Orf Virus IL-10 and VEGF-E Act Synergistically to Enhance Healing of Cutaneous Wounds in Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Recombinant Proteins
2.2. Ethics Statement
2.3. Full-Thickness Skin Wound Model
2.4. Histology
2.5. Immunohistochemistry
2.6. Quantitative PCR
2.7. Statistical Analyses
3. Results
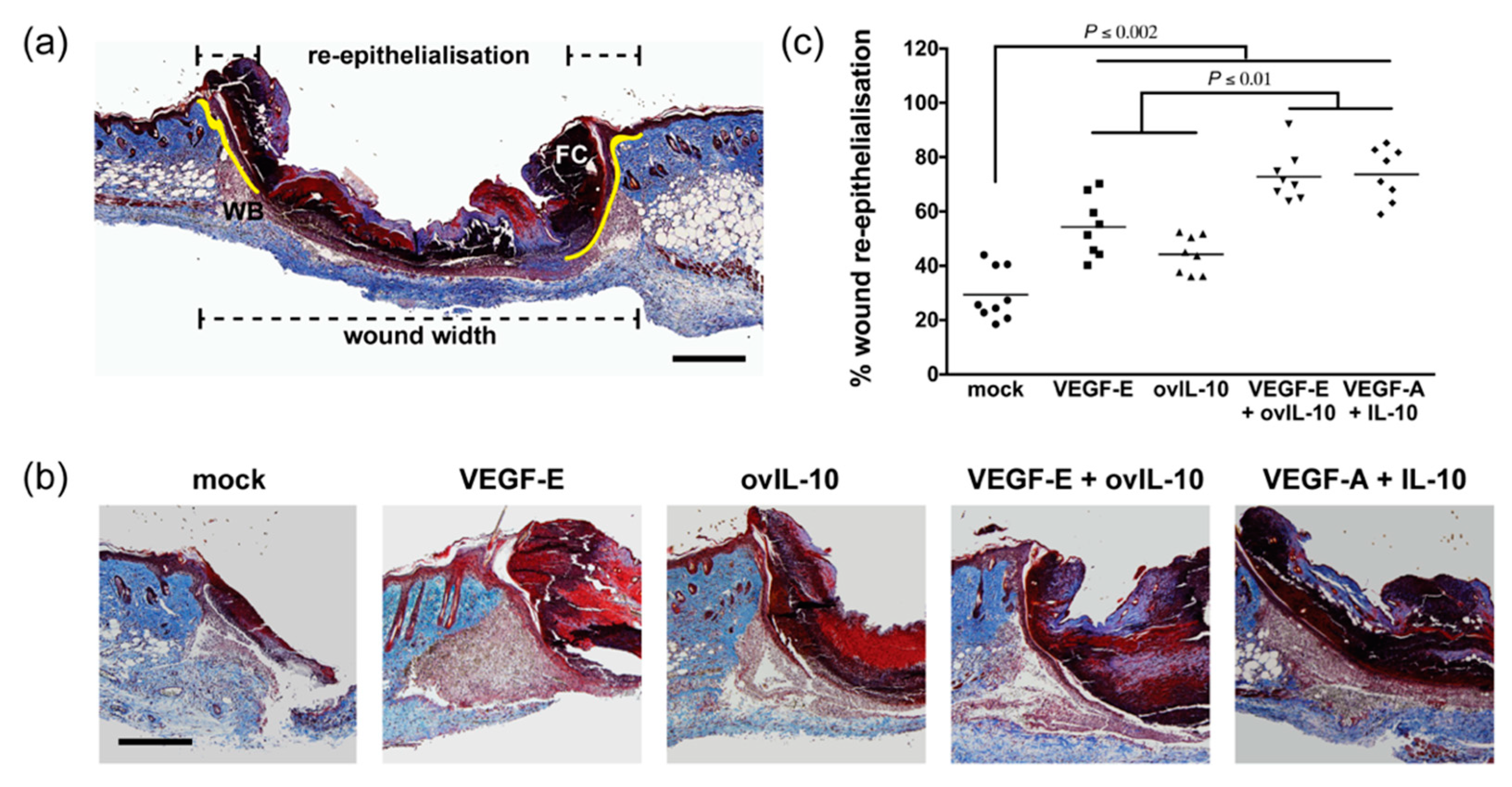
3.1. VEGF-E and ovIL-10 Synergistically Enhance Wound Closure and Re-epithelialisation
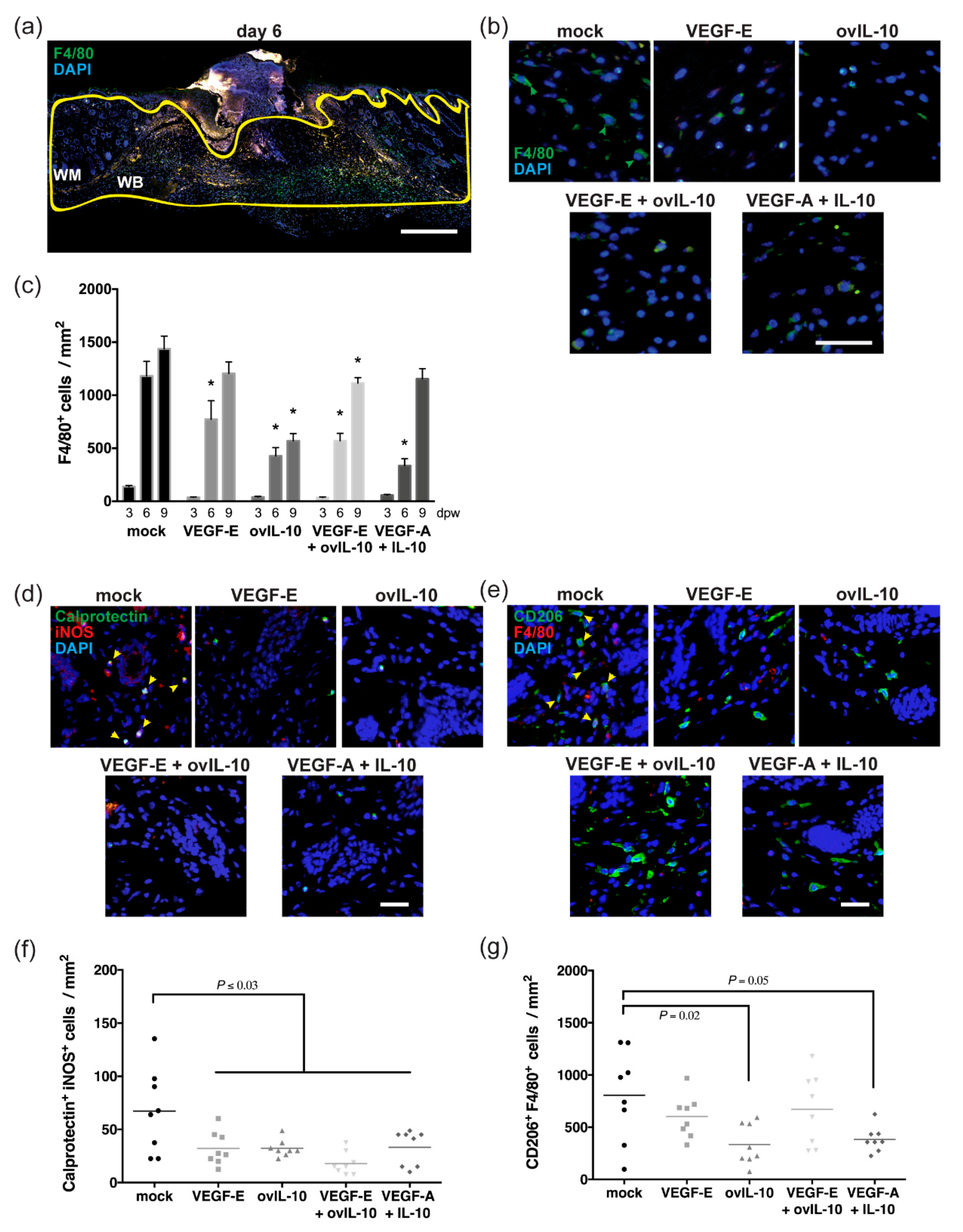
3.2. VEGF-E and ovIL-10 have Complimentary Effects on the Response of Macrophages to Wounding
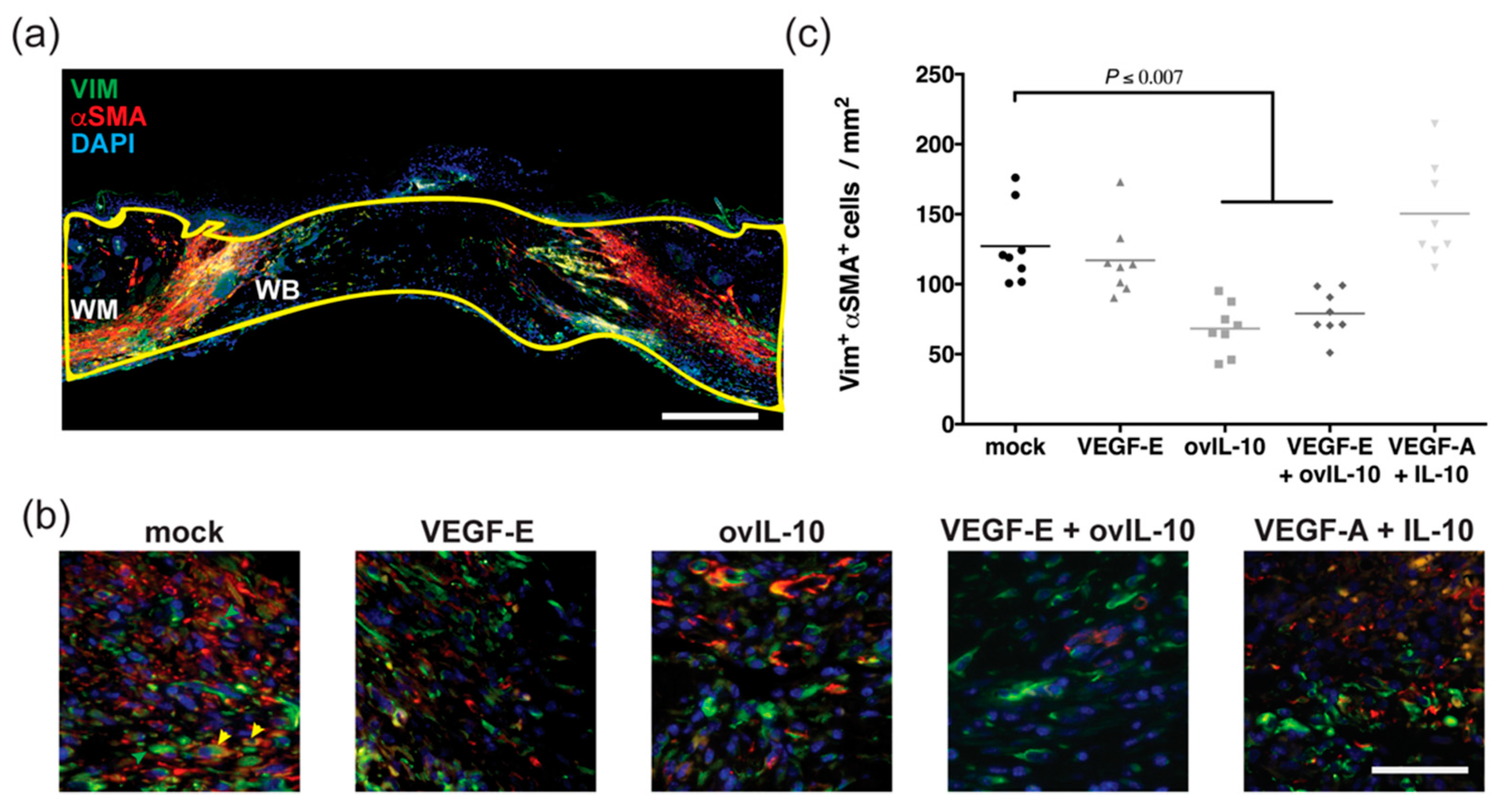
3.3. ovIL-10, with and without VEGF-E, Reduces the Response of Myofibroblasts to Wounding
3.4. VEGF-E and ovIL-10 Synergistically Enhance Wound Re-Vascularisation
3.5. VEGF-E and ovIL-10 have Complimentary Effects on Scar Size and Quality
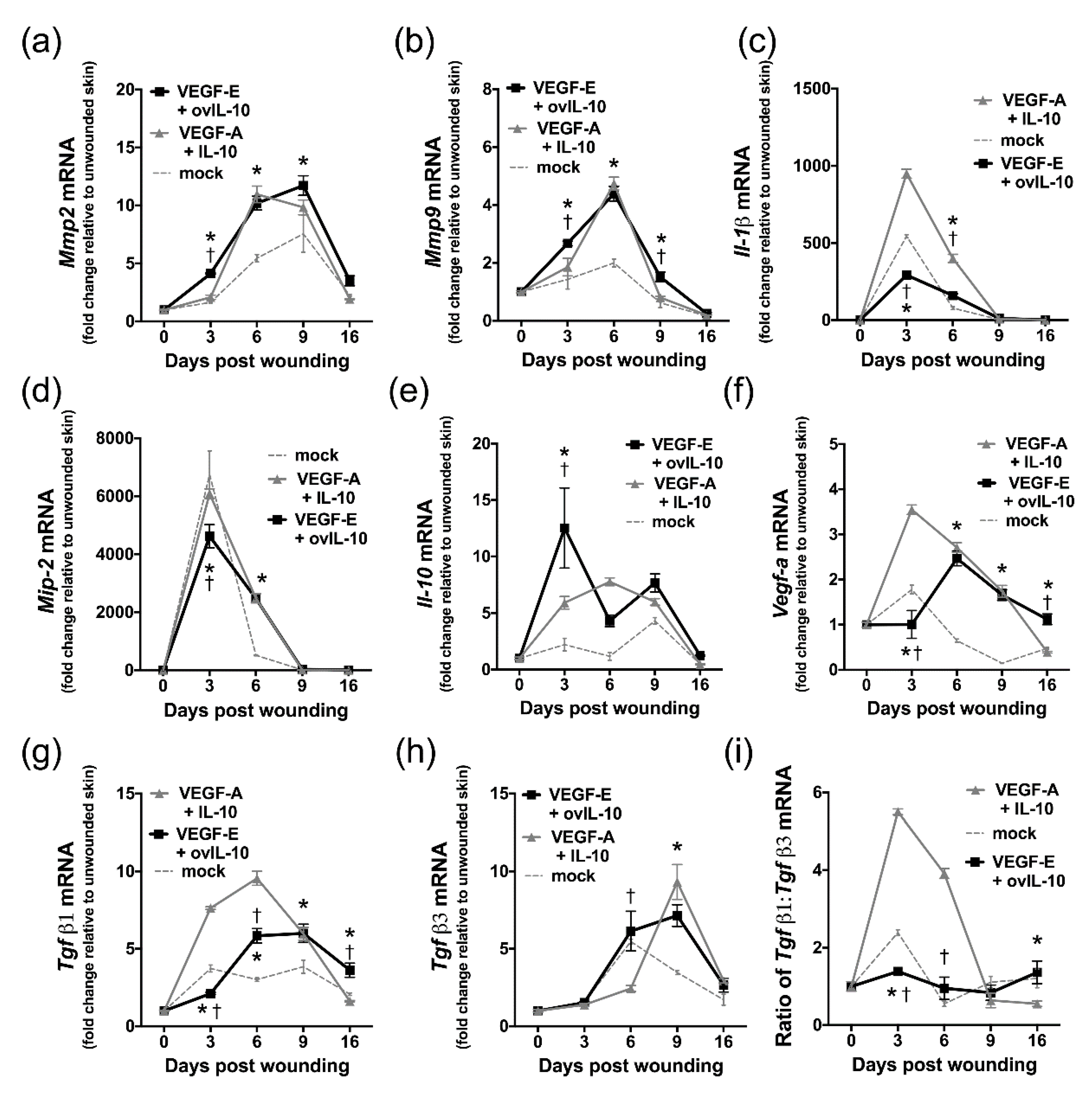
3.6. VEGF-E and ovIL-10 Alter the Genetic Expression of Key Wound Healing Mediators
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fleming, S.B.; Wise, L.; Mercer, A. Molecular Genetic Analysis of Orf Virus: A Poxvirus That Has Adapted to Skin. Viruses 2015, 7, 1505–1539. [Google Scholar] [CrossRef] [PubMed]
- Groves, R.; Wilson-Jones, E.; Macdonald, D. Human orf and milkers’ nodule: A clinicopathologic study. J. Am. Acad. Derm. 1991, 25, 706–711. [Google Scholar] [CrossRef]
- Savory, L.J.; Stacker, S.A.; Fleming, S.B.; Niven, B.E.; Mercer, A. Viral Vascular Endothelial Growth Factor Plays a Critical Role in Orf Virus Infection. J. Virol. 2000, 74, 10699–10706. [Google Scholar] [CrossRef] [PubMed]
- E Anderson, I.; Reid, H.W.; Nettleton, P.F.; McInnes, C.J.; Haig, D. Detection of cellular cytokine mRNA expression during orf virus infection in sheep: Differential interferon-γ mRNA expression by cells in primary versus reinfection skin lesions. Vet. Immunol. Immunopathol. 2001, 83, 161–176. [Google Scholar] [CrossRef]
- Haig, D.M.; McInnes, C.J.; Hutchison, G.; Seow, H.F.; Reid, H.W. Cyclosporin A abrogates the acquired immunity to cutaneous reinfection with the parapoxvirus orf virus. Immunology 1996, 89, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Yirrell, D.; Vestey, J.; Norval, M. Immune responses of patients to orf virus infection. Br. J. Derm. 1994, 130, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Gurel, M.S.; Ozardali, I.; Bitiren, M.; San, I.; Zeren, H. Giant orf on the nose. Eur. J. Derm. 2002, 12, 183–185. [Google Scholar]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Werner, S.; Grose, R. Regulation of Wound Healing by Growth Factors and Cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef]
- Lateef, Z.; Wise, L. Exploitation of receptor tyrosine kinases by viral-encoded growth factors. Growth Factors 2018, 36, 118–140. [Google Scholar] [CrossRef]
