| ROS | Reactive Oxygen Species |
| RT-PCR | Reverse transcription Polymerase Chain Reaction |
| SCD | Stearoyl-CoA Desaturase |
| T2DM | Type 2 Diabetes |
| UPR | Unfolded Protein Response |
References
- Bhatt, H.B.; Smith, R.J. Fatty liver disease in diabetes mellitus. Hepatobiliary Surg. Nutr. 2015, 4, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Touros, A.; Kim, W.R. Nonalcoholic Fatty Liver Disease and Metabolic Syndrome. Clin. Liver Dis. 2018, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Falck-Ytter, Y.; Younossi, Z.M.; Marchesini, G.; McCullough, A.J. Clinical Features and Natural History of Nonalcoholic Steatosis Syndromes. Semin. Liver Dis. 2001, 21, 17–26. [Google Scholar] [CrossRef]
- Makowski, L.; Hotamisligil, G.S. Fatty Acid Binding Proteins—The Evolutionary Crossroads of Inflammatory and Metabolic Responses. J. Nutr. 2004, 134, 2464S–2468S. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic Fatty Liver Disease. JAMA 2015, 313, 2263. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Chen, P.-J.; Cai, S.-P.; Yang, Y.; Li, W.-X.; Huang, C.; Meng, X.-M. PTP1B confers liver fibrosis by regulating the activation of hepatic stellate cells. Toxicol. Appl. Pharmacol. 2016, 292, 8–18. [Google Scholar] [CrossRef]
- Bence, K.K. Hepatic PTP1B Deficiency: The Promise of a Treatment for Metabolic Syndrome? J. Clin. Metab. Diabetes 2010, 1, 27–33. [Google Scholar]
- Panzhinskiy, E.; Ren, J.; Nair, S. Pharmacological Inhibition of Protein Tyrosine Phosphatase 1B: A Promising Strategy for the Treatment of Obesity and Type 2 Diabetes Mellitus. Curr. Med. Chem. 2013, 20, 2609–2625. [Google Scholar] [CrossRef]
- Sanderson, S.O.; Smyrk, T.C. The Use of Protein Tyrosine Phosphatase 1B and Insulin Receptor Immunostains to Differentiate Nonalcoholic from Alcoholic Steatohepatitis in Liver Biopsy Specimens. Am. J. Clin. Pathol. 2005, 123, 503–509. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Rashid-Kolvear, F.; Van Iderstine, S.C.; Le-Tien, H.; Fantus, I.G.; Lewis, G.F. Hepatic Very Low Density Lipoprotein-ApoB Overproduction Is Associated with Attenuated Hepatic Insulin Signaling and Overexpression of Protein-tyrosine Phosphatase 1B in a Fructose-fed Hamster Model of Insulin Resistance. J. Biol. Chem. 2002, 277, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.-G.; Cho, Y.-R. Liver-Specific Deletion of Protein-Tyrosine Phosphatase 1B (PTP1B) Improves Metabolic Syndrome and Attenuates Diet-Induced Endoplasmic Reticulum Stress. Diabetes 2009, 58, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Haj, F.G.; Zabolotny, J.M.; Kim, Y.-B.; Kahn, B.B.; Neel, B.G. Liver-specific Protein-tyrosine Phosphatase 1B (PTP1B) Re-expression Alters Glucose Homeostasis of PTP1B−/−Mice. J. Biol. Chem. 2005, 280, 15038–15046. [Google Scholar] [CrossRef] [PubMed]
- Zinker, B.A.; Rondinone, C.M.; Trevillyan, J.M.; Gum, R.J.; Clampit, J.E.; Waring, J.F. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11357–11362. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y. PTP1B Regulates Leptin Signal Transduction In Vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Ramponi, G.; Stefani, M. Structural, catalytic, and functional properties of low Mr phosphotyrosine protein phosphatases. Evidence of a long evolutionary history. Int. J. Biochem. Cell Biol. 1997, 29, 279–292. [Google Scholar] [CrossRef]
