Glucocerebrosidase: Functions in and Beyond the Lysosome


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Part 1: GCase and Lysosomal Glucosylceramide Degradation
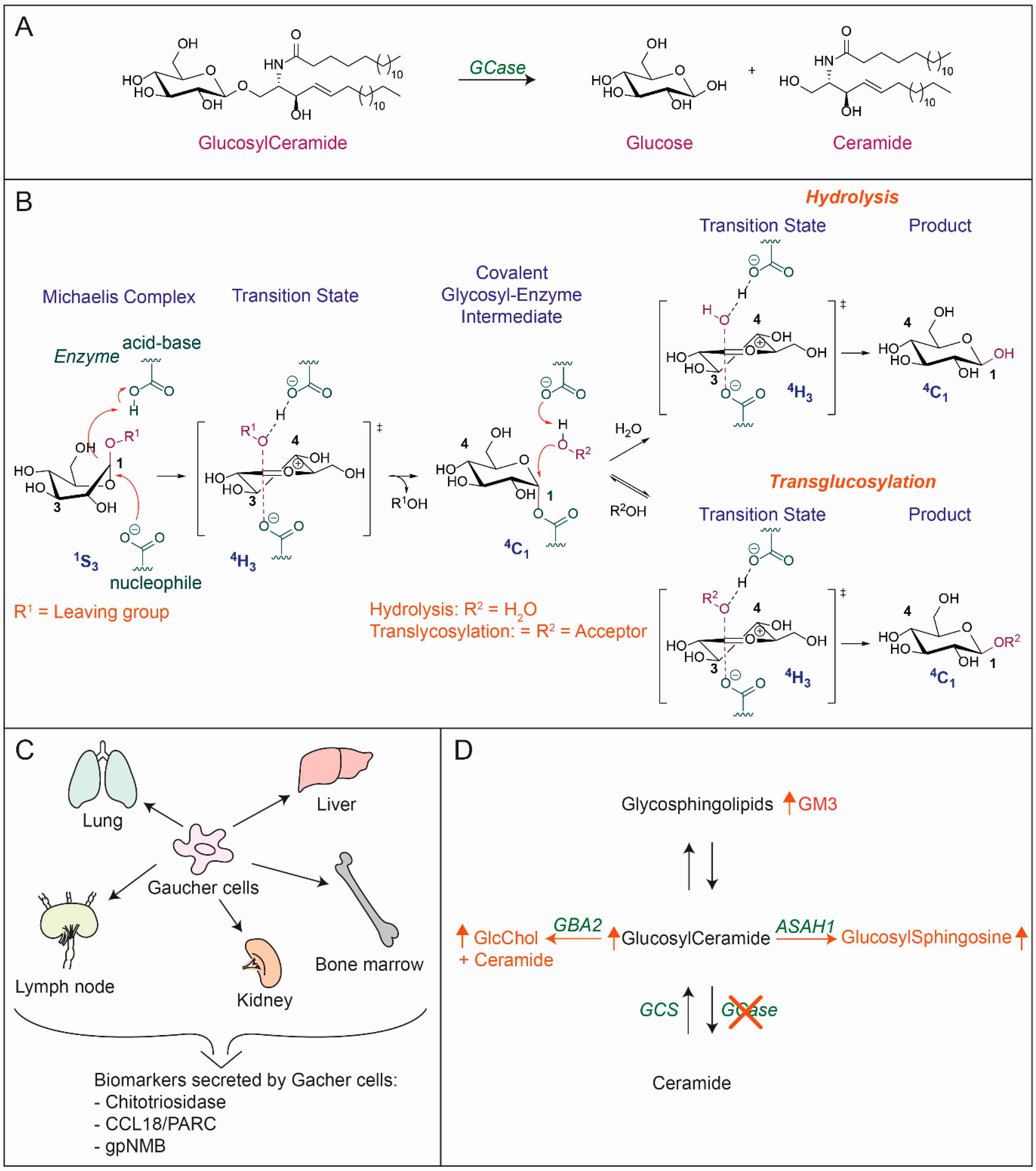
2.1. Glucosylceramide as Intermediate of Glycosphingolipids
2.2. Lysosomal Turnover of Glycosphingolipid
3. Glucocerebrosidase
3.1. GCase Protein and Life Cycle
3.2. Catalytic Activity of GCase
4. Gaucher Disease, Inherited Deficiency in GCase
4.1. Gaucher Disease, a Lysosomal Storage Disorder
4.2. Lysosomal GlcCer Deposits in Macrophages: Gaucher Cells
4.3. Therapies of Gaucher Disease: ERT, SRT, PCT/EET
5. Metabolic Adaptations to Lysosomal GCase Deficiency
5.1. Formation of Glucosylsphingosine From Accumulating GlcCer
5.2. Excessive Gangliosides
5.3. Increased Activity of Cytosol-Faced GBA2 and GlcChol
6. Part 2: GCase and Glucosylceramide Metabolism Beyond the Lysosome
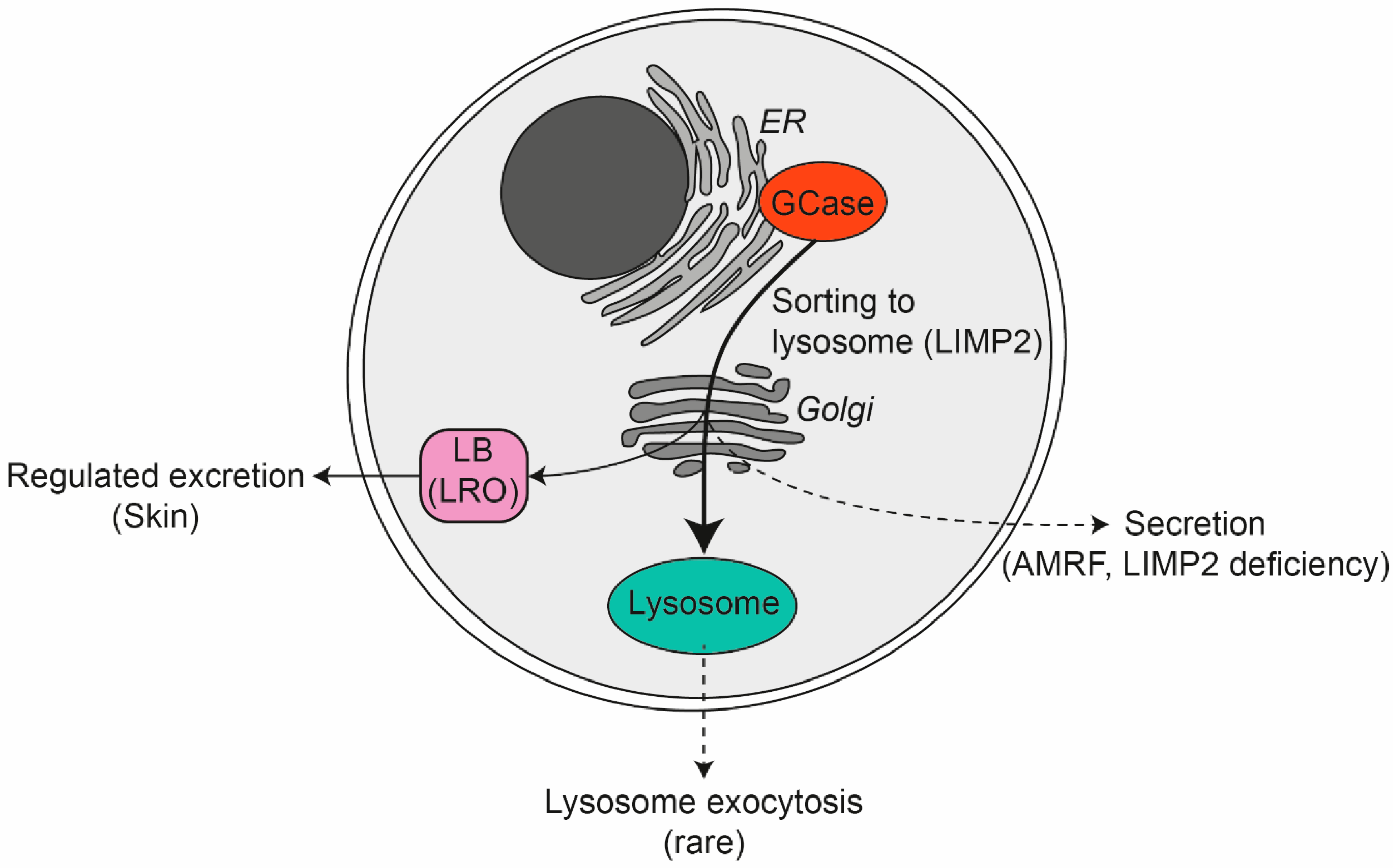
6.1. GCase: Other Locations Than Lysosomes
6.2. Lysosome-Related Organelles
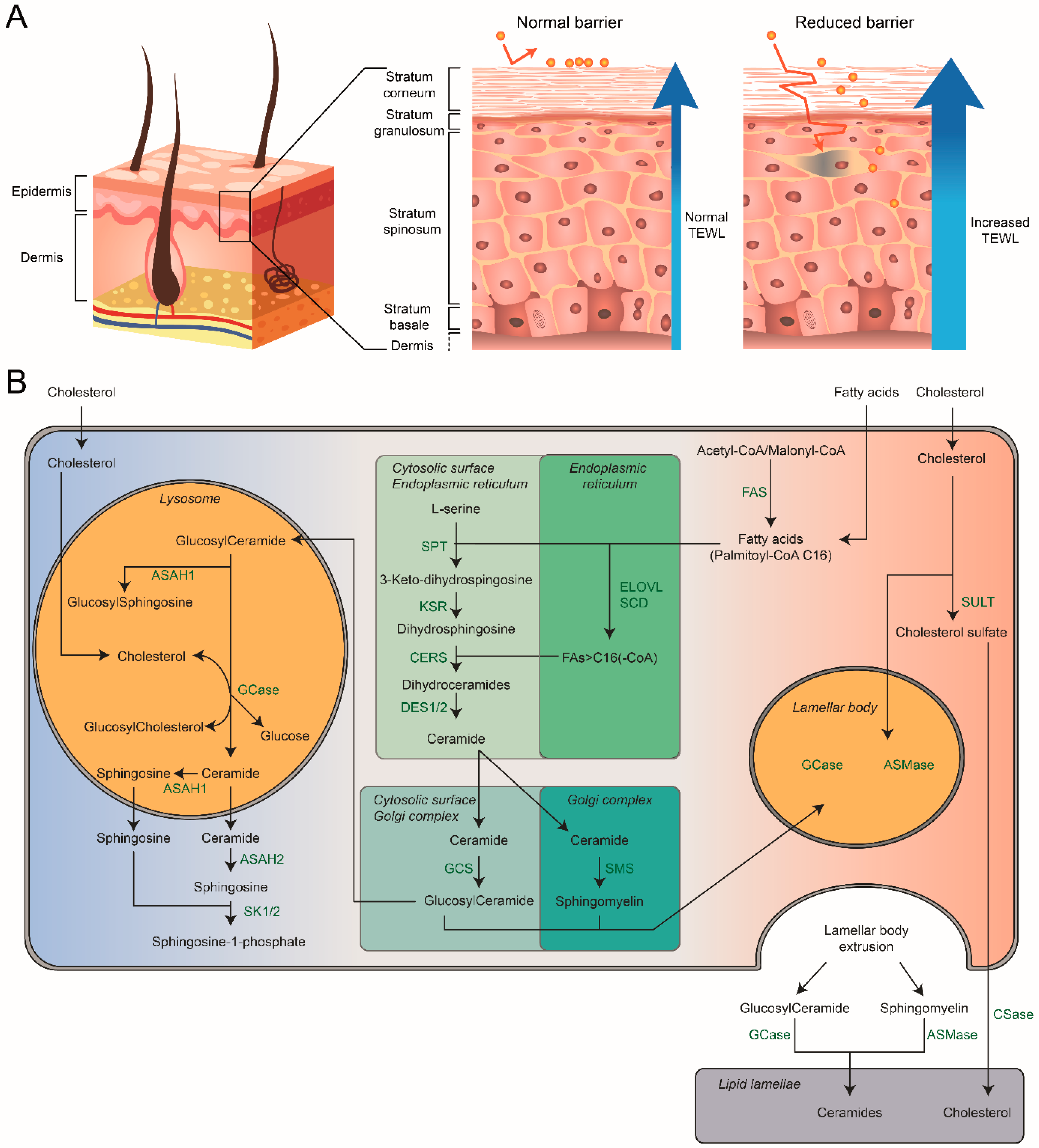
7. Composition of the Skin
7.1. Skin Differentiation and Barrier Formation
7.2. Stratum Corneum: Hydration and Skin-pH
7.3. Stratum Corneum: Composition
8. Sphingolipids of the Stratum Corneum
8.1. Role of Lamellar Bodies
8.2. Chemical Composition of Skin Sphingolipids
9. GCase: Crucial Extracellular Role in the Skin
10. Atopic Dermatitis
10.1. SC Lipids in AD
10.2. Potential Role for Glucosylsphingosine in AD Pathology
10.3. Direct Role of GCase in AD?
11. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Weinreb, N.J.; Brady, R.O.; Tappel, A.L. The lysosomal localization of sphingolipid hydrolases. Biochim. Biophys. Acta 1968, 159, 141–146. [Google Scholar] [CrossRef]
- Brady, R.O.; Kanfer, J.N.; Bradley, R.M.; Shapiro, D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J. Clin. Investig. 1966, 45, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, P.C.E. De L’epithelioma Primitif de la Rate, Hypertrophie Idiopathique de la Rate Sans Leucemie. Ph.D. Thesis, Faculté de Médecine, Paris, France, 1882. [Google Scholar]
- Beutler, E.; Grabowski, G.A. Glucosylceramide lipidosis-Gaucher disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Sriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the structural biology of Gaucher disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Sherer, D.M.; Ginns, E.I. Gaucher disease in the neonate: A distinct Gaucher phenotype is analogous to a mouse model created by targeted disruption of the glucocerebrosidase gene. Pediatr. Res. 1992, 32, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Tybulewicz, V.L.; Tremblay, M.L.; LaMarca, M.E.; Willemsen, R.; Stubblefield, B.K.; Winfield, S.; Zablocka, B.; Sidransky, E.; Martin, B.M.; Huang, S.P.; et al. Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 1992, 357, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Wolthoorn, J.; Degroote, S. The fate and function of glycosphingolipid glucosylceramide. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 869–873. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef]
- Wennekes, T.; Van Den Berg, R.J.; Boot, R.G.; Van Der Marel, G.A.; Overkleeft, H.S.; Aerts, J.M. Glycosphingolipids—Nature, function, and pharmacological modulation. Angew. Chem. Int. Ed. Engl. 2009, 48, 8848–8869. [Google Scholar] [CrossRef]
- Tidhar, R.; Futerman, A.H. The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2511–2518. [Google Scholar] [CrossRef]
- Mandon, E.C.; Ehses, I.; Rother, J.; Van Echten, G.; Sandhoff, K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 1992, 267, 11144–11148. [Google Scholar]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Lee, H.; Mesicek, J.; Fuks, Z.; Haimovitz-Friedman, A.; Kolesnick, R.N.; Futerman, A.H. Kinetic characterization of mammalian ceramide synthases: Determination of K(m) values towards sphinganine. FEBS Lett. 2007, 581, 5289–5294. [Google Scholar] [CrossRef] [PubMed]
- Fabrias, G.; Munoz-Olaya, J.; Cingolani, F.; Signorelli, P.; Casas, J.; Gagliostro, V.; Ghidoni, R. Dihydroceramide desaturase and dihydrosphingolipids: Debutant players in the sphingolipid arena. Prog. Lipid Res. 2012, 51, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Chigorno, V.; Riva, C.; Valsecchi, M.; Nicolini, M.; Brocca, P.; Sonnino, S. Metabolic processing of gangliosides by human fibroblasts in culture—Formation and recycling of separate pools of sphingosine. Eur. J. Biochem. 1997, 250, 661–669. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef]
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 4638–4643. [Google Scholar] [CrossRef]
- Van Weely, S.; Brandsma, M.; Strijland, A.; Tager, J.M.; Aerts, J.M. Demonstration of the existence of a second, non-lysosomal glucocerebrosidase that is not deficient in Gaucher disease. Biochim. Biophys. Acta 1993, 1181, 55–62. [Google Scholar] [CrossRef]
- Sandhoff, R.; Sandhoff, K. Emerging concepts of ganglioside metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef]
- Mukherjee, S.; Maxfield, F.R. Membrane domains. Annu. Rev. Cell Dev. Biol. 2004, 20, 839–866. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S.; Prinetti, A. Membrane domains and the “lipid raft” concept. Curr. Med. Chem. 2013, 20, 4–21. [Google Scholar] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Tagami, S.; Inokuchi Ji, J.; Kabayama, K.; Yoshimura, H.; Kitamura, F.; Uemura, S.; Ogawa, C.; Ishii, A.; Saito, M.; Ohtsuka, Y.; et al. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J. Biol. Chem. 2002, 277, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, M.; Aerts, J.M. Glycosphingolipids and insulin resistance. Prog. Lipid Res. 2009, 48, 196–205. [Google Scholar] [CrossRef]
- Kabayama, K.; Sato, T.; Saito, K.; Loberto, N.; Prinetti, A.; Sonnino, S.; Kinjo, M.; Igarashi, Y.; Inokuchi, J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 13678–13683. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Hashiramoto, A.; Haluzik, M.; Mizukami, H.; Beck, S.; Norton, A.; Kono, M.; Tsuji, S.; Daniotti, J.L.; Werth, N.; et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA 2003, 100, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Ottenhoff, R.; Powlson, A.S.; Grefhorst, A.; Van Eijk, M.; Dubbelhuis, P.F.; Aten, J.; Kuipers, F.; Serlie, M.J.; Wennekes, T.; et al. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 2007, 56, 1341–1349. [Google Scholar] [CrossRef]
- Coskun, U.; Grzybek, M.; Drechsel, D.; Simons, K. Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. USA 2011, 108, 9044–9048. [Google Scholar] [CrossRef]
- Nakayama, H.; Nagafuku, M.; Suzuki, A.; Iwabuchi, K.; Inokuchi, J.I. The regulatory roles of glycosphingolipid-enriched lipid rafts in immune systems. FEBS Lett. 2018, 592, 3921–3942. [Google Scholar] [CrossRef]
- Hanada, K. Sphingolipids in infectious diseases. Jpn. J. Infect. Dis. 2005, 58, 131–148. [Google Scholar] [PubMed]
- Aerts, J.; Artola, M.; Van Eijk, M.; Ferraz, M.J.; Boot, R.G. Glycosphingolipids and infection. Potential new therapeutic avenues. Front. Cell Dev. Biol. 2019, 7, 324. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, J.I.; Inamori, K.I.; Kabayama, K.; Nagafuku, M.; Uemura, S.; Go, S.; Suzuki, A.; Ohno, I.; Kanoh, H.; Shishido, F. Biology of GM3 ganglioside. Prog. Mol. Biol. Transl. Sci. 2018, 156, 151–195. [Google Scholar] [PubMed]
- Iwabuchi, K. Gangliosides in the immune system: Role of glycosphingolipids and glycosphingolipid-enriched lipid rafts in immunological functions. Methods Mol. Biol. 2018, 1804, 83–95. [Google Scholar]