- Wise, L.M.; Savory, L.J.; Dryden, N.H.; Whelan, E.M.; Fleming, S.B.; Mercer, A. Major amino acid sequence variants of viral vascular endothelial growth factor are functionally equivalent during Orf virus infection of sheep skin. Virus Res. 2007, 128, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. Vascular Hyperpermeability, Angiogenesis, and Stroma Generation. Cold Spring Harb. Perspect. Med. 2011, 2, a006544. [Google Scholar] [CrossRef] [PubMed]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Bodaan, C.J.; Wise, L.M.; Wakelin, K.; Stuart, G.S.; Real, N.C.; Mercer, A.; Riley, C.; Theoret, C. Short-term treatment of equine wounds with orf virus IL-10 and VEGF-E dampens inflammation and promotes repair processes without accelerating closure. Wound Repair Regen. 2016, 24, 966–980. [Google Scholar] [CrossRef]
- Wise, L.; Inder, M.K.; Real, N.C.; Stuart, G.S.; Fleming, S.B.; Mercer, A. The vascular endothelial growth factor (VEGF)-E encoded by orf virus regulates keratinocyte proliferation and migration and promotes epidermal regeneration. Cell. Microbiol. 2012, 14, 1376–1390. [Google Scholar] [CrossRef]
- Wise, L.; Stuart, G.S.; Real, N.C.; Fleming, S.B.; Mercer, A.A. VEGF Receptor-2 Activation Mediated by VEGF-E Limits Scar Tissue Formation Following Cutaneous Injury. Adv. Wound Care 2018, 7, 283–297. [Google Scholar] [CrossRef]
- Kiba, A.; Sagara, H.; Hara, T.; Shibuya, M. VEGFR-2-specific ligand VEGF-E induces non-edematous hyper-vascularization in mice. Biochem. Biophys. Res. Commun. 2003, 301, 371–377. [Google Scholar] [CrossRef]
- Wise, L.; Bodaan, C.J.; Stuart, G.S.; Real, N.C.; Lateef, Z.; Mercer, A.A.; Riley, C.; Theoret, C. Treatment of limb wounds of horses with orf virus IL-10 and VEGF-E accelerates resolution of exuberant granulation tissue, but does not prevent its development. PLoS ONE 2018, 13, e0197223. [Google Scholar] [CrossRef]
- Wise, L.M.; Ueda, N.; Dryden, N.H.; Fleming, S.B.; Caesar, C.; Roufail, S.; Achen, M.; Stacker, S.A.; Mercer, A. Viral Vascular Endothelial Growth Factors Vary Extensively in Amino Acid Sequence, Receptor-binding Specificities, and the Ability to Induce Vascular Permeability yet Are Uniformly Active Mitogens. J. Biol. Chem. 2003, 278, 38004–38014. [Google Scholar] [CrossRef]
- Zheng, Y.; Murakami, M.; Takahashi, H.; Yamauchi, M.; Kiba, A.; Yamaguchi, S.; Yabana, N.; Alitalo, K.; Shibuya, M. Chimeric VEGF-E(NZ7)/PlGF promotes angiogenesis via VEGFR-2 without significant enhancement of vascular permeability and inflammation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Inder, M.K.; Wise, L.M.; Fleming, S.B.; Mercer, A. The C-terminus of viral vascular endothelial growth factor-E partially blocks binding to VEGF receptor-1. FEBS J. 2007, 275, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wise, L.M.; Veikkola, T.; Mercer, A.A.; Savory, L.J.; Fleming, S.B.; Caesar, C.; Vitali, A.; Makinen, T.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor (VEGF)-like protein from orf virus NZ2 binds to VEGFR2 and neuropilin-1. Proc. Natl. Acad. Sci. USA 1999, 96, 3071–3076. [Google Scholar] [CrossRef]