- Ahmad, F.; Goldstein, B.J. Alterations in specific protein-tyrosine phosphatases accompany insulin resistance of streptozotocin diabetes. Am. J. Physiol.-Endocrinol. Metab. 1995, 268, E932–E940. [Google Scholar] [CrossRef]
- McGuire, M.C.; Fields, R.M.; Nyomba, B.L.; Raz, I.; Bogardus, C.; Tonks, N.K. Abnormal Regulation of Protein Tyrosine Phosphatase Activities in Skeletal Muscle of Insulin-Resistant Humans. Diabetes 1991, 40, 939–942. [Google Scholar] [CrossRef]
- Goldstein, B.J. Protein-Tyrosine phosphatases and the regulation of insulin action. J. Cell. Biochem. 1992, 48, 33–42. [Google Scholar] [CrossRef]
- Lucarini, N.; Antonacci, E.; Bottini, N.; Borgiani, P.; Faggioni, G.; Gloria-Bottini, F. Phosphotyrosine-Protein-Phosphatase and Diabetic Disorders. Further Studies on the Relationship between Low Molecular Weight Acid Phosphatase Genotype and Degree of Glycemic Control. Dis. Markers 1998, 14, 121–125. [Google Scholar] [CrossRef]
- Bottini, N.; MacMurray, J.; Peters, W.; Rostamkhani, M.; Comings, D.E. Association of the acid phosphatase (ACP1) gene with triglyceride levels in obese women. Mol. Genet. Metab. 2002, 77, 226–229. [Google Scholar] [CrossRef]
- Iannaccone, U.; Bergamaschi, A.; Magrini, A.; Marino, G.; Bottini, N.; Lucarelli, P. Serum glucose concentration and ACP1 genotype in healthy adult subjects. Metabolism 2005, 54, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.K.; Yu, X.X.; Watts, L.M.; Michael, M.D.; Sloop, K.W.; Rivard, A.R. Reduction of Low Molecular Weight Protein-tyrosine Phosphatase Expression Improves Hyperglycemia and Insulin Sensitivity in Obese Mice. J. Biol. Chem. 2007, 282, 14291–14299. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P.; Marra, F.; Raugei, G.; Camici, G.; Manao, G. LMW-PTP Is a Negative Regulator of Insulin-Mediated Mitotic and Metabolic Signalling. Biochem. Biophys. Res. Commun. 1997, 238, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-M.; Li, Y.-C.; Kong, L.-D.; Hu, Q.-H. Curcumin inhibits hepatic protein-tyrosine phosphatase 1B and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology 2010, 51, 1555–1566. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Gutierrez, J.A.M.; Sanz-Gonzalez, S.; Ros, M.; Burks, D.J.; Valverde, A.M. Inhibition of PTP1B Restores IRS1-Mediated Hepatic Insulin Signaling in IRS2-Deficient Mice. Diabetes 2010, 59, 588–599. [Google Scholar] [CrossRef]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef]
- Stanford, S.M.; Aleshin, A.E.; Zhang, V.; Ardecky, R.J.; Hedrick, M.P.; Zou, J. Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol. 2017, 13, 624–632. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem.-Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Bai, J.; Wang, M.X.; Chowbay, B.; Ching, C.B.; Chen, W.N. Metabolic profiling of HepG2 cells incubated with S(−) and R(+) enantiomers of anti-coagulating drug warfarin. Metabolomics 2011, 7, 353–362. [Google Scholar] [CrossRef]