- Aerts, J.; Kuo, C.L.; Lelieveld, L.T.; Boer, D.E.C.; Van Der Lienden, M.J.C.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef]
- Nagata, M.; Izumi, Y.; Ishikawa, E.; Kiyotake, R.; Doi, R.; Iwai, S.; Omahdi, Z.; Yamaji, T.; Miyamoto, T.; Bamba, T.; et al. Intracellular metabolite beta-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl. Acad. Sci. USA 2017, 114, E3285–E3294. [Google Scholar] [CrossRef]
- Van Den Bergh, F.A.; Tager, J.M. Localization of neutral glycosphingolipids in human plasma. Biochim. Biophys. Acta 1976, 441, 391–402. [Google Scholar] [CrossRef]
- Ghauharali, K.; Kallemeijn, W.; Vergeer, M.; Motazacker, M.; Van Eijk, M.; Aerts, H.; Groener, A. The role of ABCA1 in glycosphingolipid trafficking and efflux. Chem. Phys. Lipids 2009, 160, S15. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010, 584, 1700–1712. [Google Scholar] [CrossRef]
- Park, J.H.; Schuchman, E.H. Acid ceramidase and human disease. Biochim. Biophys. Acta 2006, 1758, 2133–2138. [Google Scholar] [CrossRef]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Hollak, C.; Boot, R.; Groener, A. Biochemistry of glycosphingolipid storage disorders: Implications for therapeutic intervention. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Ben Bdira, F.; Artola, M.; Overkleeft, H.S.; Ubbink, M.; Aerts, J. Distinguishing the differences in beta-glycosylceramidase folds, dynamics, and actions informs therapeutic uses. J. Lipid Res. 2018, 59, 2262–2276. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Greenblatt, H.M.; Butters, T.D.; Shaaltiel, Y.; Aviezer, D.; Silman, I.; Futerman, A.H.; Sussman, J.L. Crystal structures of complexes of N-butyl- and N-nonyl-deoxynojirimycin bound to acid beta-glucosidase: Insights into the mechanism of chemical chaperone action in Gaucher disease. J. Biol. Chem. 2007, 282, 29052–29058. [Google Scholar] [CrossRef]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef]
- Koshland, D.E. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Rye, C.S.; Withers, S.G. Glycosidase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 573–580. [Google Scholar] [CrossRef]
- Speciale, G.; Thompson, A.J.; Davies, G.J.; Williams, S.J. Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 2014, 28, 1–13. [Google Scholar] [CrossRef]
- Ardevol, A.; Rovira, C. Reaction mechanisms in carbohydrate-active enzymes: Glycoside hydrolases and glycosyltransferases. Insights from ab initio quantum mechanics/molecular mechanics dynamic simulations. J. Am. Chem. Soc. 2015, 137, 7528–7547. [Google Scholar] [CrossRef]
- Legler, G. Glycoside hydrolases: Mechanistic information from studies with reversible and irreversible inhibitors. Adv. Carbohydr. Chem. Biochem. 1990, 48, 319–384. [Google Scholar]
- Withers, S.G.; Aebersold, R. Approaches to labeling and identification of active site residues in glycosidases. Protein Sci. 1995, 4, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, S.; Umezawa, K.; Iinuma, H.; Naganawa, H.; Nakamura, H.; Iitaka, Y.; Takeuchi, T. Production, isolation and structure determination of a novel beta-glucosidase inhibitor, cyclophellitol, from Phellinus sp. J. Antibiot. 1990, 43, 49–53. [Google Scholar] [CrossRef]
- Kallemeijn, W.W.; Li, K.Y.; Witte, M.D.; Marques, A.R.; Aten, J.; Scheij, S.; Jiang, J.; Willems, L.I.; Voorn-Brouwer, T.M.; Van Roomen, C.P.; et al. Novel activity-based probes for broad-spectrum profiling of retaining beta-exoglucosidases in situ and in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 12529–12533. [Google Scholar] [CrossRef] [PubMed]
- Artola, M.; Kuo, C.L.; Lelieveld, L.T.; Rowland, R.J.; Van Der Marel, G.A.; Codee, J.D.C.; Boot, R.G.; Davies, G.J.; Aerts, J.; Overkleeft, H.S. Functionalized cyclophellitols are selective glucocerebrosidase inhibitors and induce a bona fide neuropathic Gaucher model in zebrafish. J. Am. Chem. Soc. 2019, 141, 4214–4218. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Kallemeijn, W.W.; Lelieveld, L.T.; Mirzaian, M.; Zoutendijk, I.; Vardi, A.; Futerman, A.H.; Meijer, A.H.; Spaink, H.P.; Overkleeft, H.S.; et al. In vivo inactivation of glycosidases by conduritol B epoxide and cyclophellitol as revealed by activity-based protein profiling. FEBS J. 2019, 286, 584–600. [Google Scholar] [CrossRef]
- Witte, M.D.; Kallemeijn, W.W.; Aten, J.; Li, K.Y.; Strijland, A.; Donker-Koopman, W.E.; Van Den Nieuwendijk, A.M.; Bleijlevens, B.; Kramer, G.; Florea, B.I.; et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat. Chem. Biol. 2010, 6, 907–913. [Google Scholar] [CrossRef]
- Jiang, J.; Beenakker, T.J.; Kallemeijn, W.W.; Van Der Marel, G.A.; Van Den Elst, H.; Codee, J.D.; Aerts, J.M.; Overkleeft, H.S. Comparing cyclophellitol N-alkyl and N-acyl cyclophellitol aziridines as activity-based glycosidase probes. Chemistry 2015, 21, 10861–10869. [Google Scholar] [CrossRef]
- Willems, L.I.; Beenakker, T.J.; Murray, B.; Scheij, S.; Kallemeijn, W.W.; Boot, R.G.; Verhoek, M.; Donker-Koopman, W.E.; Ferraz, M.J.; Van Rijssel, E.R.; et al. Potent and selective activity-based probes for GH27 human retaining alpha-galactosidases. J. Am. Chem. Soc. 2014, 136, 11622–11625. [Google Scholar] [CrossRef]
- Jiang, J.; Kuo, C.L.; Wu, L.; Franke, C.; Kallemeijn, W.W.; Florea, B.I.; Van Meel, E.; Van Der Marel, G.A.; Codee, J.D.; Boot, R.G.; et al. Detection of active mammalian GH31 alpha-glucosidases in health and disease using in-class, broad-spectrum activity-based probes. ACS Cent. Sci. 2016, 2, 351–358. [Google Scholar] [CrossRef]
- Jiang, J.; Kallemeijn, W.W.; Wright, D.W.; Van Den Nieuwendijk, A.; Rohde, V.C.; Folch, E.C.; Van Den Elst, H.; Florea, B.I.; Scheij, S.; Donker-Koopman, W.E.; et al. In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 alpha-l-fucosidases. Chem. Sci. 2015, 6, 2782–2789. [Google Scholar] [CrossRef]
- Artola, M.; Kuo, C.L.; McMahon, S.A.; Oehler, V.; Hansen, T.; Van Der Lienden, M.; He, X.; Van Den Elst, H.; Florea, B.I.; Kermode, A.R.; et al. New irreversible alpha-l-iduronidase inhibitors and activity-based probes. Chemistry 2018, 24, 19081–19088. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jiang, J.; Jin, Y.; Kallemeijn, W.W.; Kuo, C.L.; Artola, M.; Dai, W.; Van Elk, C.; Van Eijk, M.; Van Der Marel, G.A.; et al. Activity-based probes for functional interrogation of retaining beta-glucuronidases. Nat. Chem. Biol. 2017, 13, 867–873. [Google Scholar] [CrossRef]
- Marques, A.R.; Willems, L.I.; Herrera Moro, D.; Florea, B.I.; Scheij, S.; Ottenhoff, R.; Van Roomen, C.P.; Verhoek, M.; Nelson, J.K.; Kallemeijn, W.W.; et al. A specific activity-based probe to monitor family GH59 galactosylceramidase, the enzyme deficient in Krabbe disease. Chembiochem 2017, 18, 402–412. [Google Scholar] [CrossRef]
- Kuo, C.L.; Van Meel, E.; Kytidou, K.; Kallemeijn, W.W.; Witte, M.; Overkleeft, H.S.; Artola, M.E.; Aerts, J.M. Activity-based probes for glycosidases: Profiling and other applications. Methods Enzymol. 2018, 598, 217–235. [Google Scholar] [PubMed]
- Lahav, D.; Liu, B.; Van Den Berg, R.; Van Den Nieuwendijk, A.; Wennekes, T.; Ghisaidoobe, A.T.; Breen, I.; Ferraz, M.J.; Kuo, C.L.; Wu, L.; et al. A fluorescence polarization activity-based protein profiling assay in the discovery of potent, selective inhibitors for human nonlysosomal glucosylceramidase. J. Am. Chem. Soc. 2017, 139, 14192–14197. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymanska, A.; Groener, J.E.; Kaminski, M.L.; Lugowska, A.; Jurkiewicz, E.; Czartoryska, B. Gaucher disease due to saposin C deficiency, previously described as non-neuronopathic form—No positive effects after 2-years of miglustat therapy. Mol. Genet. Metab. 2011, 104, 627–630. [Google Scholar] [CrossRef]
