KCP10043F Represses the Proliferation of Human Non-Small Cell Lung Cancer Cells by Caspase-Mediated Apoptosis via STAT3 Inactivation


 ,
,  , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Cell Culture
2.3. Cytotoxicity Assay
2.4. Annexin V-FITC (Fluorescein Isothiocyanate) and Propidium Iodide (PI) Double Staining Assay
2.5. DAPI (4′,6-Diamidino-2-Phenylindole) Staining Assay
2.6. Terminal Deoxynucleotidyl Transferase dUTP Nick end Labeling (TUNEL) Assay
2.7. Western Blot Analysis
2.8. Mitochondrial and Cytosolic Fractionation
2.9. Measurement of Mitochondrial Membrane Potential (ΔΨm)
2.10. Transfection for Signal Transducer and Activator of Transcription (STAT3) Overexpression
2.11. Cytokine Production
2.12. Immunocytochemistry
2.13. Animals
2.14. In Vivo Tumor Xenograft Studies
2.15. Molecular Docking Analysis
2.16. Statistical Analysis
3. Results
3.1. KCP10043F Inhibits the Proliferation of A549 and NCI-H358 Human Non-Small Cell Lung Cancer (NSCLC) Cells by Inducing Apoptosis
3.2. KCP10043F Induces Caspase-Dependent Apoptosis
3.3. KCP10043F Induces the Release of Cytochrome C into the Cytosol and Loss of ΔΨm
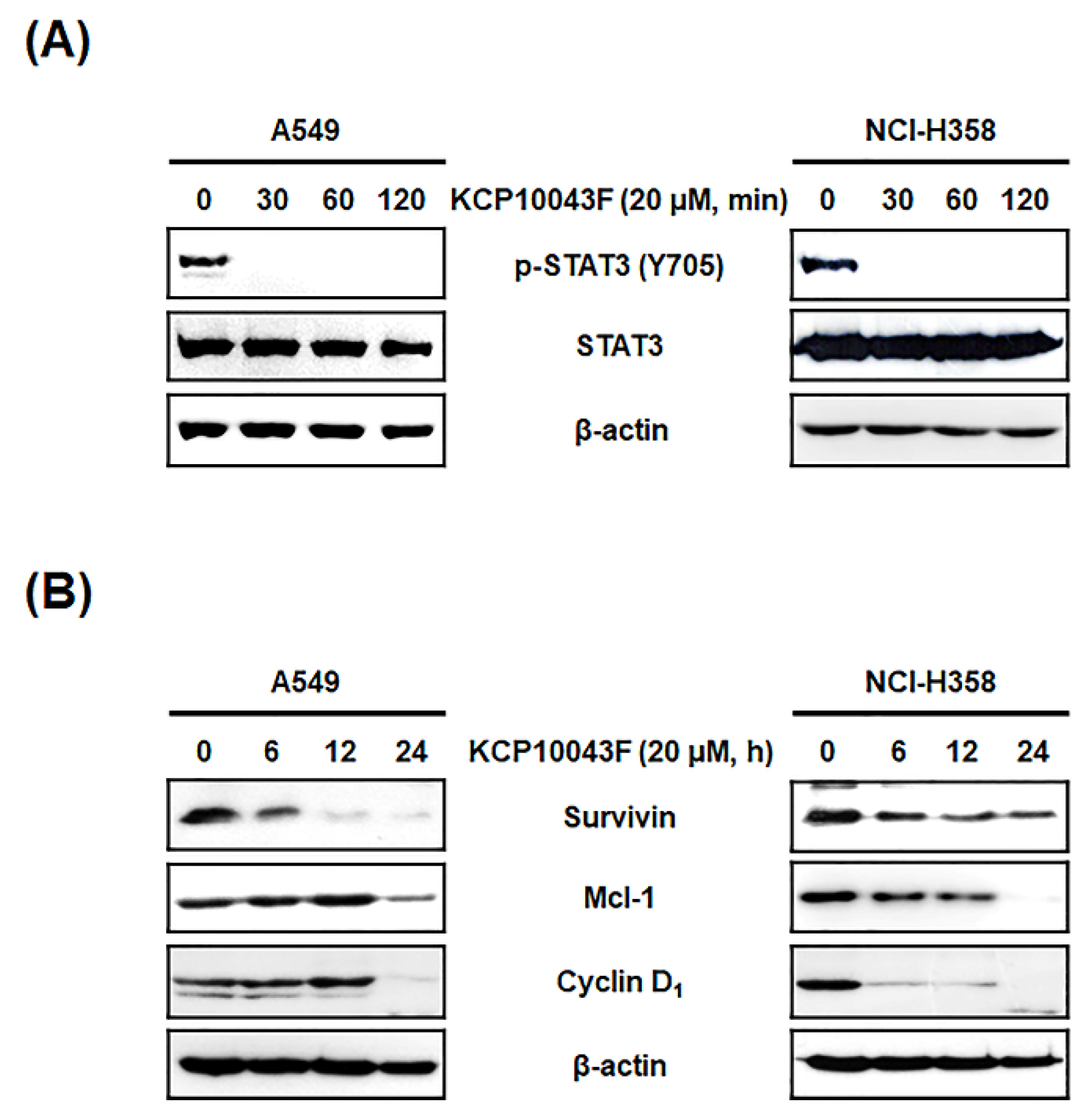
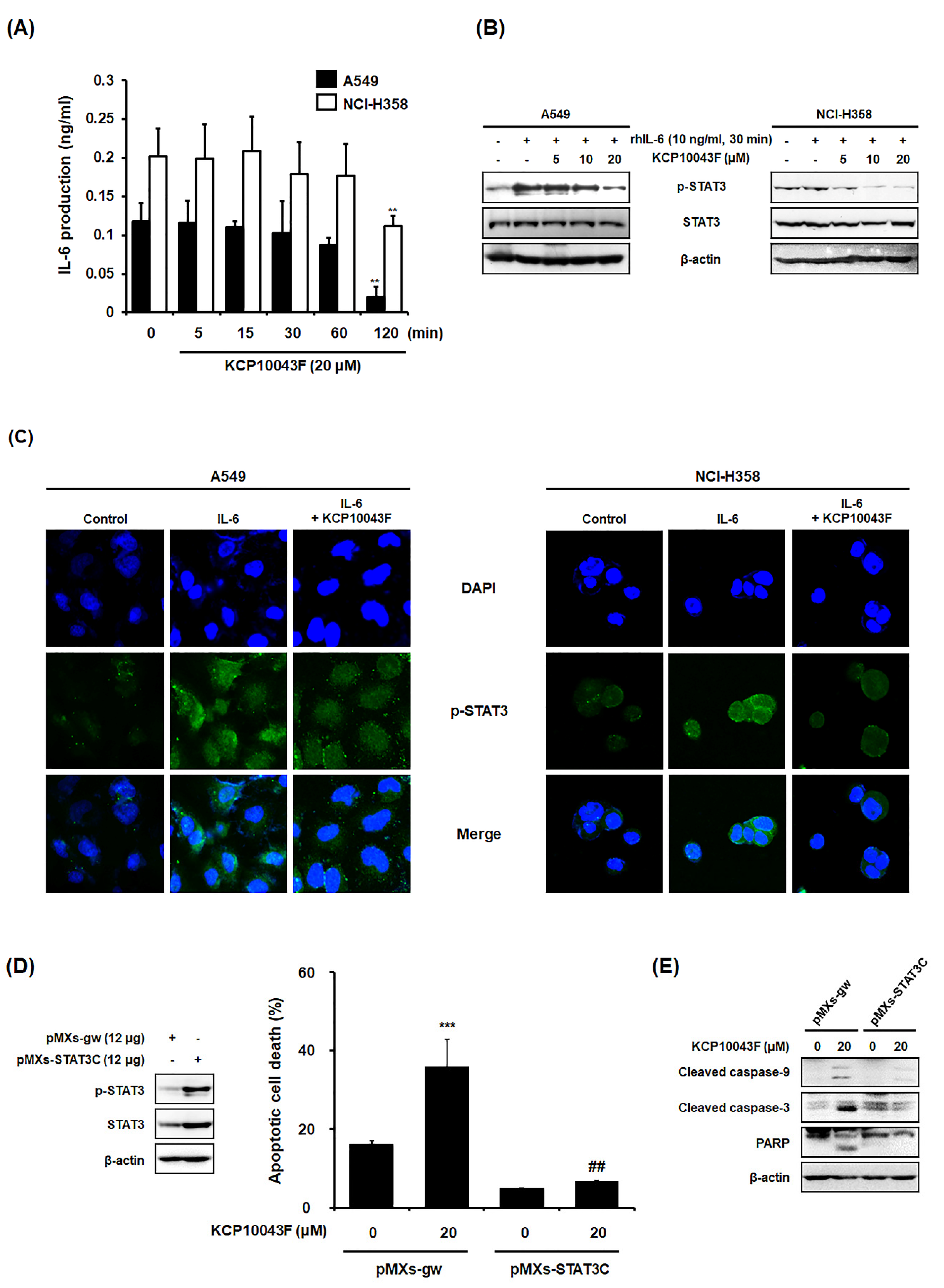
3.4. KCP10043F Affects the STAT3 Signaling Pathway in A549 and NCI-H358 Cells
3.5. KCP10043F Docks into the SH2 Domain of STAT3
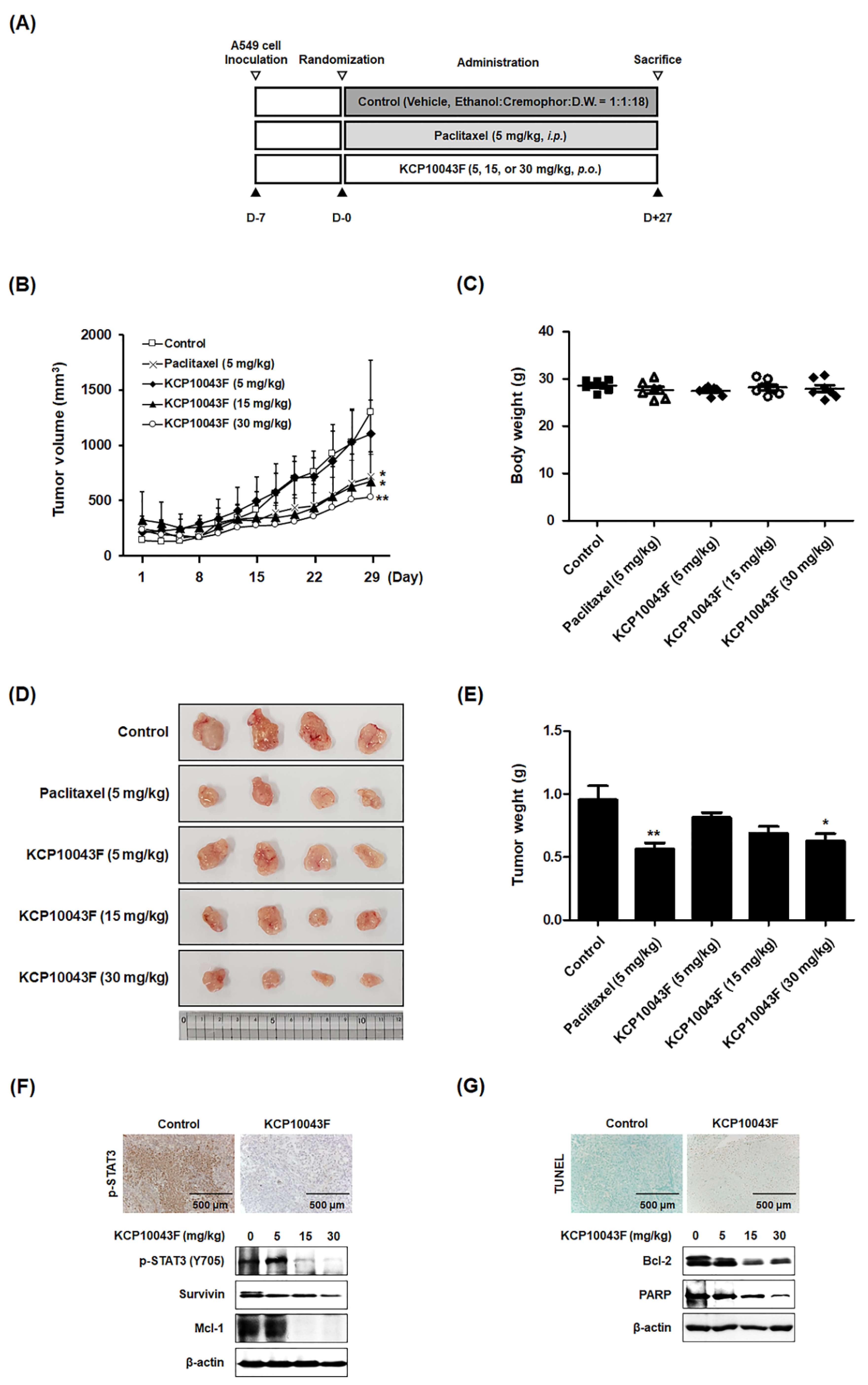
3.6. KCP10043F Suppresses Tumor Growth through Apoptotic Cell Death in A549 Xenograft Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Wangari-Talbot, J.; Hopper-Borge, E. Drug Resistance Mechanisms in Non-Small Cell Lung Carcinoma. J. Can. Res. Updates 2013, 2, 265–282. [Google Scholar] [PubMed]
- Eroglu, M.; Derry, W.B. Your neighbours matter—Non-autonomous control of apoptosis in development and disease. Cell Death Differ. 2016, 23, 1110–1118. [Google Scholar] [CrossRef]
