Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents

 and
and 
Abstract
1. Introduction
1.1. Immune Checkpoint Inhibitors
1.2. Anti-Angiogenic Agents
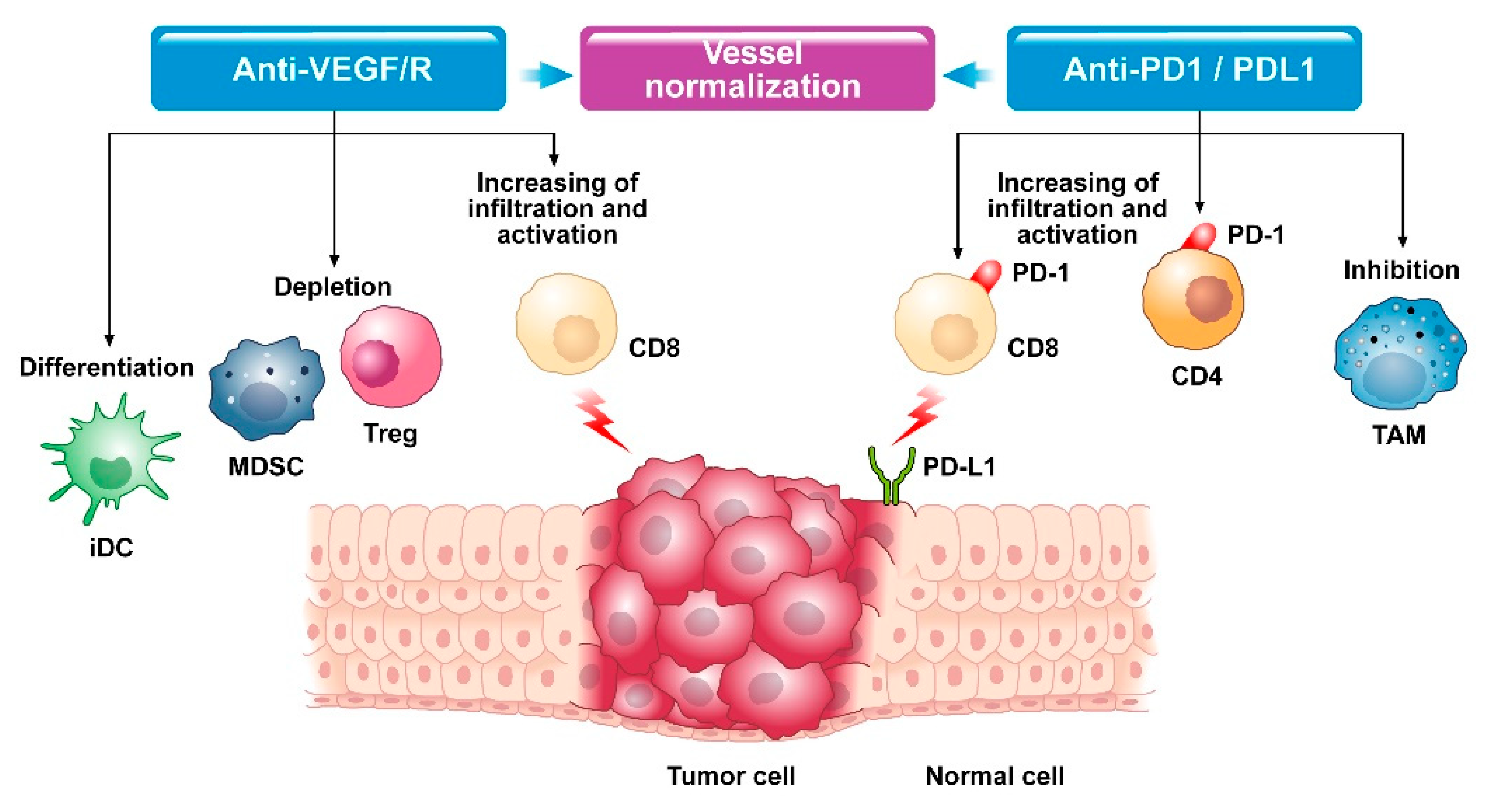
2. Biological Rationale of Combined Therapy
2.1. Crosstalk between Angiogenesis and Immune System
2.2. Modifications in Tumor Microenvironment after Exposure to Combined Therapy
2.3. Combined Therapy as a Way to Overcome Cell Resistances
2.4. A Predictive Value of PD-L1 Expression
3. Combined Therapy in Clinical Practice: Where We Are Now
3.1. Renal Cell Carcinoma (RCC)
3.2. Non Small Cell Lung Cancer (NSCLC)
3.3. Colorectal Cancer (CRC)
3.4. Gastrointestinal Malignancies
3.5. Melanoma
3.6. Breast Cancer
3.7. Urothelial Carcinoma
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 542. [Google Scholar] [CrossRef]
- Kim, H.J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Callahan, M.K.; Postow, M.A.; Wolchok, J.D. On being less tolerant: Enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Sci. Transl. Med. 2015, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Ribas, A. The evolution of checkpoint blockade as a cancer therapy: What’s here, what’s next? Curr. Opin. Immunol. 2015, 33, 23–35. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. C tla -4-M ediated I nhibition in R Egulation of T C ell R esponses: Mechanisms and manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef]
- Teft, W.A.; Kirchhof, M.G.; Madrenas, J. A molecular perspective of ctla-4 Function. Annu. Rev. Immunol. 2006, 24, 65–97. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef]
- Funt, S.A.; Page, D.B.; Wolchok, J.D.; Postow, M.A. CTLA-4 antibodies: New directions, new combinations. Oncology 2014, 11 (Suppl. 3), 28. [Google Scholar]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory t cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 129, 3384–3391. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Naboush, A.; Roman, C.A.J.; Shapira, I. Immune checkpoint inhibitors in malignancies with mismatch repair deficiency: A review of the state of the current knowledge. J. Investig. Med. 2017, 65, 754–758. [Google Scholar] [CrossRef]
- Reiss, K.A.; Forde, P.M.; Brahmer, J.R. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: A promising new anticancer strategy. Immunotherapy 2014, 6, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Eder, J.P. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology 2014, 28, 15–28. [Google Scholar] [PubMed]
- Henick, B.S.; Herbst, R.S.; Goldberg, S.B. The PD-1 pathway as a therapeutic target to overcome immune escape mechanisms in cancer. Expert Opin. Ther. Targets 2014, 18, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Gunturi, A.; McDermott, D.F. Nivolumab for the treatment of cancer. Expert Opin. Investig. Drugs 2015, 24, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Khoja, L.; Butler, M.O.; Kang, S.P.; Ebbinghaus, S.; Joshua, A.M. Pembrolizumab. J. Immunother. Cancer 2015, 3, 36. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Keck, P.J.; Hauser, S.D.; Krivi, G.; Sanzo, K.; Warren, T.; Feder, J.; Connolly, D.T. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246, 1309–1312. [Google Scholar] [CrossRef]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti–angiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor–induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Motzer, R.J.; Michaelson, M.D.; Redman, B.G.; Hudes, G.R.; Wilding, G.; Figlin, R.A.; Ginsberg, M.S.; Kim, S.T.; Baum, C.M.; DePrimo, S.E.; et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet–derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2006, 108, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to anti–angiogenic therapy in cancer–alterations to anti–VEGF pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef]
- Zarrin, B.; Zarifi, F.; Vaseghi, G.; Javanmard, S.H. Acquired tumor resistance to antiangiogenic therapy: Mechanisms at a glance. J. Res. Med. Sci. 2017, 22, 117. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 183–1813. [Google Scholar] [CrossRef]
- Formisano, L.; Jansen, V.M.; Marciano, R.; Bianco, R. From Biology to Therapy: Improvements of Therapeutic Options in Lung Cancer. Anticancer. Agents Med. Chem. 2018, 18, 1235–1240. [Google Scholar] [CrossRef]
- Kuusk, T.; Albiges, L.; Escudier, B.; Grivas, N.; Haanen, J.; Powles, T.; Bex, A. Antiangiogenic therapy combined with immune checkpoint blockade in renal cancer. Angiogenesis 2017, 18, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Tartour, É.; Sandoval, F.; Bonnefoy, J.Y.; Fridman, W.H. Cancer immunotherapy: Recent breakthroughs and perspectives. Int. J. Med. Sci. 2011, 27, 833–841. [Google Scholar]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti–angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1069–1103. [Google Scholar] [CrossRef]
- Alfaro, C.; Suarez, N.; Gonzalez, A.; Solano, S.; Erro, L.; Dubrot, J.; Palazon, A.; Hervas-Stubbs, S.; Gurpide, A.; Lopez-Picazo, J.M.; et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br. J. Cancer 2009, 100, 1111–1119. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8++ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The contribution of vascular endothelial growth factor to the induction of regulatory T- cells in malignant effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Mechanisms of Leukocyte Transendothelial Migration. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 323–344. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Damen, C.A.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res. 1996, 56, 1111–1117. [Google Scholar]