- Ben Bdira, F.; Kallemeijn, W.W.; Oussoren, S.V.; Scheij, S.; Bleijlevens, B.; Florea, B.I.; Van Roomen, C.; Ottenhoff, R.; Van Kooten, M.; Walvoort, M.T.C.; et al. Stabilization of glucocerebrosidase by active site occupancy. ACS Chem. Biol. 2017, 12, 1830–1841. [Google Scholar] [CrossRef]
- Jonsson, L.M.; Murray, G.J.; Sorrell, S.H.; Strijland, A.; Aerts, J.F.; Ginns, E.I.; Barranger, J.A.; Tager, J.M.; Schram, A.W. Biosynthesis and maturation of glucocerebrosidase in Gaucher fibroblasts. Eur. J. Biochem. 1987, 164, 171–179. [Google Scholar] [CrossRef]
- Aerts, J.M.; Schram, A.W.; Strijland, A.; Van Weely, S.; Jonsson, L.M.; Tager, J.M.; Sorrell, S.H.; Ginns, E.I.; Barranger, J.A.; Murray, G.J. Glucocerebrosidase, a lysosomal enzyme that does not undergo oligosaccharide phosphorylation. Biochim. Biophys. Acta 1988, 964, 303–308. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schroder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Rijnboutt, S.; Aerts, H.M.; Geuze, H.J.; Tager, J.M.; Strous, G.J. Mannose 6-phosphate-independent membrane association of cathepsin D, glucocerebrosidase, and sphingolipid-activating protein in HepG2 cells. J. Biol. Chem. 1991, 266, 4862–4868. [Google Scholar] [PubMed]
- Rijnboutt, S.; Kal, A.J.; Geuze, H.J.; Aerts, H.; Strous, G.J. Mannose 6-phosphate-independent targeting of cathepsin D to lysosomes in HepG2 cells. J. Biol. Chem. 1991, 266, 23586–23592. [Google Scholar] [PubMed]
- Zunke, F.; Andresen, L.; Wesseler, S.; Groth, J.; Arnold, P.; Rothaug, M.; Mazzulli, J.R.; Krainc, D.; Blanz, J.; Saftig, P.; et al. Characterization of the complex formed by beta-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc. Natl. Acad. Sci. USA 2016, 113, 3791–3796. [Google Scholar] [CrossRef] [PubMed]
- Balreira, A.; Gaspar, P.; Caiola, D.; Chaves, J.; Beirao, I.; Lima, J.L.; Azevedo, J.E.; Miranda, M.C. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum. Mol. Genet. 2008, 17, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P.; Kallemeijn, W.W.; Strijland, A.; Scheij, S.; Van Eijk, M.; Aten, J.; Overkleeft, H.S.; Balreira, A.; Zunke, F.; Schwake, M.; et al. Action myoclonus-renal failure syndrome: Diagnostic applications of activity-based probes and lipid analysis. J. Lipid Res. 2014, 55, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Zhao, S.; Tian, Q.Y.; Liu, H.; Zhao, Y.; Chen, W.C.; Grunig, G.; Torres, P.A.; Wang, B.C.; Zeng, B.; et al. Association between progranulin and Gaucher disease. EBioMedicine 2016, 11, 127–137. [Google Scholar] [CrossRef]
- Chen, Y.; Sud, N.; Hettinghouse, A.; Liu, C.J. Molecular regulations and therapeutic targets of Gaucher disease. Cytokine Growth Factor Rev. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Jian, J.; Tian, Q.Y.; Hettinghouse, A.; Zhao, S.; Liu, H.; Wei, J.; Grunig, G.; Zhang, W.; Setchell, K.D.R.; Sun, Y.; et al. Progranulin recruits HSP70 to beta-glucocerebrosidase and Is therapeutic against Gaucher disease. EBioMedicine 2016, 13, 212–224. [Google Scholar] [CrossRef]
- Tan, Y.L.; Genereux, J.C.; Pankow, S.; Aerts, J.M.; Yates, J.R., III; Kelly, J.W. ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher’s disease. Chem. Biol. 2014, 21, 967–976. [Google Scholar] [CrossRef]
- Aghion, H. La Maladie de Gaucher dans L’enfance. Ph.D. Thesis, Faculte de Medecine, Paris, France, 1934. [Google Scholar]
- Rosenberg, A.; Chargaff, E. A reinvestigation of the cerebroside deposited in Gaucher’s disease. J. Biol. Chem. 1958, 233, 1323–1326. [Google Scholar]
- Aerts, J.M.; Cox, T.M.; Roscoe, O. Brady: Physician whose pioneering discoveries in lipid biochemistry revolutionized treatment and understanding of lysosomal diseases. Blood Cells Mol. Dis. 2018, 68, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Danby, P.M.; Withers, S.G. Advances in enzymatic glycoside synthesis. ACS Chem. Biol. 2016, 11, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Ghauharali-van der Vlugt, K.; Meijer, R.; Giraldo, P.; Alfonso, P.; et al. Glucosylated cholesterol in mammalian cells and tissues: Formation and degradation by multiple cellular beta-glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Cholesterol glucosylation is catalyzed by transglucosylation reaction of beta-glucosidase 1. Biochem. Biophys. Res. Commun. 2013, 441, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Hirabayashi, Y. A novel function for glucocerebrosidase as a regulator of sterylglucoside metabolism. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Kallemeijn, W.W.; Mirzaian, M.; Herrera Moro, D.; Marques, A.; Wisse, P.; Boot, R.G.; Willems, L.I.; Overkleeft, H.S.; Aerts, J.M. Gaucher disease and Fabry disease: New markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta 2014, 1841, 811–825. [Google Scholar] [CrossRef]
- Horowitz, M.; Wilder, S.; Horowitz, Z.; Reiner, O.; Gelbart, T.; Beutler, E. The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics 1989, 4, 87–96. [Google Scholar] [CrossRef]
- Ohashi, T.; Hong, C.M.; Weiler, S.; Tomich, J.M.; Aerts, J.M.; Tager, J.M.; Barranger, J.A. Characterization of human glucocerebrosidase from different mutant alleles. J. Biol. Chem. 1991, 266, 3661–3667. [Google Scholar]
- Boot, R.G.; Hollak, C.E.; Verhoek, M.; Sloof, P.; Poorthuis, B.J.; Kleijer, W.J.; Wevers, R.A.; Van Oers, M.H.; Mannens, M.M.; Aerts, J.M.; et al. Glucocerebrosidase genotype of Gaucher patients in The Netherlands: Limitations in prognostic value. Hum. Mutat. 1997, 10, 348–358. [Google Scholar] [CrossRef]
- Diamond, J.M. Human genetics. Jewish lysosomes. Nature 1994, 368, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Boas, F.E. Linkage to Gaucher mutations in the Ashkenazi population: Effect of drift on decay of linkage disequilibrium and evidence for heterozygote selection. Blood Cells Mol. Dis. 2000, 26, 348–359. [Google Scholar] [CrossRef]
- Colombo, R. Age estimate of the N370S mutation causing Gaucher disease in Ashkenazi Jews and European populations: A reappraisal of haplotype data. Am. J. Hum. Genet. 2000, 66, 692–697. [Google Scholar] [CrossRef][Green Version]
- Meijer, A.H.; Aerts, J.M. Linking smokers’ susceptibility to tuberculosis with lysosomal storage disorders. Dev. Cell 2016, 37, 112–113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sidransky, E. Gaucher disease: Complexity in a “simple” disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, R.H.; Grant, I.R.; Halsall, D.; Cox, T.M. Twin pairs showing discordance of phenotype in adult Gaucher’s disease. QJM 2004, 97, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Van Schaik, I.N.; Aerts, J.M.; Langeveld, M.; Mannens, M.M.; Bour, L.J.; Sidransky, E.; Tayebi, N.; Fitzgibbon, E.; Hollak, C.E. A monozygotic twin pair with highly discordant Gaucher phenotypes. Blood Cells Mol. Dis. 2011, 46, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Uyama, E.; Takahashi, K.; Owada, M.; Okamura, R.; Naito, M.; Tsuji, S.; Kawasaki, S.; Araki, S. Hydrocephalus, corneal opacities, deafness, valvular heart disease, deformed toes and leptomeningeal fibrous thickening in adult siblings: A new syndrome associated with beta-glucocerebrosidase deficiency and a mosaic population of storage cells. Acta Neurol. Scand. 1992, 86, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Chabas, A.; Cormand, B.; Grinberg, D.; Burguera, J.M.; Balcells, S.; Merino, J.L.; Mate, I.; Sobrino, J.A.; Gonzalez-Duarte, R.; Vilageliu, L. Unusual expression of Gaucher’s disease: Cardiovascular calcifications in three sibs homozygous for the D409H mutation. J. Med. Genet. 1995, 32, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Abrahamov, A.; Elstein, D.; Gross-Tsur, V.; Farber, B.; Glaser, Y.; Hadas-Halpern, I.; Ronen, S.; Tafakjdi, M.; Horowitz, M.; Zimran, A. Gaucher’s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet 1995, 346, 1000–1003. [Google Scholar] [CrossRef]