- Zheng, Y.; Watanabe, M.; Kuraishi, T.; Hattori, S.; Kai, C.; Shibuya, M. Chimeric VEGF-ENZ7/PlGF specifically binding to VEGFR-2 accelerates skin wound healing via enhancement of neovascularization. Arter. Thromb. Vasc. Biol. 2007, 27, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Kieran, I.; Knock, A.; Bush, J.; So, K.; Metcalfe, A.; Hobson, R.; Mason, T.; O’Kane, S.; Ferguson, M. Interleukin-10 reduces scar formation in both animal and human cutaneous wounds: Results of two preclinical and phase II randomized control studies. Wound Repair Regen. 2013, 21, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Haig, D.; Thomson, J.; McInnes, C.J.; McCaughan, C.; Imlach, W.L.; Mercer, A.; Fleming, S. Orf virus immuno-modulation and the host immune response. Vet. Immunol. Immunopathol. 2002, 87, 395–399. [Google Scholar] [CrossRef]
- Imlach, W.L.; McCaughan, C.A.; Mercer, A.; Haig, D.; Fleming, S.B. Orf virus-encoded interleukin-10 stimulates the proliferation of murine mast cells and inhibits cytokine synthesis in murine peritoneal macrophages. J. Gen. Virol. 2002, 83, 1049–1058. [Google Scholar] [CrossRef]
- Wise, L.; McCaughan, C.; Tan, C.K.; Mercer, A.; Fleming, S.B. Orf virus interleukin-10 inhibits cytokine synthesis in activated human THP-1 monocytes, but only partially impairs their proliferation. J. Gen. Virol. 2007, 88, 1677–1682. [Google Scholar] [CrossRef]
- Wise, L.; Stuart, G.S.; Real, N.C.; Fleming, S.B.; Mercer, A. Orf virus IL-10 accelerates wound healing while limiting inflammation and scarring. Wound Repair Regen. 2014, 22, 356–367. [Google Scholar] [CrossRef]
- Bennett, J.R.; Lateef, Z.; Fleming, S.B.; Mercer, A.; Wise, L. Orf virus IL-10 reduces monocyte, dendritic cell and mast cell recruitment to inflamed skin. Virus Res. 2016, 213, 230–237. [Google Scholar] [CrossRef]
- Van Zuijlen, P.P.; De Vries, H.; Lamme, E.N.; Coppens, J.E.; Van Marle, J.; Kreis, R.W.; Middelkoop, E. Morphometry of dermal collagen orientation by Fourier analysis is superior to multi-observer assessment. J. Pathol. 2002, 198, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Khorasani, H.; Zheng, Z.; Nguyen, C.; Zara, J.; Zhang, X.; Wang, J.; Ting, K.; Soo, C. A Quantitative Approach to Scar Analysis. Am. J. Pathol. 2011, 178, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Biej, M.; Kaminska, N.; Matuszczak, S.; Cichoń, T.; Pamuła-Piłat, J.; Czapla, J.; Smolarczyk, R.; Skwarzyńska, D.; Kulik, K.; Szala, S. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef] [PubMed]
- Lateef, Z.; Stuart, G.; Jones, N.; Mercer, A.; Fleming, S.B.; Wise, L. The Cutaneous Inflammatory Response to Thermal Burn Injury in a Murine Model. Int. J. Mol. Sci. 2019, 20, 538. [Google Scholar] [CrossRef]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramírez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in Wound Healing: A Comprehensive Review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef]
- Di Peppe, S.R.; Mangoni, A.; Zambruno, G.; Spinetti, G.; Melillo, G.; Napolitano, M.; Capogrossi, M.C. Adenovirus-mediated VEGF165 gene transfer enhances wound healing by promoting angiogenesis in CD1 diabetic mice. Gene 2002, 9, 1271–1277. [Google Scholar]