- Guo, W.; Wong, S.; Xie, W.; Lei, T.; Luo, Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, E576–E586. [Google Scholar] [CrossRef] [PubMed]
- Dyntar, D.; Eppenberger-Eberhardt, M.; Maedler, K.; Pruschy, M.; Eppenberger, H.M.; Spinas, G.A. Glucose and Palmitic Acid Induce Degeneration of Myofibrils and Modulate Apoptosis in Rat Adult Cardiomyocytes. Diabetes 2001, 50, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Cell Deficit and Increased -Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Grill, V. Long term exposure to fatty acids and ketones inhibits Bcell functions in human pancreatic islets of Langerhans. J. Clin. Endocrinol. Metab. 1995, 80, 1584–1890. [Google Scholar] [PubMed]
- El-Assaad, W.; Buteau, J.; Peyot, M.-L.; Nolan, C.; Roduit, R.; Hardy, S. Saturated Fatty Acids Synergize with Elevated Glucose to Cause Pancreatic β-Cell Death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Agouni, A.; Mody, N.; Owen, C.; Czopek, A.; Zimmer, D.; Bentires-Alj, M. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem. J. 2011, 438, 369–378. [Google Scholar] [CrossRef]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar] [CrossRef]
- Anderson, S.M.; Cheesman, H.K.; Peterson, N.D.; Salisbury, J.E.; Soukas, A.A.; Pukkila-Worley, R. The fatty acid oleate is required for innate immune activation and pathogen defense in Caenorhabditis elegans. PLoS Pathog. 2019, 15, e1007893. [Google Scholar] [CrossRef]
- Baylin, A.; Kabagambe, E.K.; Siles, X.; Campos, H. Adipose tissue biomarkers of fatty acid intake. Am. J. Clin. Nutr. 2002, 76, 750–757. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Cao, H.; King, I.B.; Lemaitre, R.N.; Song, X.; Siscovick, D.S. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am. J. Clin. Nutr. 2010, 92, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Ntambi, J.M. Role of stearoyl-CoA desaturase in human metabolic disease. Future Lipidol. 2008, 3, 163–173. [Google Scholar] [CrossRef]
- Rahman, S.M.; Dobrzyn, A.; Dobrzyn, P.; Lee, S.-H.; Miyazaki, M.; Ntambi, J.M. Stearoyl-CoA desaturase 1 deficiency elevates insulin-signaling components and down-regulates protein-tyrosine phosphatase 1B in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 11110–11115. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.T. Probing the role of stearoyl-CoA desaturase_1 in hepatic insulin resistance. J. Clin. Investig. 2006, 116, 1478–1481. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Juarez, R. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Investig. 2006, 116, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Di Marzo, V. Arachidonic acid and eicosanoids as targets and effectors in second messenger interactions. Prostaglandins Leukot. Essent. Fat. Acids 1995, 53, 239–254. [Google Scholar] [CrossRef]
- González-Rodríguez, Á.; Valdecantos, M.P.; Rada, P.; Addante, A.; Barahona, I.; Rey, E. Dual role of protein tyrosine phosphatase 1B in the progression and reversion of non-alcoholic steatohepatitis. Mol. Metab. 2018, 7, 132–146. [Google Scholar] [CrossRef]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic Palmitate But Not Oleate Exposure Induces Endoplasmic Reticulum Stress, Which May Contribute to INS-1 Pancreatic β-Cell Apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef]