- Zhang, C.K.; Stein, P.B.; Liu, J.; Wang, Z.; Yang, R.; Cho, J.H.; Gregersen, P.K.; Aerts, J.M.; Zhao, H.; Pastores, G.M.; et al. Genome-wide association study of N370S homozygous Gaucher disease reveals the candidacy of CLN8 gene as a genetic modifier contributing to extreme phenotypic variation. Am. J. Hematol. 2012, 87, 377–383. [Google Scholar] [CrossRef]
- Di Ronza, A.; Bajaj, L.; Sharma, J.; Sanagasetti, D.; Lotfi, P.; Adamski, C.J.; Collette, J.; Palmieri, M.; Amawi, A.; Popp, L.; et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 2018, 20, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Velayati, A.; DePaolo, J.; Gupta, N.; Choi, J.H.; Moaven, N.; Westbroek, W.; Goker-Alpan, O.; Goldin, E.; Stubblefield, B.K.; Kolodny, E.; et al. A mutation in SCARB2 is a modifier in Gaucher disease. Hum. Mutat. 2011, 32, 1232–1238. [Google Scholar] [CrossRef]
- Alfonso, P.; Navascues, J.; Navarro, S.; Medina, P.; Bolado-Carrancio, A.; Andreu, V.; Irun, P.; Rodriguez-Rey, J.C.; Pocovi, M.; Espana, F.; et al. Characterization of variants in the glucosylceramide synthase gene and their association with type 1 Gaucher disease severity. Hum. Mutat. 2013, 34, 1396–1403. [Google Scholar] [CrossRef]
- Siebert, M.; Westbroek, W.; Chen, Y.C.; Moaven, N.; Li, Y.; Velayati, A.; Saraiva-Pereira, M.L.; Martin, S.E.; Sidransky, E. Identification of miRNAs that modulate glucocerebrosidase activity in Gaucher disease cells. RNA Biol. 2014, 11, 1291–1300. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Nalls, M.A.; Duran, R.; Lopez, G.; Kurzawa-Akanbi, M.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M.; Theuns, J.; Crosiers, D.; Cras, P.; et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013, 70, 727–735. [Google Scholar] [CrossRef]
- Siebert, M.; Sidransky, E.; Westbroek, W. Glucocerebrosidase is shaking up the synucleinopathies. Brain 2014, 137, 1304–1322. [Google Scholar] [CrossRef]
- Mullin, S.; Beavan, M.; Bestwick, J.; McNeill, A.; Proukakis, C.; Cox, T.; Hughes, D.; Mehta, A.; Zetterberg, H.; Schapira, A.H.V. Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov. Disord. 2019, 34, 1365–1373. [Google Scholar] [CrossRef]
- Isacson, O.; Brekk, O.R.; Hallett, P.J. Novel results and concepts emerging from lipid cell biology relevant to degenerative brain aging and disease. Front. Neurol. 2019, 10, 1053. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200. [Google Scholar] [CrossRef]
- Huebecker, M.; Moloney, E.B.; Van Der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained systemic glucocerebrosidase inhibition induces brain alpha-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid. Redox Signal. 2015, 23, 550–564. [Google Scholar] [CrossRef]
- Brekk, O.R.; Moskites, A.; Isacson, O.; Hallett, P.J. Lipid-dependent deposition of alpha-synuclein and Tau on neuronal Secretogranin II-positive vesicular membranes with age. Sci. Rep. 2018, 8, 15207. [Google Scholar] [CrossRef]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. Expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef] [PubMed]
- Stojkovska, I.; Krainc, D.; Mazzulli, J.R. Molecular mechanisms of α-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res. 2018, 373, 51–60. [Google Scholar] [CrossRef]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Westbroek, W.; Goldin, E.; Moaven, N.; Sidransky, E.; Lee, J.C. Alpha-synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J. Biol. Chem. 2011, 286, 28080–28088. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, M.; Smith, L.; Schapira, A.H.V. The biochemical basis of interactions between Glucocerebrosidase and alpha-synuclein in GBA1 mutation carriers. J. Neurochem. 2020, e14968. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [PubMed]
- Cabasso, O.; Paul, S.; Dorot, O.; Maor, G.; Krivoruk, O.; Pasmanik-Chor, M.; Mirzaian, M.; Ferraz, M.; Aerts, J.; Horowitz, M. Drosophila melanogaster mutated in its GBA1b ortholog recapitulates neuronopathic Gaucher disease. J. Clin. Med. 2019, 8, 1420. [Google Scholar] [CrossRef] [PubMed]
- Bussink, A.P.; Van Eijk, M.; Renkema, G.H.; Aerts, J.M.; Boot, R.G. The biology of the Gaucher cell: The cradle of human chitinases. Int. Rev. Cytol. 2006, 252, 71–128. [Google Scholar]
- Boven, L.A.; Van Meurs, M.; Boot, R.G.; Mehta, A.; Boon, L.; Aerts, J.M.; Laman, J.D. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am. J. Clin. Pathol. 2004, 122, 359–369. [Google Scholar] [CrossRef]
- Hollak, C.E.; Van Weely, S.; Van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef]
- Boot, R.G.; Renkema, G.H.; Strijland, A.; Van Zonneveld, A.J.; Aerts, J.M. Cloning of a cDNA encoding chitotriosidase, a human chitinase produced by macrophages. J. Biol. Chem. 1995, 270, 26252–26256. [Google Scholar] [CrossRef]
- Aguilera, B.; Ghauharali-van der Vlugt, K.; Helmond, M.T.; Out, J.M.; Donker-Koopman, W.E.; Groener, J.E.; Boot, R.G.; Renkema, G.H.; Van Der Marel, G.A.; Van Boom, J.H.; et al. Transglycosidase activity of chitotriosidase: Improved enzymatic assay for the human macrophage chitinase. J. Biol. Chem. 2003, 278, 40911–40916. [Google Scholar] [CrossRef]
- Schoonhoven, A.; Rudensky, B.; Elstein, D.; Zimran, A.; Hollak, C.E.; Groener, J.E.; Aerts, J.M. Monitoring of Gaucher patients with a novel chitotriosidase assay. Clin. Chim. Acta 2007, 381, 136–139. [Google Scholar] [CrossRef]
- Boot, R.G.; Renkema, G.H.; Verhoek, M.; Strijland, A.; Bliek, J.; De Meulemeester, T.M.; Mannens, M.M.; Aerts, J.M. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J. Biol. Chem. 1998, 273, 25680–25685. [Google Scholar] [CrossRef]
- Deegan, P.B.; Moran, M.T.; McFarlane, I.; Schofield, J.P.; Boot, R.G.; Aerts, J.M.; Cox, T.M. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol. Dis. 2005, 35, 259–267. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; De Fost, M.; Hollak, C.E.; Maas, M.; Bleijlevens, B.; Van Breemen, M.J.; Van Meurs, M.; Boven, L.A.; Laman, J.D.; et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood 2004, 103, 33–39. [Google Scholar] [CrossRef]
- Kramer, G.; Wegdam, W.; Donker-Koopman, W.; Ottenhoff, R.; Gaspar, P.; Verhoek, M.; Nelson, J.; Gabriel, T.; Kallemeijn, W.; Boot, R.G.; et al. Elevation of glycoprotein nonmetastatic melanoma protein B in type 1 Gaucher disease patients and mouse models. FEBS Open Bio 2016, 6, 902–913. [Google Scholar] [CrossRef]
- Murugesan, V.; Liu, J.; Yang, R.; Lin, H.; Lischuk, A.; Pastores, G.; Zhang, X.; Chuang, W.L.; Mistry, P.K. Validating glycoprotein non-metastatic melanoma B (gpNMB, osteoactivin), a new biomarker of Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 47–53. [Google Scholar] [CrossRef]
- Zigdon, H.; Savidor, A.; Levin, Y.; Meshcheriakova, A.; Schiffmann, R.; Futerman, A.H. Identification of a biomarker in cerebrospinal fluid for neuronopathic forms of Gaucher disease. PLoS ONE 2015, 10, e0120194. [Google Scholar] [CrossRef]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef]
- Dahl, M.; Doyle, A.; Olsson, K.; Mansson, J.E.; Marques, A.R.A.; Mirzaian, M.; Aerts, J.M.; Ehinger, M.; Rothe, M.; Modlich, U.; et al. Lentiviral gene therapy using cellular promoters cures type 1 Gaucher disease in mice. Mol. Ther. 2015, 23, 835–844. [Google Scholar] [CrossRef]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef]
- Aerts, J.M.; Hollak, C.E. Plasma and metabolic abnormalities in Gaucher’s disease. Baillieres Clin. Haematol. 1997, 10, 691–709. [Google Scholar] [CrossRef]