- Michaels, J.; Dobryansky, M.; Galiano, R.D.; Bhatt, K.A.; Ashinoff, R.; Ceradini, D.J.; Gurtner, G.C. Topical vascular endothelial growth factor reverses delayed wound healing secondary to angiogenesis inhibitor administration. Wound Repair Regen. 2005, 13, 506–512. [Google Scholar] [CrossRef]
- Xue, M.; Le, N.T.; Jackson, C. Targeting matrix metalloproteases to improve cutaneous wound healing. Expert Opin. Targets 2006, 10, 143–155. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Derm. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Eming, S.A.; Werner, S.; Bugnon, P.; Wickenhauser, C.; Siewe, L.; Utermöhlen, O.; Davidson, J.M.; Krieg, T.; Roers, A. Accelerated Wound Closure in Mice Deficient for Interleukin-10. Am. J. Pathol. 2007, 170, 188–202. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Jetten, N.; Roumans, N.; Gijbels, M.J.; Romano, A.; Post, M.J.; De Winther, M.P.J.; Van Der Hulst, R.R.W.J.; Xanthoulea, S. Wound Administration of M2-Polarized Macrophages Does Not Improve Murine Cutaneous Healing Responses. PLoS ONE 2014, 9, 102994. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, K.; Dohi, M.; Okunishi, K.; Tanaka, R.; Miyazaki, J.-I.; Yamamoto, K. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-β in the lung. Thorax 2006, 61, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; Ten Dijke, P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Wang, Y.H.; Ling, T.Y.; Chuang, S.S.; Johnson, F.E.; Huang, S.S. Synthetic TGF-beta antagonist accelerates wound healing and reduces scarring. FASEB J. 2002, 16, 1269–1270. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Bagchi, R.A.; Ludke, A.L.; Sharma, A.K.; Singal, P.K. Akt Regulates IL-10 Mediated Suppression of TNFα-Induced Cardiomyocyte Apoptosis by Upregulating Stat3 Phosphorylation. PLoS ONE 2011, 6, 25009. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef]
- Gordon, A.; Kozin, E.D.; Keswani, S.G.; Vaikunth, S.S.; Katz, A.B.; Zoltick, P.W.; Favata, M.; Radu, A.P.; Soslowsky, L.J.; Herlyn, M.; et al. Permissive environment in postnatal wounds induced by adenoviral-mediated overexpression of the anti-inflammatory cytokine interleukin-10 prevents scar formation. Wound Repair Regen. 2008, 16, 70–79. [Google Scholar] [CrossRef]
- Liechty, K.W.; Kim, H.B.; Adzick, N.; Crombleholme, T.M. Fetal wound repair results in scar formation in interleukin-10–deficient mice in a syngeneic murine model of scarless fetal wound repair. J. Pediatr. Surg. 2000, 35, 866–873. [Google Scholar] [CrossRef]