- Baig, S.; Seevasant, I.; Mohamad, J.; Mukheem, A.; Huri, H.Z.; Kamarul, T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis. 2016, 7, e2058. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-E.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Lee, H.; Jeong, A.J.; Ye, S.-K. Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep. 2019, 52, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, Y.; Li, Y.; Lv, T.; Liu, J.; Wang, X. Prognostic significance of STAT3 expression and its correlation with chemoresistance of non-small cell lung cancer cells. Acta. Histochem. 2012, 114, 151–158. [Google Scholar] [CrossRef]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.-C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta. 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Meng, X.; Shi, H.; Li, W.; Ming, Z.; Zhong, Y.; Deng, W.; Zhang, Q.; Fan, N.; Niu, Z.; et al. The role of JAK/STAT3 signaling pathway on apoptosis of lung adenocarcinoma cell line PC-9 induced by icotinib. Am. J. Transl. Res. 2016, 8, 1730–1737. [Google Scholar] [PubMed]
- Kopecky, B.J.; Liang, R.; Bao, J. T-type calcium channel blockers as neuroprotective agents. Pflugers Arch. 2014, 466, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, F.M.; Tscherter, A.; Errington, A.C.; Renger, J.J.; Shin, H.-S.; Uebele, V.N.; Crunelli, V.; Lambert, R.C.; Leresche, N. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J. Neurosci. 2010, 30, 99–109. [Google Scholar] [CrossRef]
- Toyota, M.; Ho, C.; Ohe-Toyota, M.; Baylin, S.B.; Issa, J.-P. Inactivation of CACNA1G, a T-type calcium channel gene, by aberrant methylation of its 5’ CpG island in human tumors. Cancer Res. 1999, 59, 4535–4541. [Google Scholar]
- Panner, A.; Cribbs, L.L.; Zainelli, G.M.; Origitano, T.C.; Singh, S.; Wurster, R.D. Variation of T-type calcium channel protein expression affects cell division of cultured tumor cells. Cell Calcium 2005, 37, 105–119. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Gray, L.S.; Dziegielewski, J. T-type calcium channels blockers as new tools in cancer therapies. Pflugers Arch. 2014, 466, 801–810. [Google Scholar] [CrossRef]