- Aerts, J.M.; Kallemeijn, W.W.; Wegdam, W.; Joao Ferraz, M.; Van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids, and inhibodies. J. Inherit. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef]
- Vissers, J.P.; Langridge, J.I.; Aerts, J.M. Analysis and quantification of diagnostic serum markers and protein signatures for Gaucher disease. Mol. Cell. Proteom. 2007, 6, 755–766. [Google Scholar] [CrossRef]
- Hollak, C.E.; Levi, M.; Berends, F.; Aerts, J.M.; Van Oers, M.H. Coagulation abnormalities in type 1 Gaucher disease are due to low-grade activation and can be partly restored by enzyme supplementation therapy. Br. J. Haematol. 1997, 96, 470–476. [Google Scholar] [CrossRef]
- Brady, R.O. Enzyme replacement therapy: Conception, chaos and culmination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 915–919. [Google Scholar] [CrossRef]
- Maas, M.; Van Kuijk, C.; Stoker, J.; Hollak, C.E.; Akkerman, E.M.; Aerts, J.F.; Den Heeten, G.J. Quantification of bone involvement in Gaucher disease: MR imaging bone marrow burden score as an alternative to Dixon quantitative chemical shift MR imaging—Initial experience. Radiology 2003, 229, 554–561. [Google Scholar] [CrossRef]
- Aerts, J.M.; Hollak, C.E.; Boot, R.G.; Groener, J.E.; Maas, M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 2006, 29, 449–456. [Google Scholar] [CrossRef]
- Platt, F.M.; Jeyakumar, M.; Andersson, U.; Priestman, D.A.; Dwek, R.A.; Butters, T.D.; Cox, T.M.; Lachmann, R.H.; Hollak, C.; Aerts, J.M.; et al. Inhibition of substrate synthesis as a strategy for glycolipid lysosomal storage disease therapy. J. Inherit. Metab. Dis. 2001, 24, 275–290. [Google Scholar] [CrossRef]
- Heitner, R.; Elstein, D.; Aerts, J.; Van Weely, S.; Zimran, A. Low-dose N-butyldeoxynojirimycin (OGT 918) for type I Gaucher disease. Blood Cells Mol. Dis. 2002, 28, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Balwani, M.; Baris, H.N.; Turkia, H.B.; Burrow, T.A.; Charrow, J.; Cox, G.F.; Danda, S.; Dragosky, M.; Drelichman, G.; et al. Safety, efficacy, and authorization of eliglustat as a first-line therapy in Gaucher disease type 1. Blood Cells Mol. Dis. 2018, 71, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J.A.; Larsen, S.D. The development and use of small molecule inhibitors of glycosphingolipid metabolism for lysosomal storage diseases. J. Lipid Res. 2014, 55, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Van Dussen, L.; Hendriks, E.J.; Groener, J.E.; Boot, R.G.; Hollak, C.E.; Aerts, J.M. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J. Inherit. Metab. Dis. 2014, 37, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, G.H.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Zimran, A.; Altarescu, G.; Elstein, D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 134–137. [Google Scholar] [CrossRef]
- Narita, A.; Shirai, K.; Itamura, S.; Matsuda, A.; Ishihara, A.; Matsushita, K.; Fukuda, C.; Kubota, N.; Takayama, R.; Shigematsu, H.; et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann. Clin. Transl. Neurol. 2016, 3, 200–215. [Google Scholar] [CrossRef]
- Fog, C.K.; Zago, P.; Malini, E.; Solanko, L.M.; Peruzzo, P.; Bornaes, C.; Magnoni, R.; Mehmedbasic, A.; Petersen, N.H.T.; Bembi, B.; et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 2018, 38, 142–153. [Google Scholar] [CrossRef]
- Aerts, J.M.; Ferraz, M.J.; Mirzaian, M.; Gaspar, P.; Oussoren, S.V.; Wisse, P.; Kuo, C.L.; Lelieveld, L.T.; Kytidou, K.; Hazeu, M.D.; et al. Lysosomal storage diseases. In For Better or Worse: Adapting to Defective Lysosomal Glycosphingolipid; John Wiley & Sons, Ltd.: Chichester, UK, 2017; pp. 1–13. [Google Scholar]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef]
- Raghavan, S.S.; Mumford, R.A.; Kanfer, J.N. Isolation and characterization of glucosylsphingosine from Gaucher’s spleen. J. Lipid Res. 1974, 15, 484–490. [Google Scholar]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef]
- Dekker, N.; Van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; Van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [PubMed]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Gold, H.; Donker-Koopman, W.E.; Verhoek, M.; Overkleeft, H.S.; Boot, R.G.; Kramer, G.; Dekker, N.; et al. Mass spectrometric quantification of glucosylsphingosine in plasma and urine of type 1 Gaucher patients using an isotope standard. Blood Cells Mol. Dis. 2015, 54, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, L.T.; Mirzaian, M.; Kuo, C.L.; Artola, M.; Ferraz, M.J.; Peter, R.E.A.; Akiyama, H.; Greimel, P.; Van Den Berg, R.; Overkleeft, H.S.; et al. Role of beta-glucosidase 2 in aberrant glycosphingolipid metabolism: Model of glucocerebrosidase deficiency in zebrafish. J. Lipid Res. 2019, 60, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Groener, J.E.; Poorthuis, B.J.; Kuiper, S.; Helmond, M.T.; Hollak, C.E.; Aerts, J.M. HPLC for simultaneous quantification of total ceramide, glucosylceramide, and ceramide trihexoside concentrations in plasma. Clin. Chem. 2007, 53, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J.; et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal immunoglobulin against lysolipids in the origin of myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef]
- Pandey, M.K.; Grabowski, G.A.; Kohl, J. An unexpected player in Gaucher disease: The multiple roles of complement in disease development. Semin. Immunol. 2018, 37, 30–42. [Google Scholar] [CrossRef]
- Smith, N.J.; Fuller, M.; Saville, J.T.; Cox, T.M. Reduced cerebral vascularization in experimental neuronopathic Gaucher disease. J. Pathol. 2018, 244, 120–128. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.; Gaspar, P.; Mirzaian, M.; Van Roomen, C.; Ottenhoff, R.; Alfonso, P.; Irun, P.; Giraldo, P.; Wisse, P.; et al. Lyso-glycosphingolipid abnormalities in different murine models of lysosomal storage disorders. Mol. Genet. Metab. 2016, 117, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Kuchar, L.; Sikora, J.; Gulinello, M.E.; Poupetova, H.; Lugowska, A.; Malinova, V.; Jahnova, H.; Asfaw, B.; Ledvinova, J. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal. Biochem. 2017, 525, 73–77. [Google Scholar] [CrossRef]
- Suzuki, K. Twenty five years of the: “psychosine hypothesis”: A personal perspective of its history and present status. Neurochem. Res. 1998, 23, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; Van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Choi, L.; Vernon, J.; Kopach, O.; Minett, M.S.; Mills, K.; Clayton, P.T.; Meert, T.; Wood, J.N. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci. Lett. 2015, 594, 163–168. [Google Scholar] [CrossRef]
- Sanchez-Nino, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef]
- Kaissarian, N.; Kang, J.; Shu, L.; Ferraz, M.J.; Aerts, J.M.; Shayman, J.A. Dissociation of globotriaosylceramide and impaired endothelial function in alpha-galactosidase-A deficient EA.hy926 cells. Mol. Genet. Metab. 2018, 125, 338–344. [Google Scholar] [CrossRef]
- Rombach, S.M.; Twickler, T.B.; Aerts, J.M.; Linthorst, G.E.; Wijburg, F.A.; Hollak, C.E. Vasculopathy in patients with Fabry disease: Current controversies and research directions. Mol. Genet. Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Ghauharali-van der Vlugt, K.; Langeveld, M.; Poppema, A.; Kuiper, S.; Hollak, C.E.; Aerts, J.M.; Groener, J.E. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin. Chim. Acta 2008, 389, 109–113. [Google Scholar] [CrossRef]