- Peranteau, W.H.; Zhang, L.; Muvarak, N.; Badillo, A.T.; Radu, A.; Zoltick, P.W.; Liechty, K.W. IL-10 Overexpression Decreases Inflammatory Mediators and Promotes Regenerative Healing in an Adult Model of Scar Formation. J. Investig. Derm. 2008, 128, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Morise, Z.; Eppihimer, M.; Granger, D.N.; Anderson, D.C.; Grisham, M.B. Effects of lipopolysaccharide on endothelial cell adhesion molecule expression in interleukin-10 deficient mice. Inflammation 1999, 23, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Mtairag, M.E.; Chollet-Martin, S.; Oudghiri, M.; Laquay, N.; Jacob, M.-P.; Michel, J.-B.; Feldman, L.J. Effects of interleukin-10 on monocyte/endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovasc. Res. 2001, 49, 882–890. [Google Scholar] [CrossRef]
- Czepluch, F.S.; Olieslagers, S.; Van Hulten, R.; Voo, S.A.; Waltenberger, J. VEGF-A-induced chemotaxis of CD16+ monocytes is decreased secondary to lower VEGFR-1 expression. Atherosclerosis 2011, 215, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Outtz, H.H.; Wu, J.K.; Wang, X.; Kitajewski, J. Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J. Immunol. 2010, 185, 4363–4373. [Google Scholar] [CrossRef] [PubMed]
- Okizaki, S.; Ito, Y.; Hosono, K.; Oba, K.; Ohkubo, H.; Kojo, K.; Nishizawa, N.; Shibuya, M.; Shichiri, M.; Majima, M. Vascular Endothelial Growth Factor Receptor Type 1 Signaling Prevents Delayed Wound Healing in Diabetes by Attenuating the Production of IL-1beta by Recruited Macrophages. Am. J. Pathol. 2016, 186, 1481–1498. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476. [Google Scholar] [CrossRef]
- Wheeler, K.C.; Jena, M.; Pradhan, B.; Nayak, N.; Das, S.; Hsu, C.-D.; Wheeler, D.S.; Chen, K.; Nayak, N.R. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS ONE 2018, 13, e0191040. [Google Scholar] [CrossRef]
- Linde, N.; Lederle, W.; Depner, S.; Van Rooijen, N.; Gutschalk, C.M.; Mueller, M.M. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J. Pathol. 2012, 227, 17–28. [Google Scholar] [CrossRef]
- Darby, I.; Zakuan, N.; Billet, F.; Desmoulière, A. The myofibroblast, a key cell in normal and pathological tissue repair. Cell. Mol. Life Sci. 2015, 73, 1145–1157. [Google Scholar] [CrossRef]
- Pakyari, M.; Farrokhi, A.; Maharlooei, M.K.; Ghahary, A. Critical Role of Transforming Growth Factor Beta in Different Phases of Wound Healing. Adv. Wound Care 2013, 2, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Sapudom, J.; Wu, X.; Chkolnikov, M.; Ansorge, M.; Anderegg, U.; Pompe, T. Fibroblast fate regulation by time dependent TGF-beta1 and IL-10 stimulation in biomimetic 3D matrices. Biomater. Sci. 2017, 5, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, J.; Guan, H.; Cai, W.; Bai, X.; Fang, X.; Hu, X.; Wang, Y.; Wang, H.; Zheng, Z.; et al. Anti-Fibrotic Actions of Interleukin-10 against Hypertrophic Scarring by Activation of PI3K/AKT and STAT3 Signaling Pathways in Scar-Forming Fibroblasts. PLoS ONE 2014, 9, e98228. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.-H.; Guan, H.; Shi, S.; Cai, W.-X.; Bai, X.-Z.; Hu, X.-L.; Fang, X.-B.; Liu, J.-Q.; Tao, K.; Zhu, X.-X.; et al. Protection against TGF-β1-induced fibrosis effects of IL-10 on dermal fibroblasts and its potential therapeutics for the reduction of skin scarring. Arch. Derm. Res. 2013, 305, 341–352. [Google Scholar] [CrossRef]