- Gray, L.S.; Schiff, D.; Macdonald, T.L. A model for the regulation of T-type Ca2+ channels in proliferation: Roles in stem cells and cancer. Expert Rev. Anticancer Ther. 2013, 13, 589–595. [Google Scholar] [CrossRef]
- Das, A.; Pushparaj, C.; Herreros, J.; Nager, M.; Vilella, R.; Portero, M.; Pamplona, R.; Matias-Guiu, X.; Marti, R.M.; Canti, C. T-type calcium channel blockers inhibit autophagy and promote apoptosis of malignant melanoma cells. Pigment Cell Melanoma Res. 2013, 26, 874–885. [Google Scholar] [CrossRef]
- Kang, H.B.; Rim, H.-K.; Park, J.Y.; Choi, H.W.; Choi, D.L.; Seo, J.-H.; Chung, K.-S.; Huh, G.; Kim, J.; Choo, D.J.; et al. In vivo evaluation of oral anti-tumoral effect of 3,4-dihydroquinazoline derivative on solid tumor. Bioorg. Med. Chem. Lett. 2012, 22, 1198–1201. [Google Scholar] [CrossRef]
- Rim, H.-K.; Cho, S.; Shin, D.-H.; Chung, K.-S.; Cho, Y.-W.; Choi, J.-H.; Lee, J.Y.; Lee, K.-T. T-type Ca2+ channel blocker, KYS05090 induces autophagy and apoptosis in A549 cells through inhibiting glucose uptake. Molecules 2014, 19, 9864–9875. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.S.; Sohn, J.M.; Leem, D.G.; Park, B.; Nam, J.H.; Shin, D.H.; Shin, J.S.; Kim, H.J.; Lee, K.-T.; Lee, J.Y. In vitro synergistic anticancer activity of the combination of T-type calcium channel blocker and chemotherapeutic agent in A549 cells. Bioorg. Med. Chem. Lett. 2016, 26, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Chung, K.-S.; Shin, J.-S.; Lee, J.-H.; Gil, H.-S.; Lee, H.-H.; Choi, E.; Choi, J.-H.; Hassan, A.H.E.; Lee, Y.S.; et al. The Anti-Proliferative Activity of the Hybrid TMS-TMF-4f Compound Against Human Cervical Cancer Involves Apoptosis Mediated by STAT3 Inactivation. Cancers 2019, 11, 1927. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.A.; Prewitt, J.M.; Mendelsohn, M.L. Analysis of tumor growth curves. J. Natl. Cancer Inst. 1968, 40, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Park, H.R.; Yoon, Y.M.; Kim, H.I.; Yoo, S.Y.; Lee, K.W.; Lee, Y.S. Antiproliferative 3-deoxysphingomyelin analogs: Design, synthesis, biological evaluation and molecular docking of pyrrolidine-based 3-deoxysphingomyelin analogs as anticancer agents. Bioorg. Chem. 2019, 84, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.K.; Hassan, A.H.E.; Jeong, H.; Kwon, Y.; Choi, J.G.; Oh, M.S.; Park, K.D.; Kim, Y.K.; Roh, E.J. First-in-class DAPK1/CSF1R dual inhibitors: Discovery of 3,5-dimethoxy-N-(4-(4-methoxyphenoxy)-2-((6-morpholinopyridin-3-yl)amino)pyrimidi n-5-yl)benzamide as a potential anti-tauopathies agent. Eur. J. Med. Chem. 2019, 162, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.-M. Targeting apoptosis pathways in cancer therapy. Curr. Cancer Drug Targets 2004, 4, 569–576. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 2014, 6, 708–722. [Google Scholar] [CrossRef]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Ray, R.M.; Johnson, L.R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 2005, 392, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Mao, X.; Mertens, C.; Krishnaraj, R.; Qin, J.; Mandal, P.K.; Romanowski, M.J.; McMurray, J.S.; Chen, X. Crystal structure of unphosphorylated STAT3 core fragment. Biochem. Biophys. Res. Commun. 2008, 374, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Beebe, J.D.; Liu, J.-Y.; Zhang, J.T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther. 2018, 191, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Panner, A.; Wurster, R.D. T-type calcium channels and tumor proliferation. Cell Calcium 2006, 40, 253–259. [Google Scholar] [CrossRef]
