An Overview on Primary Sclerosing Cholangitis

 , ,
, ,  and
and
Abstract
1. Introduction
2. Epidemiology
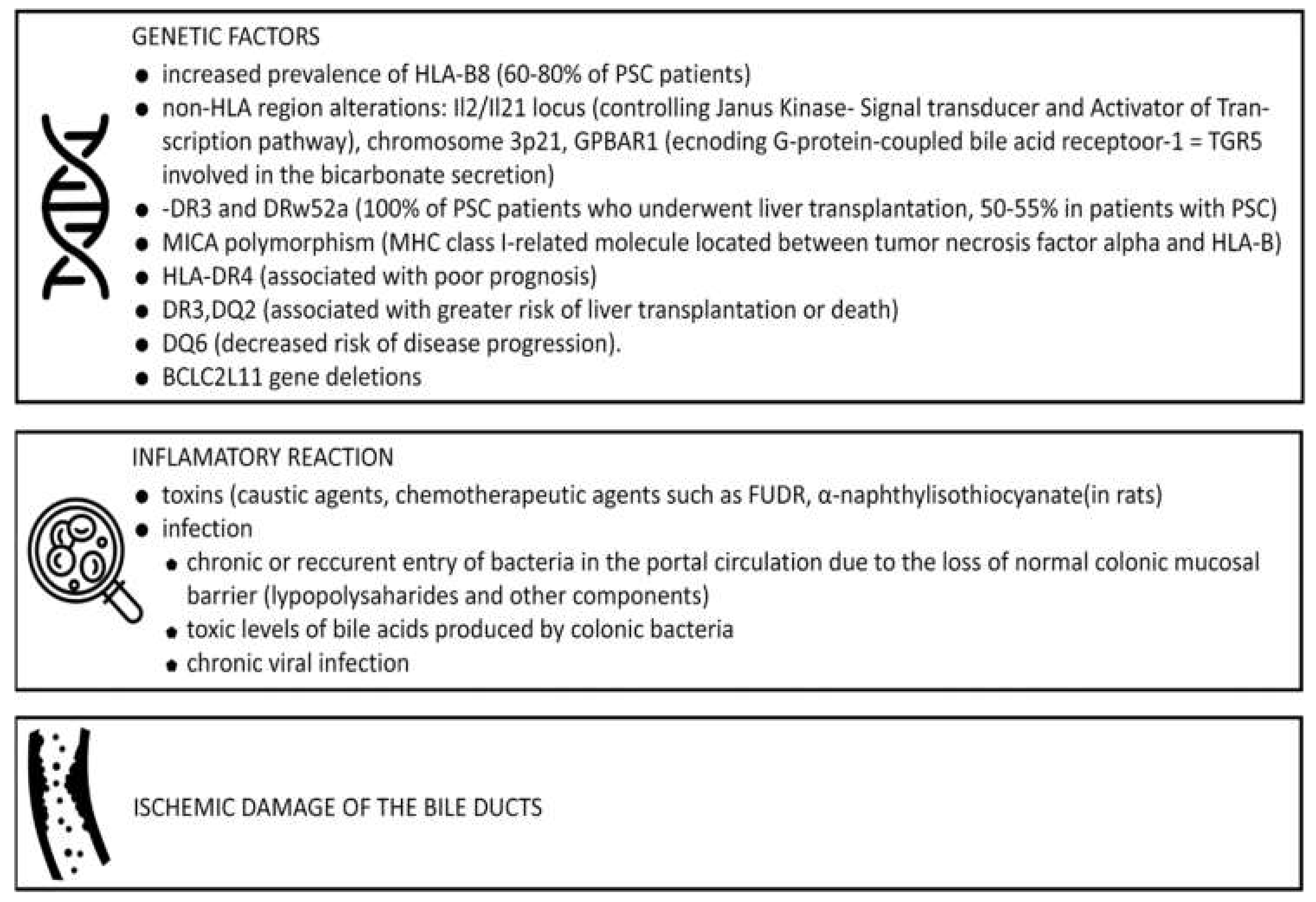
3. Pathogenesis
4. Natural History and Prognostic Models
5. Diagnosis
5.1. Signs and Symptoms
5.2. Laboratory Tests
5.3. Immunological and Serological Markers
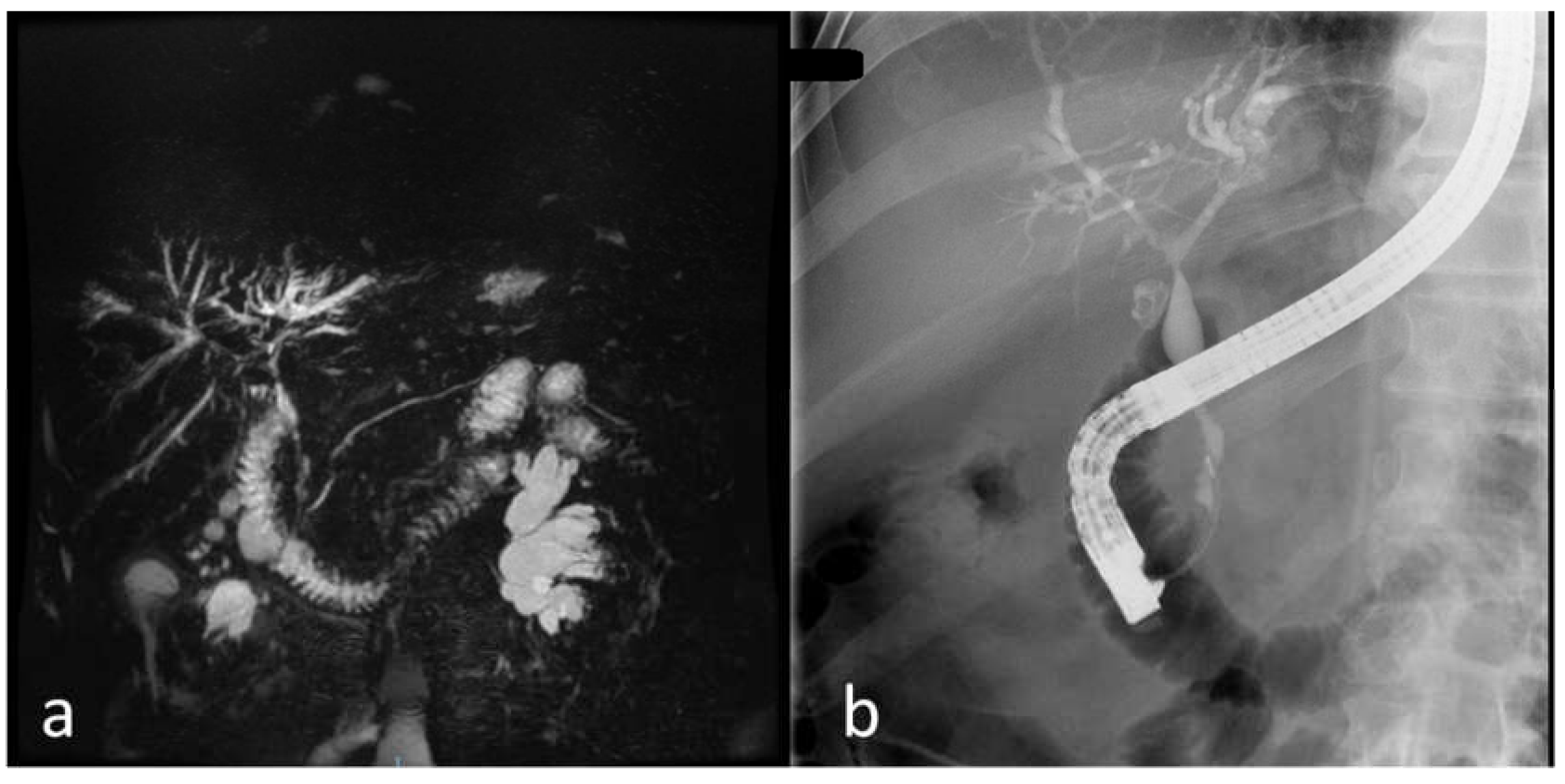