- Yamamoto, T.; Eckes, B.; Krieg, T. Effect of Interleukin-10 on the Gene Expression of Type I Collagen, Fibronectin, and Decorin in Human Skin Fibroblasts: Differential Regulation by Transforming Growth Factor-β and Monocyte Chemoattractant Protein-1. Biochem. Biophys. Res. Commun. 2001, 281, 200–205. [Google Scholar] [CrossRef]
- Dulauroy, S.; Di Carlo, S.E.; Langa, F.; Eberl, G.; Peduto, L. Lineage tracing and genetic ablation of ADAM12+ perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat. Med. 2012, 18, 1262–1270. [Google Scholar] [CrossRef]
- Kantari-Mimoun, C.; Castells, M.; Klose, R.; Meinecke, A.-K.; Lemberger, U.J.; Rautou, P.-E.; Pinot-Roussel, H.; Badoual, C.; Schrödter, K.; Österreicher, C.H.; et al. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology 2015, 61, 2042–2055. [Google Scholar] [CrossRef]
- Stockmann, C.; Kerdiles, Y.M.; Nomaksteinsky, M.; Weidemann, A.; Takeda, N.; Doedens, A.; Torres-Collado, A.X.; Iruela-Arispe, L.; Nizet, V.; Johnson, R.S. Loss of myeloid cell-derived vascular endothelial growth factor accelerates fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 4329–4334. [Google Scholar] [CrossRef]
- Yang, L.; Kwon, J.; Popov, Y.V.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenteroogy 2014, 146, 1339–1350. [Google Scholar] [CrossRef]
- Bodnar, R.J.; Wells, A. Differential regulation of pericyte function by the CXC receptor 3. Wound Repair Regen. 2015, 23, 785–796. [Google Scholar] [CrossRef]
- Lindblom, P.; Gerhardt, H.; Liebner, S.; Abramsson, A.; Enge, M.; Hellstrom, M.; Bäckström, G.; Fredriksson, S.; Landegren, U.; Nyström, H.C.; et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genome Res. 2003, 17, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E. The role of pericytes in angiogenesis. Int. J. Dev. Biol. 2011, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.N.; Tassiulas, I.; Hu, Y.; Mecklenbräuker, I.; Tarakhovsky, A.; Ivashkiv, L.B. IFN-α Priming Results in a Gain of Proinflammatory Function by IL-10: Implications for Systemic Lupus Erythematosus Pathogenesis. J. Immunol. 2004, 172, 6476–6481. [Google Scholar] [CrossRef]
- Wilgus, T.A.; Ferreira, A.M.; Oberyszyn, T.; Bergdall, V.K.; DiPietro, L.A. Regulation of scar formation by vascular endothelial growth factor. Lab. Investig. 2008, 88, 579–590. [Google Scholar] [CrossRef]
- Pufe, T.; Harde, V.; Petersen, W.; Goldring, M.B.; Tillmann, B.; Mentlein, R. Vascular endothelial growth factor (VEGF) induces matrix metalloproteinase expression in immortalized chondrocytes. J. Pathol. 2004, 202, 367–374. [Google Scholar] [CrossRef]
- Wang, H.; A Keiser, J. Vascular endothelial growth factor upregulates the expression of matrix metalloproteinases in vascular smooth muscle cells: Role of flt-1. Circ. Res. 1998, 83, 832–840. [Google Scholar] [CrossRef]
- Anderson, E.M.; Silva, E.A.; Hao, Y.; Martinick, K.D.; Vermillion, S.A.; Stafford, A.G.; Doherty, E.G.; Wang, L.; Doherty, E.J.; Grossman, P.M.; et al. VEGF and IGF Delivered from Alginate Hydrogels Promote Stable Perfusion Recovery in Ischemic Hind Limbs of Aged Mice and Young Rabbits. J. Vasc. Res. 2017, 54, 288–298. [Google Scholar] [CrossRef]