- Taylor, J.T.; Zeng, X.-B.; Pottle, J.E.; Lee, K.; Wang, A.R.; Yi, S.G.; Scruggs, J.A.S.; Sikka, S.S.; Li, M. Calcium signaling and T-type calcium channels in cancer cell cycling. World J. Gastroenterol. 2008, 14, 4984–4991. [Google Scholar] [CrossRef]
- Choi, D.L.; Jang, S.J.; Cho, S.; Choi, H.-E.; Rim, H.-K.; Lee, K.-T.; Lee, J.Y. Inhibition of cellular proliferation and induction of apoptosis in human lung adenocarcinoma A549 cells by T-type calcium channel antagonist. Bioorg. Med. Chem. Lett. 2014, 24, 1565–1570. [Google Scholar] [CrossRef]
- Jang, S.J.; Choi, H.W.; Choi, D.L.; Cho, S.; Rim, H.-K.; Choi, H.-E.; Kim, K.-S.; Huang, M.; Rhim, H.; Lee, K.-T.; et al. In vitro cytotoxicity on human ovarian cancer cells by T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2013, 23, 6656–6662. [Google Scholar] [CrossRef]
- Rim, H.-K.; Lee, H.-W.; Choi, I.S.; Park, J.Y.; Choi, H.W.; Choi, J.-H.; Cho, Y.-W.; Lee, J.Y.; Lee, K.-T. T-type Ca2+ channel blocker, KYS05047 induces G1 phase cell cycle arrest by decreasing intracellular Ca2+ levels in human lung adenocarcinoma A549 cells. Bioorg. Med. Chem. Lett. 2012, 22, 7123–7126. [Google Scholar] [CrossRef]
- Jung, S.Y.; Lee, S.H.; Kang, H.B.; Park, H.A.; Chang, S.K.; Kim, J.; Choo, D.J.; Oh, C.R.; Kim, Y.D.; Seo, J.H.; et al. Antitumor activity of 3,4-dihydroquinazoline dihydrochloride in A549 xenograft nude mice. Bioorg. Med. Chem. Lett. 2010, 20, 6633–6636. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.-C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Ghibelli, L.; Diederich, M. Multistep and multitask Bax activation. Mitochondrion 2010, 10, 604–613. [Google Scholar] [CrossRef]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: Direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral. Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef]