5.4. Imaging
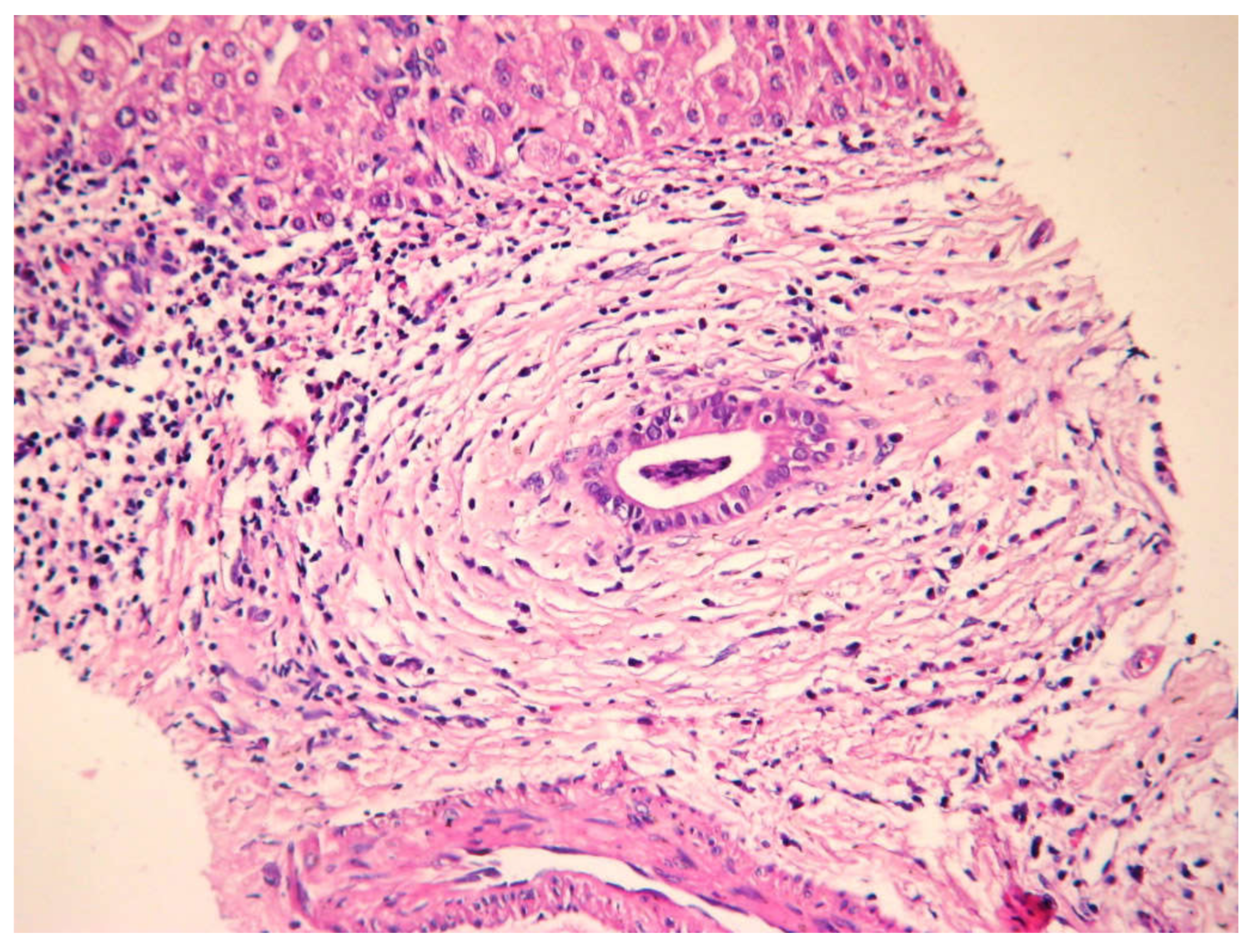
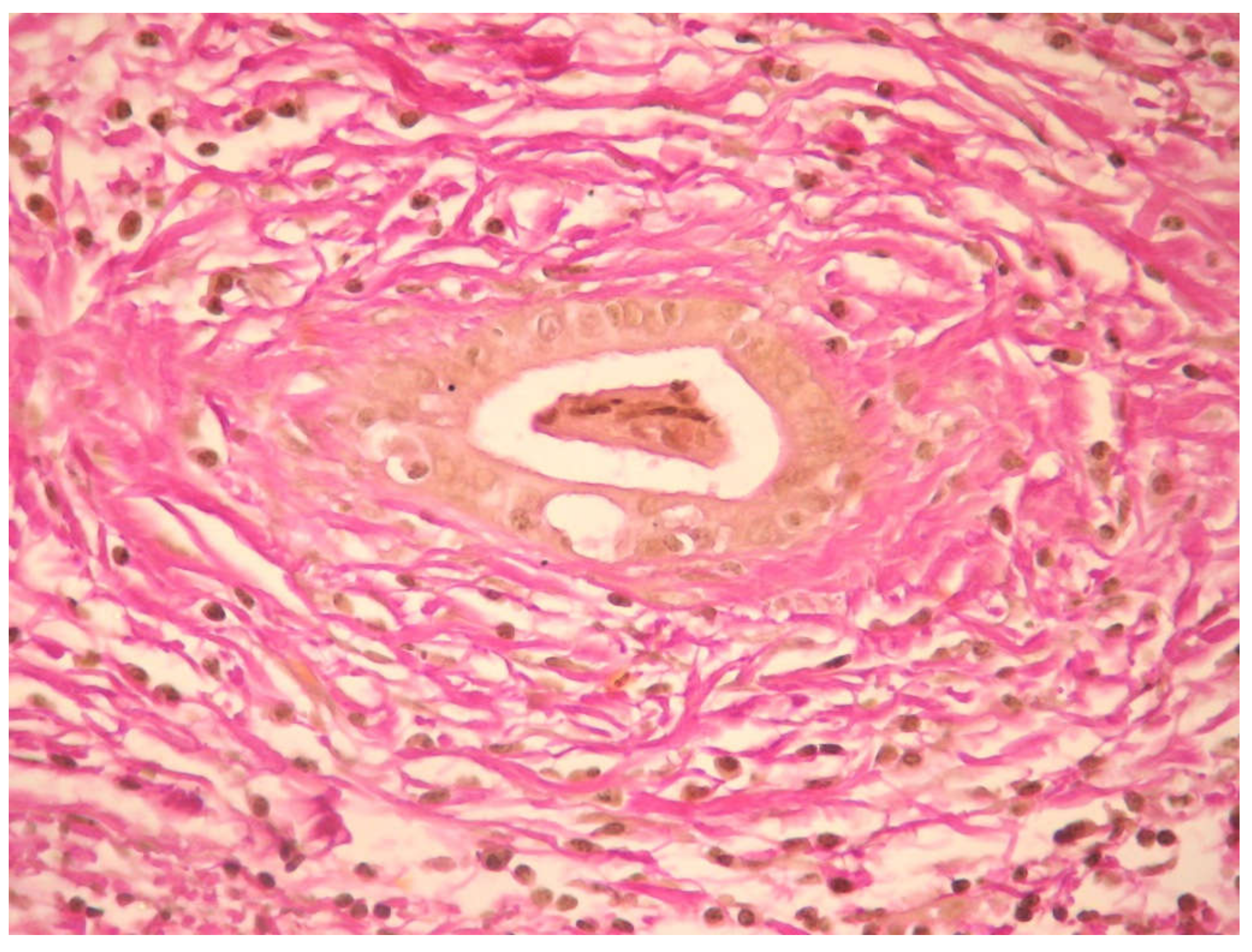
5.5. Histology
5.6. Noninvasive Tests for Assessing Liver Fibrosis
6. Differential Diagnosis
7. Disease-Related Complications
8. Management of PSC
8.1. Medical Treatment of Underlying Disease
8.1.1. Ursodeoxycholic Acid and nonUDCA
8.1.2. Immunosuppressive Drugs
8.1.3. Antibiotics and Probiotics
8.2. Endoscopic Management
8.3. Percutaneous Treatment
8.4. Surgical Therapy in PSC
8.5. Novel Therapies
9. PSC follow-Up
10. Special Situations
10.1. PSC-AIH Overlap Syndrome
10.2. PSC in Children
10.3. Immunoglobulin G4-Related Sclerosing Cholangitis
10.4. Small Duct PSC
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Feldman, M.; Lawrence, J.B. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease; Tenth, Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1166–1183. [Google Scholar]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Uptodate. Available online: www.uptodate.com (accessed on 3 April 2019).