- Langeveld, M.; Ghauharali, K.J.; Sauerwein, H.P.; Ackermans, M.T.; Groener, J.E.; Hollak, C.E.; Aerts, J.M.; Serlie, M.J. Type I Gaucher disease, a glycosphingolipid storage disorder, is associated with insulin resistance. J. Clin. Endocrinol. Metab. 2008, 93, 845–851. [Google Scholar] [CrossRef]
- Yildiz, Y.; Matern, H.; Thompson, B.; Allegood, J.C.; Warren, R.L.; Ramirez, D.M.; Hammer, R.E.; Hamra, F.K.; Matern, S.; Russell, D.W. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Investig. 2006, 116, 2985–2994. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; Donker-Koopman, W.; Strijland, A.; Van Marle, J.; Overkleeft, H.S.; Wennekes, T.; Aerts, J.M. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J. Biol. Chem. 2007, 282, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.B.; Eleuch-Fayache, G.; Schottlaender, L.V.; Nehdi, H.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Hernandez, D.G.; Sailer, A.; Liu, G.; et al. Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am. J. Hum. Genet. 2013, 92, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Schule, R.; Smets, K.; Rastetter, A.; Boukhris, A.; Loureiro, J.L.; Gonzalez, M.A.; Mundwiller, E.; Deconinck, T.; Wessner, M.; et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Kancheva, D.; Atkinson, D.; De Rijk, P.; Zimon, M.; Chamova, T.; Mitev, V.; Yaramis, A.; Maria Fabrizi, G.; Topaloglu, H.; Tournev, I.; et al. Novel mutations in genes causing hereditary spastic paraplegia and Charcot-Marie-Tooth neuropathy identified by an optimized protocol for homozygosity mapping based on whole-exome sequencing. Genet. Med. 2016, 18, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Sultana, S.; Reichbauer, J.; Schule, R.; Mochel, F.; Synofzik, M.; Van Der Spoel, A.C. Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem. Biophys. Res. Commun. 2015, 465, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Woeste, M.A.; Stern, S.; Raju, D.N.; Grahn, E.; Dittmann, D.; Gutbrod, K.; Dormann, P.; Hansen, J.N.; Schonauer, S.; Marx, C.E.; et al. Species-specific differences in nonlysosomal glucosylceramidase GBA2 function underlie locomotor dysfunction arising from loss-of-function mutations. J. Biol. Chem. 2019, 294, 3853–3871. [Google Scholar] [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef]
- Bowman, S.L.; Bi-Karchin, J.; Le, L.; Marks, M.S. The road to lysosome-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic 2019, 20, 404–435. [Google Scholar] [CrossRef]
- Delevoye, C.; Marks, M.S.; Raposo, G. Lysosome-related organelles as functional adaptations of the endolysosomal system. Curr. Opin. Cell Biol. 2019, 59, 147–158. [Google Scholar] [CrossRef]
- Bahadori, R.; Rinner, O.; Schonthaler, H.B.; Biehlmaier, O.; Makhankov, Y.V.; Rao, P.; Jagadeeswaran, P.; Neuhauss, S.C. The Zebrafish fade out mutant: A novel genetic model for Hermansky-Pudlak syndrome. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4523–4531. [Google Scholar] [CrossRef]
- Ellis, K.; Bagwell, J.; Bagnat, M. Notochord vacuoles are lysosome-related organelles that function in axis and spine morphogenesis. J. Cell Biol. 2013, 200, 667–679. [Google Scholar] [CrossRef]
- Diaz-Tellez, A.; Zampedri, C.; Ramos-Balderas, J.L.; Garcia-Hernandez, F.; Maldonado, E. Zebrafish scarb2a insertional mutant reveals a novel function for the Scarb2/Limp2 receptor in notochord development. Dev. Dyn. 2016, 245, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Van Dijk, S.M.; Wolthoorn, J.; Neumann, S.; Theos, A.C.; De Maziere, A.M.; Klumperman, J.; Van Meer, G.; Sprong, H. Glycolipid-dependent sorting of melanosomal from lysosomal membrane proteins by lumenal determinants. Traffic 2008, 9, 951–963. [Google Scholar] [CrossRef] [PubMed]
- McLean, W.H.; Hull, P.R. Breach delivery: Increased solute uptake points to a defective skin barrier in atopic dermatitis. J. Investig. Dermatol. 2007, 127, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Yousef, H.; Alhajj, M.; Sharma, S. Anatomy, Skin (Integument), Epidermis; StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Eckhart, L.; Lippens, S.; Tschachler, E.; Declercq, W. Cell death by cornification. Biochim. Biophys. Acta 2013, 1833, 3471–3480. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, A.V.; Harding, C.R. Moisturization and skin barrier function. Dermatol. Ther. 2004, 17, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tezuka, T. The content of free amino acids in the stratum corneum is increased in senile xerosis. Arch. Dermatol. Res. 2004, 295, 448–452. [Google Scholar] [CrossRef]
- Voegeli, D. The role of emollients in the care of patients with dry skin. Nurs. Stand. 2007, 22, 64–68. [Google Scholar] [CrossRef]
- Fluhr, J.W.; Elias, P.M. Stratum corneum pH: Formation and function of the “acid mantle”. Exog. Dermatol. 2002, 1, 163–175. [Google Scholar] [CrossRef]
- Proksch, E. pH in nature, humans and skin. J. Dermatol. 2018, 45, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M. Epidermal lipids, membranes, and keratinization. Int. J. Dermatol. 1981, 20. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Duschl, J.; Steinbacher, P.; Salzmann, M.; Bischof, J.; Schuller, M.; Wimmer, H.; Peer, T.; Bauer, J.W.; Richter, K. Age-related changes in the composition of the cornified envelope in human skin. Exp. Dermatol. 2013, 22, 329–335. [Google Scholar] [CrossRef]
- Meguro, S.; Arai, Y.; Masukawa, Y.; Uie, K.; Tokimitsu, I. Relationship between covalently bound ceramides and transepidermal water loss (TEWL). Arch. Dermatol. Res. 2000, 292, 463–468. [Google Scholar] [CrossRef]
- Elias, P.M.; Gruber, R.; Crumrine, D.; Menon, G.; Williams, M.L.; Wakefield, J.S.; Holleran, W.M.; Uchida, Y. Formation and functions of the corneocyte lipid envelope (CLE). Biochim. Biophys. Acta 2014, 1841, 314–318. [Google Scholar] [CrossRef]
- Rabionet, M.; Gorgas, K.; Sandhoff, R. Ceramide synthesis in the epidermis. Biochim. Biophys. Acta 2014, 1841, 422–434. [Google Scholar] [CrossRef]
- Lampe, M.A.; Burlingame, A.L.; Whitney, J.; Williams, M.L.; Brown, B.E.; Roitman, E.; Elias, P.M. Human stratum corneum lipids: Characterization and regional variations. J. Lipid Res. 1983, 24, 120–130. [Google Scholar]
- Janssens, M.; Van Smeden, J.; Gooris, G.S.; Bras, W.; Portale, G.; Caspers, P.J.; Vreeken, R.J.; Kezic, S.; Lavrijsen, A.P.; Bouwstra, J.A. Lamellar lipid organization and ceramide composition in the stratum corneum of patients with atopic eczema. J. Investig. Dermatol. 2011, 131, 2136–2138. [Google Scholar] [CrossRef]
- Imokawa, G.; Abe, A.; Jin, K.; Higaki, Y.; Kawashima, M.; Hidano, A. Decreased level of ceramides in stratum corneum of atopic dermatitis: An etiologic factor in atopic dry skin? J. Investig. Dermatol. 1991, 96, 523–526. [Google Scholar] [CrossRef]
- Motta, S.; Monti, M.; Sesana, S.; Caputo, R.; Carelli, S.; Ghidoni, R. Ceramide composition of the psoriatic scale. Biochim. Biophys. Acta 1993, 1182, 147–151. [Google Scholar] [CrossRef]
- Coderch, L.; Lopez, O.; De la Maza, A.; Parra, J.L. Ceramides and skin function. Am. J. Clin. Dermatol. 2003, 4, 107–129. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Williams, M.L.; Crumrine, D.; Schmuth, M. Inherited clinical disorders of lipid metabolism. Curr. Probl. Dermatol. 2010, 39, 30–88. [Google Scholar] [PubMed]
- Wertz, P. Epidermal lamellar granules. Skin. Pharmacol. Physiol. 2018, 31, 262–268. [Google Scholar] [CrossRef]
- Mitsutake, S.; Suzuki, C.; Akiyama, M.; Tsuji, K.; Yanagi, T.; Shimizu, H.; Igarashi, Y. ABCA12 dysfunction causes a disorder in glucosylceramide accumulation during keratinocyte differentiation. J. Dermatol. Sci. 2010, 60, 128–129. [Google Scholar] [CrossRef]