- Lin, L.; Liu, A.; Peng, Z.; Lin, H.-J.; Li, P.-K.; Li, C.; Lin, J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011, 71, 7226–7237. [Google Scholar] [CrossRef] [PubMed]
- Pancotti, F.; Roncuzzi, L.; Maggiolini, M.; Gasperi-Campani, A. Caveolin-1 silencing arrests the proliferation of metastatic lung cancer cells through the inhibition of STAT3 signaling. Cell Signal 2012, 24, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.-R.; Yang, Z.-M. Regulation and function of signal transducer and activator of transcription 3. World J. Biol. Chem. 2014, 5, 231–239. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-H.; Lee, H.-H.; Ryu, K.D.; Kim, M.; Ko, D.; Chung, K.-S.; Hassan, A.H.E.; Lee, S.H.; Lee, J.Y.; Lee, K.-T. KCP10043F Represses the Proliferation of Human Non-Small Cell Lung Cancer Cells by Caspase-Mediated Apoptosis via STAT3 Inactivation. J. Clin. Med. 2020, 9, 704. https://doi.org/10.3390/jcm9030704
Lee J-H, Lee H-H, Ryu KD, Kim M, Ko D, Chung K-S, Hassan AHE, Lee SH, Lee JY, Lee K-T. KCP10043F Represses the Proliferation of Human Non-Small Cell Lung Cancer Cells by Caspase-Mediated Apoptosis via STAT3 Inactivation. Journal of Clinical Medicine. 2020; 9(3):704. https://doi.org/10.3390/jcm9030704
Chicago/Turabian StyleLee, Jeong-Hun, Hwi-Ho Lee, Ki Deok Ryu, Misong Kim, Dohyeong Ko, Kyung-Sook Chung, Ahmed H.E. Hassan, Seung Hyeun Lee, Jae Yeol Lee, and Kyung-Tae Lee. 2020. "KCP10043F Represses the Proliferation of Human Non-Small Cell Lung Cancer Cells by Caspase-Mediated Apoptosis via STAT3 Inactivation" Journal of Clinical Medicine 9, no. 3: 704. https://doi.org/10.3390/jcm9030704
APA StyleLee, J.-H., Lee, H.-H., Ryu, K. D., Kim, M., Ko, D., Chung, K.-S., Hassan, A. H. E., Lee, S. H., Lee, J. Y., & Lee, K.-T. (2020). KCP10043F Represses the Proliferation of Human Non-Small Cell Lung Cancer Cells by Caspase-Mediated Apoptosis via STAT3 Inactivation. Journal of Clinical Medicine, 9(3), 704. https://doi.org/10.3390/jcm9030704