- Lindor, K.; Knowdley, K.V.; Harrison, E.M. ACG Clinical Guideline: Primary Sclerosing Cholangitis. Am. J. Gastroenterol. 2015, 110, 646–659. [Google Scholar] [CrossRef]
- Chapman, M.H.; Thorburn, D.; Hirschfield, G.M.; Webster, G.G.J.; Rushbrook, S.M.; Alexander, G.; Collier, J.; Dyson, J.K.; Jones, D.E.; Patanwala, I.; et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut 2019, 68, 1356–1378. [Google Scholar] [CrossRef]
- Joo, M.; Abreu-e-Lima, P.; Farraye, F.; Smith, T.; Swaroop, P.; Gardner, L.; Lauwers, G.Y.; Odze, R.D. Pathologic features of ulcerative colitis in patients with primary sclerosing cholangitis: A case-control study. Am. J. Surg. Pathol. 2009, 33, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Bowlus, C.L.; Kim, J.K.; Lindor, K.D. AGA Clinical Practice Update on Surveillance for Hepatobiliary Cancers in Patients with Primary Sclerosing Cholangitis: Expert Review. Clin. Gastroenterol. Hepatol. 2019, 17, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.G.; Juran, B.D.; Mucha, S.; Folseraas, T.; Jostins, L.; Melum, E.; Kumasaka, N.; Atkinson, E.J.; Schlicht, E.M.; Liu, J.Z.; et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat. Genet. 2017, 49, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Krones, E.; Marschall, H.U.; Fickert, P. Future Medical Treatment in PSC. Curr. Hepatol. Rep. 2019, 18, 96–106. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Melum, E.; Franke, A. The utility of genome-wide association studies in hepatology. Hepatology 2010, 51, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, M.M.; Folseraas, T. Pathogenesis, diagnosis and treatment of premalignant and malignant stages of cholangiocarcinoma in primary sclerosing cholangitis. Liver Int. 2019, 39, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, D.K.; Camilleri, M.; Fitz, J.G.; Kalloo, A.N.; Shanahan, F.; Wang, T.C. Yamada’s Textbook of Gastroenterology, 6th ed.; John Wiley and Sons: Hoboken, NJ, USA, 2016; pp. 1835–1948. [Google Scholar]
- Aabakken, L.; Karlsen, H. Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guidelines. J. Hepatol. 2017, 66, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Jarnagin, W.R.; Allen, P.; Chapman, W.; Angelica, M.I.; DeMatteo, R.P.; Gian Do, R.K.; Vauthey, J.N. Blumgart’s Surgery of the Liver, Pancreas and Biliary Tract, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 663–674. [Google Scholar]
- Singh, S.; Khanna, S.; Pardi, D.S.; Loftus, E.V., Jr.; Talwalkar, J.A. Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: A systematic review and meta-analysis. Inflamm. Bowel Dis. 2013, 19, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, E.; Trottier, J.; Milkiewicz, M.; Raszeja-Wyszomirska, J.; Hirschfield, G.M.; Barbier, O.; Milkiewicz, P. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology 2014, 60, 931. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Lindor, K.D. Ursodeoxycholic acid in primary sclerosing cholangitis: If withdrawal is bad, then administration is good (right?). Hepatology 2014, 60, 785. [Google Scholar] [CrossRef]
- Tabibian, J.H.; O’Hara, S.P.; Lindor, K.D. Primary sclerosing cholangitis and the microbiota: Current knowledge and perspectives on etiopathogenesis and emerging therapies. Scand. J. Gastroenterol. 2014, 49, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Farkkila, M.; Karvonen, A.L.; Nurmi, H.; Nuutinen, H.; Taavitsainen, M.; Pikkarainen, P.; Kärkkäinen, P. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: A randomized placebocontrolled trial. Hepatology 2004, 40, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.R.; Blanco, P.G.; Keach, J.C.; Petz, J.L.; Zaman, M.M.; Bhaskar, K.R.; Cluette-Brown, J.E.; Gautam, S.; Sheth, S.; Afdhal, N.H.; et al. The safety and effi cacy of oral docosahexaenoic acid supplementation for the treatment of primary sclerosing cholangitis—A pilot study. Aliment. Pharmacol. Ther. 2012, 35, 255–265. [Google Scholar] [CrossRef]
- Davies, Y.K.; Cox, K.M.; Abdullah, B.A.; Safta, A.; Terry, A.B.; Cox, K.L. Long-term treatment of primary sclerosing cholangitis in children with oral vancomycin: An immunomodulating antibiotic. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 61–67. [Google Scholar] [CrossRef]