- Chen, R.R.; Silva, E.A.; Yuen, W.W.; Mooney, D. Spatio–temporal VEGF and PDGF Delivery Patterns Blood Vessel Formation and Maturation. Pharm. Res. 2006, 24, 258–264. [Google Scholar] [CrossRef]
- Kempen, D.; Lu, L.; Heijink, A.; Hefferan, T.E.; Creemers, L.B.; Maran, A.; Yaszemski, M.J.; Dhert, W. Effect of local sequential VEGF and BMP-2 delivery on ectopic and orthotopic bone regeneration. Biomaterials 2009, 30, 2816–2825. [Google Scholar] [CrossRef]
- Ko, J.; Jun, H.; Chung, H.; Yoon, C.; Kim, T.; Kwon, M.; Lee, S.; Jung, S.; Kim, M.; Park, J.H. Comparison of EGF with VEGF Non-Viral Gene Therapy for Cutaneous Wound Healing of Streptozotocin Diabetic Mice. Diabetes Metab. J. 2011, 35, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Peattie, R.A.; Nayate, A.; Firpo, M.; Shelby, J.; Fisher, R.; Prestwich, G. Stimulation of in vivo angiogenesis by cytokine-loaded hyaluronic acid hydrogel implants. Biomaterials 2004, 25, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-H.; Lee, J.; Ahn, D.-G.; Lee, K.Y. Co-delivery of Vascular Endothelial Growth Factor and Angiopoietin-1 Using Injectable Microsphere/Hydrogel Hybrid Systems for Therapeutic Angiogenesis. Pharm. Res. 2013, 30, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Eijkelkamp, N.; Steen-Louws, C.; Hartgring, S.A.Y.; Willemen, H.L.D.M.; Prado, J.; Lafeber, F.P.J.G.; Heijnen, C.J.; Hack, C.E.; Van Roon, J.A.G.; Kavelaars, A. IL4-10 Fusion Protein Is a Novel Drug to Treat Persistent Inflammatory Pain. J. Neurosci. 2016, 36, 7353–7363. [Google Scholar] [CrossRef] [PubMed]
- Canesso, M.C.C.; Vieira, A.; Castro, T.B.R.; Schirmer, B.G.A.; Cisalpino, D.; Martins, F.; Rachid, M.A.; Nicoli, J.; Teixeira, M.M.; Barcelos, L.S. Skin Wound Healing Is Accelerated and Scarless in the Absence of Commensal Microbiota. J. Immunol. 2014, 193, 5171–5180. [Google Scholar] [CrossRef] [PubMed]
- Crandall, M.A. World Wound Care Markets 2008; Kalorama Information: New York, NY, USA, 2008. [Google Scholar]
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic Scarring and Keloids: Pathomechanisms and Current and Emerging Treatment Strategies. Mol. Med. 2010, 17, 113–125. [Google Scholar] [CrossRef]
- Lauer, G.; Sollberg, S.; Cole, M.; Krieg, T.; Eming, S.A.; Flamme, I.; Stürzebecher, J.; Mann, K. Expression and Proteolysis of Vascular Endothelial Growth Factor is Increased in Chronic Wounds. J. Investig. Derm. 2000, 115, 12–18. [Google Scholar] [CrossRef]
- Lundberg, J.E.; Roth, T.P.; Dunn, R.M.; Doyle, J.W. Comparison of IL-10 levels in chronic venous insufficiency ulcers and autologous donor tissue. Arch. Derm. Res. 1998, 290, 669–673. [Google Scholar] [CrossRef]
- Medina, A.; Scott, P.G.; Ghahary, A.; Tredget, E.E. Pathophysiology of Chronic Nonhealing Wounds. J. Burn. Care Rehabil. 2005, 26, 306–319. [Google Scholar] [CrossRef]