- Yuzefovych, L.; Wilson, G.; Rachek, L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: Role of oxidative stress. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E1096–E1105. [Google Scholar] [CrossRef]
- Zhang, Q.; Kong, X.; Yuan, H.; Guan, H.; Li, Y.; Niu, Y. Mangiferin Improved Palmitate-Induced-Insulin Resistance by Promoting Free Fatty Acid Metabolism in HepG2 and C2C12 Cells via PPAR α: Mangiferin Improved Insulin Resistance. J. Diabetes Res. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- González-Rodriguez, Á.; Escribano, Ó.; Alba, J.; Rondinone, C.M.; Benito, M.; Valverde, Á.M. Levels of protein tyrosine phosphatase 1B determine susceptibility to apoptosis in serum-deprived hepatocytes. J. Cell. Physiol. 2007, 212, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, Z.; Wang, L. Small interference RNA against PTP-1B reduces hypoxia/reoxygenation induced apoptosis of rat cardiomyocytes. Apoptosis 2008, 13, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Paliouras, G.N.; Cheng, A.; Dubé, N.; Tremblay, M.L.; Park, M. Protein-tyrosine Phosphatase 1B Deficiency Protects against Fas-induced Hepatic Failure. J. Biol. Chem. 2006, 281, 221–228. [Google Scholar] [CrossRef] [PubMed]
- MohammadTaghvaei, N.; Meshkani, R.; Taghikhani, M.; Larijani, B.; Adeli, K. Palmitate Enhances Protein Tyrosine Phosphatase 1B (PTP1B) Gene Expression at Transcriptional Level in C2C12 Skeletal Muscle Cells. Inflammation 2011, 34, 43–48. [Google Scholar] [CrossRef]
- Shi, D.; Li, J.; Jiang, B.; Guo, S.; Su, H.; Wang, T. Bromophenols as inhibitors of protein tyrosine phosphatase 1B with antidiabetic properties. Bioorganic Med. Chem. Lett. 2012, 22, 2827–2832. [Google Scholar] [CrossRef]
- Shao, J.; Gao, Y.; Yuan, Z. Free fatty acids promoting PTP1B expression in rat skeletal muscle and hepatic cells. Chin. Med. J. 1998, 78, 753–755. [Google Scholar]
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 Type I/Type II model. Semin. Immunol. 2003, 15, 185–193. [Google Scholar] [CrossRef]
- Alicka, M.; Kornicka-Garbowska, K.; Roecken, M.; Marycz, K. Inhibition of the Low Molecular Weight Protein Tyrosine Phosphatase (LMPTP) as a Potential Therapeutic Strategy for Hepatic Progenitor Cells Lipotoxicity—Short Communication. Int. J. Mol. Sci. 2019, 20, 5873. [Google Scholar] [CrossRef]
- Rachek, L.I.; Musiyenko, S.I.; LeDoux, S.P.; Wilson, G.L. Palmitate Induced Mitochondrial Deoxyribonucleic Acid Damage and Apoptosis in L6 Rat Skeletal Muscle Cells. Endocrinology 2007, 148, 293–299. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Solodushko, V.A.; Wilson, G.L.; Rachek, L.I. Protection from Palmitate-Induced Mitochondrial DNA Damage Prevents from Mitochondrial Oxidative Stress, Mitochondrial Dysfunction, Apoptosis, and Impaired Insulin Signaling in Rat L6 Skeletal Muscle Cells. Endocrinology 2012, 153, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, X.; Zhu, Y.; Li, X.; Xu, Q.; Lin, H. Tetramethylpyrazine protects palmitate-induced oxidative damage and mitochondrial dysfunction in C2C12 myotubes. Life Sci. 2011, 88, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Coll, T.; Jove, M.; Rodriguez-Calvo, R.; Eyre, E.; Palomer, X.; Sanchez, R.M. Palmitate-Mediated Downregulation of Peroxisome Proliferator-Activated Receptor-Coactivator 1 in Skeletal Muscle Cells Involves MEK1/2 and Nuclear Factor- B Activation. Diabetes 2006, 55, 2779–2787. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.K.; Kashyap, S.; Bajaj, M.; Cusi, K.; Mandarino, S.J.; Finlayson, J. Lipid Infusion Decreases the Expression of Nuclear Encoded Mitochondrial Genes and Increases the Expression of Extracellular Matrix Genes in Human Skeletal Muscle. J. Biol. Chem. 2005, 280, 10290–10297. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, F.; Ge, X.; Yan, T.; Chen, X.; Shi, X. SIRT1 Improves Insulin Sensitivity under Insulin-Resistant Conditions by Repressing PTP1B. Cell Metab. 2007, 6, 307–319. [Google Scholar] [CrossRef]
- Taheripak, G.; Bakhtiyari, S.; Rajabibazl, M.; Pasalar, P.; Meshkani, R. Protein tyrosine phosphatase 1B inhibition ameliorates palmitate-induced mitochondrial dysfunction and apoptosis in skeletal muscle cells. Free Radic. Biol. Med. 2013, 65, 1435–1446. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Gu, F.; Nguyên, D.T.; Stuible, M.; Dubé, N.; Tremblay, M.L.; Chevet, E. Protein-tyrosine Phosphatase 1B Potentiates IRE1 Signaling during Endoplasmic Reticulum Stress. J. Biol. Chem. 2004, 279, 49689–49693. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence 5′–3′ | Amplicon Length (bp) | Accession No. |
|---|---|---|---|---|
| Mnf1 | F: R: | GTTGCCGGGTGATAGTTGGA TGCCACCTTCATGTGTCTCC | 146 | NM_033540.3 |
| Fis1 | F: R: | TGGTGCGGAGCAAGTACAAT TGCCCACGAGTCCATCTTTC | 132 | NM_016068.3 |
| Pink1 | F: R: | GCTTGGGACCTCTCTTGGAT CGAAGCCATCTTGAACACAA | 142 | NM_032409.3 |
| Mff | F: R: | TCTCAGCCAACCACCTCTGA TGAGAGCCACTTTTGTCCCC | 109 | NM_001277061.2 |
| Atf6 | F: R: | ACCTCCTTGTCAGCCCCTAA CACTCCCTGAGTTCCTGCTG | 150 | NM_007348.4 |
| Perk | F: R: | TGCTCCCACCTCAGCGAC TTTCAGGATCCAAGGCAGCA | 124 | NM_004836.6 |
| Chop | F: R: | TAAAGATGAGCGGGTGGCAG GGATAATGGGGAGTGGCTGG | 103 | NM_001195053.1 |
| Ire1 | F: R: | CGGCCTCGGGATTTTTGGA AGAAAGGCAGGCTCTTCCAC | 110 | NM_001433.5 |
| Xbp1 | F: R: | TTACGCGAGAAAACTCATGGCC GGGTCCAAGTTGAACAGAATGC | 281 (unspliced) 255 (spliced) | XM_014742035.2 |
| p53 | F: R: | AGATAGCGATGGTCTGGC TTGGGCAGTGCTCGCTTAGT | 381 | NM_001126118.1 |
| Bax | F: R: | ACCAAGAAGCTGAGCGAGTGTC ACAAAGATGGTCACGGTCTGCC | 356 | XM_011527191.1 |
| Bcl-2 | F: R: | ATCGCCCTGTGGATGACTGAG CAGCCAGGAGAAATCAAACAGAGG | 129 | NM_000633.2 |
| p21 | F: R: | AGAAGAGGCTGGTGGCTATTT CCCGCCATTAGCGCATCAC | 169 | NM_001220777.1 |
| Casp3 | F: R: | CTCTGGTTTTCGGTGGGTGT CTTCCATGTATGATCTTTGGTTCC | 136 | NM_004346.4 |
| Casp9 | F: R: | CAGGCCCCATATGATCGAGG CTGGCCTGTGTCCTCTAAGC | 142 | NM_032996.3 |
| GAPDH | F: R: | GTCAGTGGTGGACCTGACCT CACCACCCTGTTGCTGTAGC | 256 | NM_001289746.1 |