- Akiyama, M. The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim. Biophys. Acta 2014, 1841, 435–440. [Google Scholar] [CrossRef]
- Akiyama, M.; Sugiyama-Nakagiri, Y.; Sakai, K.; McMillan, J.R.; Goto, M.; Arita, K.; Tsuji-Abe, Y.; Tabata, N.; Matsuoka, K.; Sasaki, R.; et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J. Clin. Investig. 2005, 115, 1777–1784. [Google Scholar] [CrossRef]
- Schurer, N.Y.; Elias, P.M. The biochemistry and function of stratum corneum lipids. Adv. Lipid Res. 1991, 24, 27–56. [Google Scholar]
- Feingold, K.R. Lamellar bodies: The key to cutaneous barrier function. J. Investig. Dermatol. 2012, 132, 1951–1953. [Google Scholar] [CrossRef]
- Geeraert, L.; Mannaerts, G.P.; Van Veldhoven, P.P. Conversion of dihydroceramide into ceramide: Involvement of a desaturase. Biochem. J. 1997, 327, 125–132. [Google Scholar] [CrossRef]
- Michel, C.; Van Echten-Deckert, G.; Rother, J.; Sandhoff, K.; Wang, E.; Merrill, A.H., Jr. Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide. J. Biol. Chem. 1997, 272, 22432–22437. [Google Scholar] [CrossRef]
- Savile, C.K.; Fabrias, G.; Buist, P.H. Dihydroceramide delta (4) desaturase initiates substrate oxidation at C-4. J. Am. Chem. Soc. 2001, 123, 4382–4385. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Identification of the human sphingolipid C4-hydroxylase, hDES2, and its up-regulation during keratinocyte differentiation. FEBS Lett. 2004, 563, 93–97. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. The role of sphingolipid metabolism in cutaneous permeability barrier formation. Biochim. Biophys. Acta 2014, 1841, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Jang, W.H.; Seo, J.A.; Park, M.; Lee, T.R.; Park, Y.H.; Kim, D.K.; Lim, K.M. Decrease of ceramides with very long-chain fatty acids and downregulation of elongases in a murine atopic dermatitis model. J. Investig. Dermatol. 2012, 132, 476–479. [Google Scholar] [CrossRef]
- Uchida, Y. The role of fatty acid elongation in epidermal structure and function. Dermatoendocrinology 2011, 3, 65–69. [Google Scholar] [CrossRef]
- Hanley, K.; Ng, D.C.; He, S.S.; Lau, P.; Min, K.; Elias, P.M.; Bikle, D.D.; Mangelsdorf, D.J.; Williams, M.L.; Feingold, K.R. Oxysterols induce differentiation in human keratinocytes and increase Ap-1-dependent involucrin transcription. J. Investig. Dermatol. 2000, 114, 545–553. [Google Scholar] [CrossRef]
- Denning, M.F.; Kazanietz, M.G.; Blumberg, P.M.; Yuspa, S.H. Cholesterol sulfate activates multiple protein kinase C isoenzymes and induces granular cell differentiation in cultured murine keratinocytes. Cell Growth Differ. 1995, 6, 1619–1626. [Google Scholar]
- Hanley, K.; Wood, L.; Ng, D.C.; He, S.S.; Lau, P.; Moser, A.; Elias, P.M.; Bikle, D.D.; Williams, M.L.; Feingold, K.R. Cholesterol sulfate stimulates involucrin transcription in keratinocytes by increasing Fra-1, Fra-2, and Jun D. J. Lipid Res. 2001, 42, 390–398. [Google Scholar]
- Elias, P.M.; Williams, M.L.; Holleran, W.M.; Jiang, Y.J.; Schmuth, M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: Inherited disorders of lipid metabolism. J. Lipid Res. 2008, 49, 697–714. [Google Scholar] [CrossRef]
- Feingold, K.R.; Jiang, Y.J. The mechanisms by which lipids coordinately regulate the formation of the protein and lipid domains of the stratum corneum: Role of fatty acids, oxysterols, cholesterol sulfate and ceramides as signaling molecules. Dermato Endocrinol. 2011, 3, 113–118. [Google Scholar] [CrossRef][Green Version]
- Elias, P.M.; Crumrine, D.; Rassner, U.; Hachem, J.P.; Menon, G.K.; Man, W.; Choy, M.H.; Leypoldt, L.; Feingold, K.R.; Williams, M.L. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J. Investig. Dermatol. 2004, 122, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Denda, M.; Nakanishi, J.; Nomura, J.; Koyama, J. Cholesterol sulfate inhibits proteases that are involved in desquamation of stratum corneum. J. Investig. Dermatol. 1998, 111, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.; Squier, C.A. Variations in lipids in different layers of porcine epidermis. J. Investig. Dermatol. 1986, 87, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, A.W.; Wertz, P.W.; Downing, D.T.; Mackenzie, I.C. Lipid composition of cohesive and desquamated corneocytes from mouse ear skin. J. Investig. Dermatol. 1986, 86, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M. Corneocyte lipid envelope (CLE), the key structure for skin barrier function and ichthyosis pathogenesis. J. Dermatol. Sci. 2017, 88, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Thematic review series: Skin lipids. The role of epidermal lipids in cutaneous permeability barrier homeostasis. J. Lipid Res. 2007, 48, 2531–2546. [Google Scholar] [CrossRef]
- Greene, S.L.; Muller, S.A. Netherton’s syndrome. Report of a case and review of the literature. J. Am. Acad. Dermatol. 1985, 13, 329–337. [Google Scholar] [CrossRef]
- Traupe, H. The Ichthyoses: A Guide to Clinical Diagnosis, Genetic Counseling, and Therapy; Springer: Berlin, Germany; New York, NY, USA, 1989. [Google Scholar]
- Stone, D.L.; Carey, W.F.; Christodoulou, J.; Sillence, D.; Nelson, P.; Callahan, M.; Tayebi, N.; Sidransky, E. Type 2 Gaucher disease: The collodion baby phenotype revisited. Arch. Dis. Child. Fetal Neonatal Ed. 2000, 82, F163–F166. [Google Scholar] [CrossRef]
- Holleran, W.M.; Takagi, Y.; Menon, G.K.; Legler, G.; Feingold, K.R.; Elias, P.M. Processing of epidermal glucosylceramides is required for optimal mammalian cutaneous permeability barrier function. J. Clin. Investig. 1993, 91, 1656–1664. [Google Scholar] [CrossRef]
- Holleran, W.M.; Takagi, Y.; Menon, G.K.; Jackson, S.M.; Lee, J.M.; Feingold, K.R.; Elias, P.M. Permeability barrier requirements regulate epidermal beta-glucocerebrosidase. J. Lipid Res. 1994, 35, 905–912. [Google Scholar]
- Holleran, W.M.; Ginns, E.I.; Menon, G.K.; Grundmann, J.U.; Fartasch, M.; McKinney, C.E.; Elias, P.M.; Sidransky, E. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J. Clin. Investig. 1994, 93, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.; Holleran, W.M.; Potratz, A.; Vielhaber, G.; Elias, P.M.; Suzuki, K.; Sandhoff, K. Sphingolipid activator proteins are required for epidermal permeability barrier formation. J. Biol. Chem. 1999, 274, 11038–11045. [Google Scholar] [CrossRef] [PubMed]
- Freinkel, R.K.; Traczyk, T.N. Lipid composition and acid hydrolase content of lamellar granules of fetal rat epidermis. J. Investig. Dermatol. 1985, 85, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Grayson, S.; Johnson-Winegar, A.G.; Wintroub, B.U.; Isseroff, R.R.; Epstein, E.H., Jr.; Elias, P.M. Lamellar body-enriched fractions from neonatal mice: Preparative techniques and partial characterization. J. Investig. Dermatol. 1985, 85, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Madison, K.C.; Sando, G.N.; Howard, E.J.; True, C.A.; Gilbert, D.; Swartzendruber, D.C.; Wertz, P.W. Lamellar granule biogenesis: A role for ceramide glucosyltransferase, lysosomal enzyme transport, and the Golgi. J. Investig. Dermatol. Symp. Proc. 1998, 3, 80–86. [Google Scholar] [CrossRef]
- Takagi, Y.; Kriehuber, E.; Imokawa, G.; Elias, P.M.; Holleran, W.M. Beta-glucocerebrosidase activity in mammalian stratum corneum. J. Lipid Res. 1999, 40, 861–869. [Google Scholar] [PubMed]
- Schmuth, M.; Schoonjans, K.; Yu, Q.C.; Fluhr, J.W.; Crumrine, D.; Hachem, J.P.; Lau, P.; Auwerx, J.; Elias, P.M.; Feingold, K.R. Role of peroxisome proliferator-activated receptor alpha in epidermal development in utero. J. Investig. Dermatol. 2002, 119, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Hachem, J.P.; Crumrine, D.; Fluhr, J.; Brown, B.E.; Feingold, K.R.; Elias, P.M. pH directly regulates epidermal permeability barrier homeostasis, and stratum corneum integrity/cohesion. J. Investig. Dermatol. 2003, 121, 345–353. [Google Scholar] [CrossRef]