- Hanft, J.; Pollak, R.; Barbul, A.; Gils, C.; Kwon, P.; Gray, S.; Lynch, C.; Semba, C.; Breen, T. Phase I trial on the safety of topical rhVEGF on chronic neuropathic diabetic foot ulcers. J. Wound Care 2008, 17, 30–37. [Google Scholar] [CrossRef]
- Chen, W.; Fu, X.; Sun, X.; Sun, T.; Zhao, Z.; Sheng, Z. Analysis of differentially expressed genes in keloids and normal skin with cDNA microarray. J. Surg. Res. 2003, 113, 208–216. [Google Scholar] [CrossRef]
- Da Silva, I.R.; Tiveron, L.C.R.D.C.; Da Silva, M.V.; Peixoto, A.B.; Carneiro, C.A.X.; Dos Reis, M.A.; Furtado, P.C.; Rodrigues, B.R.; Rodrigues, V.; Rodrigues, D.B.R. In Situ Cytokine Expression and Morphometric Evaluation of Total Collagen and Collagens Type I and Type III in Keloid Scars. Mediat. Inflamm. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Ma, S.; Wen, H. Upregulation of proinflammatory genes in skin lesions may be the cause of keloid formation (Review). Biomed. Rep. 2013, 1, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Gira, A.K.; Brown, L.F.; Washington, C.V.; Cohen, C.; Arbiser, J.L. Keloids demonstrate high-level epidermal expression of vascular endothelial growth factor. J. Am. Acad. Derm. 2004, 50, 850–853. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef]
- Salem, A.; Assaf, M.; Helmy, A.; Nofal, A.; Ibrahim, S.; Eldeeb, F.; Youssef, C. Role of vascular endothelial growth factor in keloids: A clinicopathologic study. Int. J. Derm. 2009, 48, 1071–1077. [Google Scholar] [CrossRef]
- Broek, L.J.V.D.; Van Der Veer, W.M.; De Jong, E.H.; Gibbs, S.; Niessen, F.B. Suppressed inflammatory gene expression during human hypertrophic scar compared to normotrophic scar formation. Exp. Derm. 2015, 24, 623–629. [Google Scholar] [CrossRef]
- Van Der Veer, W.M.; Niessen, F.B.; Ferreira, J.A.; Zwiers, P.J.; De Jong, E.H.; Middelkoop, E.; Molema, G. Time course of the angiogenic response during normotrophic and hypertrophic scar formation in humans. Wound Repair Regen. 2011, 19, 292–301. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wise, L.M.; Stuart, G.S.; Jones, N.C.; Fleming, S.B.; Mercer, A.A. Orf Virus IL-10 and VEGF-E Act Synergistically to Enhance Healing of Cutaneous Wounds in Mice. J. Clin. Med. 2020, 9, 1085. https://doi.org/10.3390/jcm9041085
Wise LM, Stuart GS, Jones NC, Fleming SB, Mercer AA. Orf Virus IL-10 and VEGF-E Act Synergistically to Enhance Healing of Cutaneous Wounds in Mice. Journal of Clinical Medicine. 2020; 9(4):1085. https://doi.org/10.3390/jcm9041085
Chicago/Turabian StyleWise, Lyn M., Gabriella S. Stuart, Nicola C. Jones, Stephen B. Fleming, and Andrew A. Mercer. 2020. "Orf Virus IL-10 and VEGF-E Act Synergistically to Enhance Healing of Cutaneous Wounds in Mice" Journal of Clinical Medicine 9, no. 4: 1085. https://doi.org/10.3390/jcm9041085
APA StyleWise, L. M., Stuart, G. S., Jones, N. C., Fleming, S. B., & Mercer, A. A. (2020). Orf Virus IL-10 and VEGF-E Act Synergistically to Enhance Healing of Cutaneous Wounds in Mice. Journal of Clinical Medicine, 9(4), 1085. https://doi.org/10.3390/jcm9041085