- Trikudanathan, G.; Navaneethan, U.; Njei, B.; Vargo, J.J.; Parsi, M.A. Diagnostic yield of bile duct brushings for cholangiocarcinoma in primary sclerosing cholangitis: A systematic review and meta-analysis. Gastrointest. Endosc. 2014, 79, 783–789. [Google Scholar] [CrossRef]
- Goldberg, D.; Bittermann, T.; Makar, G. Lack of standardization in exception points for patients with primary sclerosing cholangitis and bacterial cholangitis. Am. J. Transpl. 2012, 12, 1603–1609. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Bowlus, C.L.; Levy, C.; Vuppalanchi, R.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.S.; et al. The AESOP trial: A randomized, double-blind, placebo-controlled, phase 2 study of obeticholic acid in patients with primary sclerosing cholangitis. Hepatology 2017, 66, 1254A–1255A. [Google Scholar]
- Corpechot, C.; Chazouilleres, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.; Deutschmann, K.; Sommerfeld, A.; Klindt, C.; Kluge, S.; Kubitz, R.; Ullmer, C.; Knoefel, W.T.; Herebian, D.; Mayatepek, E.; et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut 2016, 65, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.D.; Chapman, R.W.; Keshav, S.; Montano-Loza, A.J.; Mason, A.L.; Kremer, A.E.; Vetter, M.; Krijger, M.; Ponsioen, C.Y.; Trivedi, P.; et al. Effects of Vedolizumab in Patients with Primary Sclerosing Cholangitis and Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.T.; Rothe, J.A.; Hetzel, J.T.; Cohen, R.D.; Hanauer, S.B. Are dysplasia and colorectal cancer endoscopically visible in patients with ulcerative colitis? Gastrointest. Endosc. 2007, 65, 998–1004. [Google Scholar] [CrossRef]
- Boonstra, K.; Culver, E.L.; de Buy Wenniger, L.M.; van Heerde, M.J.; van Erpecum, K.J.; Poen, A.C.; van Nieuwkerk, K.M.J.; Spanier, B.W.M.; Witteman, B.J.M.; Tuynman, H.A.R.E.; et al. Serum immunoglobulin G4 and immunoglobulin G1 for distinguishing immunoglobulin G4-associated cholangitis from primary sclerosing cholangitis. Hepatology 2014, 59, 1954–1963. [Google Scholar] [CrossRef]
- Hart, P.A.; Topazian, M.D.; Witzig, T.E.; Clain, J.E.; Gleeson, F.C.; Klebig, R.R.; Levy, M.J.; Pearson, R.K.; Petersen, B.T.; Smyrk, T.C.; et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: The Mayo Clinic experience. Gut 2013, 62, 1607–1615. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Asymptomatic phase | no clinical symptoms cholangiographic abnormalities normal biochemistry |
| Biochemical phase | asymptomatic biochemical abnormalities |
| Symptomatic phase | fatigue pruritus and excoriations jaundice fever, chills, night sweats, and right upper quadrant pain * diarrhea and bloody stools ** |
| Decompensated cirrhosis | jaundice ascites muscle atrophy spider telangiectasias peripheral edema hepatosplenomegaly xanthelasma hepatic encephalopathy variceal bleeding |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlăduţ, C.; Ciocîrlan, M.; Bilous, D.; Șandru, V.; Stan-Ilie, M.; Panic, N.; Becheanu, G.; Jinga, M.; Costache, R.S.; Costache, D.O.; et al. An Overview on Primary Sclerosing Cholangitis. J. Clin. Med. 2020, 9, 754. https://doi.org/10.3390/jcm9030754
Vlăduţ C, Ciocîrlan M, Bilous D, Șandru V, Stan-Ilie M, Panic N, Becheanu G, Jinga M, Costache RS, Costache DO, et al. An Overview on Primary Sclerosing Cholangitis. Journal of Clinical Medicine. 2020; 9(3):754. https://doi.org/10.3390/jcm9030754
Chicago/Turabian StyleVlăduţ, Cătălina, Mihai Ciocîrlan, Dana Bilous, Vasile Șandru, Mădălina Stan-Ilie, Nikola Panic, Gabriel Becheanu, Mariana Jinga, Raluca S. Costache, Daniel O. Costache, and et al. 2020. "An Overview on Primary Sclerosing Cholangitis" Journal of Clinical Medicine 9, no. 3: 754. https://doi.org/10.3390/jcm9030754
APA StyleVlăduţ, C., Ciocîrlan, M., Bilous, D., Șandru, V., Stan-Ilie, M., Panic, N., Becheanu, G., Jinga, M., Costache, R. S., Costache, D. O., & Diculescu, M. (2020). An Overview on Primary Sclerosing Cholangitis. Journal of Clinical Medicine, 9(3), 754. https://doi.org/10.3390/jcm9030754