| FFAS | HEPG2_HE (µG) | HEPG2_FFAS (µG) | HEPG2_MSI-1436 (µG) | HEPG2_COMP 23 (µG) |
|---|---|---|---|---|
| TETRADECANOIC ACID, METHYL ESTER; ME. C14:0 | 3.00 ± 0.05 | 2.16 ± 0.01 *** | 2.85 ± 0.02 **,### | 2.96 ± 0.03 ### |
| PENTADECANOIC ACID, METHYL ESTER; ME. C15:0 | 1.05 ± 0.00 | 0.60 ± 0.01 *** | 0.83 ± 0.01 ***,### | 1.07 ± 0.02 ### |
| HEXADECANOIC ACID, METHYL ESTER; ME. C16:0 | 21.37 ± 0.21 | 26.86 ± 0.24 *** | 27.86 ± 0.34 ***,# | 28.50 ± 0.26 ***,## |
| 9-HEXADECENOIC ACID, METHYL ESTER, (E)-; ME. C18:1N(9Z) | 1.08 ± 0.10 | 0.64 ± 0.05 *** | 0.95 ± 0.06 ## | 1.14 ± 0.03 ### |
| 9-HEXADECENOIC ACID, METHYL ESTER, (Z)-; ME. C18:1N(9Z) | 2.28 ± 0.20 | 1.80 ± 0.13 * | 2.77 ± 0.16 *,### | 2.55 ± 0.03 ## |
| OCTADECANOIC ACID, METHYL ESTER; ME. C18:0 | 11.66 ± 0.06 | 15.38 ± 0.07 *** | 14.23 ± 0.06 ***,### | 15.29 ± 0.06 *** |
| 9-OCTADECENOIC ACID (Z)-, METHYL ESTER; ME. C18:1N(9Z) | 5.10 ± 0.43 | 6.23 ± 0.45 | 7.92 ± 0.53 **,# | 8.27 ± 0.03 ***,## |
| 9-OCTADECENOIC ACID, METHYL ESTER, (9E); ME. C18:1N(9E) | 2.82 ± 0.06 | 1.28 ± 0.02 *** | 2.40 ± 0.02 ***,### | 2.09 ± 0.02 ***,### |
| 9,11-OCTADECADIENOIC ACID, METHYL ESTER, (9Z,11Z); ME. C18:2N7(9Z, 11Z) | 0.35 ± 0.01 | 0.16 ± 0.01 | 0.29 ± 0.02 | 0.18 ± 0.12 |
| 9,11-OCTADECADIENOIC ACID, METHYL ESTER, (9Z,11Z); ME. C18:2N7(9Z, 11Z) | 0.65 ± 0.01 | 1.75 ± 0.02 *** | 1.65 ± 0.03 ***,## | 1.51 ± 0.01 ***,### |
| 11-EICOSENOIC ACID, METHYL ESTER. (11E); ME. C20:1N(11E) | 0.12 ± 0.02 | 0.00 ± 0.00 ** | 0.04 ± 0.05 | 0.00 ± 0.00 ** |
| 5,8,11-EICOSATRIENOIC ACID, METHYL ESTER, (5Z,8Z,11Z); ME. C2:3N(5Z,8Z,11Z) | 0.48 ± 0.02 | 0.44 ± 0.02 | 0.64 ± 0.04 **,### | 0.61 ± 0.02 **,## |
| 5,8,11,14-EICOSATETRAENOIC ACID, METHYL ESTER, (ALL-Z) (ME. C20:4N6(5,8,11,14—ALLZ) | 0.67 ± 0.03 | 0.69 ± 0.02 | 0.91 ± 0.05 ***,### | 0.74 ± 0.01 *,# |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourebaba, L.; Łyczko, J.; Alicka, M.; Bourebaba, N.; Szumny, A.; Fal, A.M.; Marycz, K. Inhibition of Protein-Tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-Challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress. J. Clin. Med. 2020, 9, 1294. https://doi.org/10.3390/jcm9051294
Bourebaba L, Łyczko J, Alicka M, Bourebaba N, Szumny A, Fal AM, Marycz K. Inhibition of Protein-Tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-Challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress. Journal of Clinical Medicine. 2020; 9(5):1294. https://doi.org/10.3390/jcm9051294
Chicago/Turabian StyleBourebaba, Lynda, Jacek Łyczko, Michalina Alicka, Nabila Bourebaba, Antoni Szumny, Andrzej M. Fal, and Krzysztof Marycz. 2020. "Inhibition of Protein-Tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-Challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress" Journal of Clinical Medicine 9, no. 5: 1294. https://doi.org/10.3390/jcm9051294
APA StyleBourebaba, L., Łyczko, J., Alicka, M., Bourebaba, N., Szumny, A., Fal, A. M., & Marycz, K. (2020). Inhibition of Protein-Tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-Challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress. Journal of Clinical Medicine, 9(5), 1294. https://doi.org/10.3390/jcm9051294