- Van Smeden, J.; Dijkhoff, I.M.; Helder, R.W.J.; Al-Khakany, H.; Boer, D.E.C.; Schreuder, A.; Kallemeijn, W.W.; Absalah, S.; Overkleeft, H.S.; Aerts, J.; et al. In situ visualization of glucocerebrosidase in human skin tissue: Zymography versus activity-based probe labeling. J. Lipid Res. 2017, 58, 2299–2309. [Google Scholar] [CrossRef]
- Schmuth, M.; Man, M.Q.; Weber, F.; Gao, W.; Feingold, K.R.; Fritsch, P.; Elias, P.M.; Holleran, W.M. Permeability barrier disorder in Niemann-Pick disease: Sphingomyelin-ceramide processing required for normal barrier homeostasis. J. Investig. Dermatol. 2000, 115, 459–466. [Google Scholar] [CrossRef]
- Jensen, J.M.; Schutze, S.; Forl, M.; Kronke, M.; Proksch, E. Roles for tumor necrosis factor receptor p55 and sphingomyelinase in repairing the cutaneous permeability barrier. J. Clin. Investig. 1999, 104, 1761–1770. [Google Scholar] [CrossRef]
- Oyoshi, M.K.; He, R.; Kumar, L.; Yoon, J.; Geha, R.S. Cellular and molecular mechanisms in atopic dermatitis. Adv. Immunol. 2009, 102, 135–226. [Google Scholar]
- Wollenberg, A.; Rawer, H.C.; Schauber, J. Innate immunity in atopic dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 272–281. [Google Scholar] [CrossRef]
- David Boothe, W.; Tarbox, J.A.; Tarbox, M.B. Atopic dermatitis: Pathophysiology. Adv. Exp. Med. Biol. 2017, 1027, 21–37. [Google Scholar]
- Leung, D.Y.; Bieber, T. Atopic dermatitis. Lancet 2003, 361, 151–160. [Google Scholar] [CrossRef]
- Seguchi, T.; Cui, C.Y.; Kusuda, S.; Takahashi, M.; Aisu, K.; Tezuka, T. Decreased expression of filaggrin in atopic skin. Arch. Dermatol. Res. 1996, 288, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.; Kroboth, K.; Schurch, N.J.; Sandilands, A.; Sherstnev, A.; O’Regan, G.M.; Watson, R.M.; McLean, W.H.; Barton, G.J.; Irvine, A.D.; et al. Filaggrin-stratified transcriptomic analysis of pediatric skin identifies mechanistic pathways in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 82–91. [Google Scholar] [CrossRef]
- Jungersted, J.M.; Scheer, H.; Mempel, M.; Baurecht, H.; Cifuentes, L.; Hogh, J.K.; Hellgren, L.I.; Jemec, G.B.; Agner, T.; Weidinger, S. Stratum corneum lipids, skin barrier function and filaggrin mutations in patients with atopic eczema. Allergy 2010, 65, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Farwanah, H.; Raith, K.; Neubert, R.H.; Wohlrab, J. Ceramide profiles of the uninvolved skin in atopic dermatitis and psoriasis are comparable to those of healthy skin. Arch. Dermatol. Res. 2005, 296, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, J.; Narita, H.; Kondo, N.; Hotta, M.; Takagi, Y.; Masukawa, Y.; Kitahara, T.; Takema, Y.; Koyano, S.; Yamazaki, S.; et al. Changes in the ceramide profile of atopic dermatitis patients. J. Investig. Dermatol. 2010, 130, 2511–2514. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Serizawa, S.; Ito, M.; Sato, Y. Stratum corneum lipid abnormalities in atopic dermatitis. Arch. Dermatol. Res. 1991, 283, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Bleck, O.; Abeck, D.; Ring, J.; Hoppe, U.; Vietzke, J.P.; Wolber, R.; Brandt, O.; Schreiner, V. Two ceramide subfractions detectable in Cer (AS) position by HPTLC in skin surface lipids of non-lesional skin of atopic eczema. J. Investig. Dermatol. 1999, 113, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Wertz, P.; Giannetti, A.; Seidenari, S. Ceramide and cholesterol composition of the skin of patients with atopic dermatitis. Acta Derm. Venereol. 1998, 78, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Janssens, M.; Van Smeden, J.; Gooris, G.S.; Bras, W.; Portale, G.; Caspers, P.J.; Vreeken, R.J.; Hankemeier, T.; Kezic, S.; Wolterbeek, R.; et al. Increase in short-chain ceramides correlates with an altered lipid organization and decreased barrier function in atopic eczema patients. J. Lipid Res. 2012, 53, 2755–2766. [Google Scholar] [CrossRef]
- Van Smeden, J.; Janssens, M.; Kaye, E.C.; Caspers, P.J.; Lavrijsen, A.P.; Vreeken, R.J.; Bouwstra, J.A. The importance of free fatty acid chain length for the skin barrier function in atopic eczema patients. Exp. Dermatol. 2014, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Macheleidt, O.; Kaiser, H.W.; Sandhoff, K. Deficiency of epidermal protein-bound omega-hydroxyceramides in atopic dermatitis. J. Investig. Dermatol. 2002, 119, 166–173. [Google Scholar] [CrossRef]
- Ohno, Y.; Suto, S.; Yamanaka, M.; Mizutani, Y.; Mitsutake, S.; Igarashi, Y.; Sassa, T.; Kihara, A. ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18439–18444. [Google Scholar] [CrossRef]
- Danso, M.; Boiten, W.; Van Drongelen, V.; Gmelig Meijling, K.; Gooris, G.; El Ghalbzouri, A.; Absalah, S.; Vreeken, R.; Kezic, S.; Van Smeden, J.; et al. Altered expression of epidermal lipid bio-synthesis enzymes in atopic dermatitis skin is accompanied by changes in stratum corneum lipid composition. J. Dermatol. Sci. 2017, 88, 57–66. [Google Scholar] [CrossRef]
- Imokawa, G. A possible mechanism underlying the ceramide deficiency in atopic dermatitis: Expression of a deacylase enzyme that cleaves the N-acyl linkage of sphingomyelin and glucosylceramide. J. Dermatol. Sci. 2009, 55, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Arikawa, J.; Okamoto, R.; Kawashima, M.; Takagi, Y.; Ohguchi, K.; Imokawa, G. Abnormal expression of the novel epidermal enzyme, glucosylceramide deacylase, and the accumulation of its enzymatic reaction product, glucosylsphingosine, in the skin of patients with atopic dermatitis. Lab. Investig. 2003, 83, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Hara, J.; Okamoto, R.; Kawashima, M.; Imokawa, G. The skin of atopic dermatitis patients contains a novel enzyme, glucosylceramide sphingomyelin deacylase, which cleaves the N-acyl linkage of sphingomyelin and glucosylceramide. Biochem. J. 2000, 350, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, K.M.; Noh, M.; Yoo, H.J.; Lee, C.H. Glucosylsphingosine induces itch-scratch responses in mice. Biomol. Ther. 2010, 18, 316–320. [Google Scholar] [CrossRef][Green Version]
- Afzal, R.; Shim, W.S. Glucosylsphingosine activates serotonin receptor 2a and 2b: Implication of a novel itch signaling pathway. Biomol. Ther. 2017, 25, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Higaki, Y.; Takagi, Y.; Higuchi, K.; Yada, Y.; Kawashima, M.; Imokawa, G. Analysis of beta-glucocerebrosidase and ceramidase activities in atopic and aged dry skin. Acta Derm. Venereol. 1994, 74, 337–340. [Google Scholar] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boer, D.E.C.; van Smeden, J.; Bouwstra, J.A.; Aerts, J.M.F.G. Glucocerebrosidase: Functions in and Beyond the Lysosome. J. Clin. Med. 2020, 9, 736. https://doi.org/10.3390/jcm9030736
Boer DEC, van Smeden J, Bouwstra JA, Aerts JMFG. Glucocerebrosidase: Functions in and Beyond the Lysosome. Journal of Clinical Medicine. 2020; 9(3):736. https://doi.org/10.3390/jcm9030736
Chicago/Turabian StyleBoer, Daphne E.C., Jeroen van Smeden, Joke A. Bouwstra, and Johannes M.F.G Aerts. 2020. "Glucocerebrosidase: Functions in and Beyond the Lysosome" Journal of Clinical Medicine 9, no. 3: 736. https://doi.org/10.3390/jcm9030736
APA StyleBoer, D. E. C., van Smeden, J., Bouwstra, J. A., & Aerts, J. M. F. G. (2020). Glucocerebrosidase: Functions in and Beyond the Lysosome. Journal of Clinical Medicine, 9(3), 736. https://doi.org/10.3390/jcm9030736