- Eppihimer, M.J.; Gunn, J.; Freeman, G.J.; Greenfield, E.A.; Chernova, T.; Erickson, J.; Leonard, J.P. Expression and regulation of the PD-L1 immunoinhibitory molecule on micro vascular endothelial cells. Microcirculation 2002, 9, 133–145. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Noman, M.Z.; Messai, Y.; Muret, J.; Hasmim, M.; Chouaib, S. Crosstalk between CTC, Immune System and Hypoxic Tumor Microenvironment. Cancer Microenviron. 2014, 7, 153–160. [Google Scholar] [CrossRef]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia enhances immunosuppression by inhibiting CD4+ Effector T cell function and promoting treg activity. Cell. Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Tosetti, F.; Benelli, R.; Noonan, D.M. Tumor inflammatory angiogenesis and its chemoprevention. Cancer Res. 2005, 65, 10637–10641. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Coussens, L.M. The inflammatory tumor microenvironment and its impact on cancer development. Contrib. Microbiol. 2006, 13, 118–137. [Google Scholar]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-J.; Chang, W.-A.; Huang, M.-S.; Kuo, P.-L. Tumor Microenvironment: A New Treatment Target for Cancer. ISRN Biochem. 2014, 15, 8–17. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Sunderkotter, C.; Steinbrink, K.; Goebeler, M.; Bhardwaj, R.; Sorg, C. Macrophages and angiogenesis. J. Leukoc. Biol. 1994, 55, 410–422. [Google Scholar] [CrossRef]
- Kimura, Y.N.; Watari, K.; Fotovati, A.; Hosoi, F.; Yasumoto, K.; Izumi, H.; Kohno, K.; Umezawa, K.; Iguchi, H.; Shirouzu, K.; et al. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci. 2007, 98, 2009–2018. [Google Scholar] [CrossRef]
- Lewis, C.E.; Leek, R.; Harris, A.; McGee O’D, J. Cytokine regulation of angiogenesis in breast cancer: The role of tumor-associated macrophages. In Proceedings of the 2nd International Cytokine Conference, Banff, AB, Canada, 1–5 October 1994. [Google Scholar]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9—Expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- Neufeld, G.; Kessler, O. Pro-angiogenic cytokines and their role in tumor angiogenesis. Cancer Metastasis Rev. 2006, 25, 373–385. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Badoual, C.; Sandoval, F.; Pere, H.; Hans, S.; Gey, A.; Merillon, N.; Van Ryswick, C.; Quintin-Colonna, F.; Bruneval, P.; Brasnu, D.; et al. Better understanding tumor-host interaction in head and neck cancer to improve the design and development of immunotherapeutic strategies. Head Neck 2010, 32, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 118, e5. [Google Scholar] [CrossRef] [PubMed]
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol. 2017, 343, 103713. [Google Scholar] [CrossRef]
- Helfen, A.; Roth, J.; Ng, T.; Eisenblaetter, M. In vivo imaging of pro- and antitumoral cellular components of the tumor microenvironment. J. Nucl. Med. 2018, 59, 183–188. [Google Scholar] [CrossRef]
- Sica, A.; Massarotti, M. Myeloid suppressor cells in cancer and autoimmunity. J. Autoimmun. 2017, 87, 117–125. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Manning, E.A.; Ullman, J.G.M.; Leatherman, J.M.; Asquith, J.M.; Hansen, T.R.; Armstrong, T.D.; Hicklin, D.J.; Jaffee, E.M.; Emens, L.A. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin. Cancer Res. 2007, 13, 3951–3959. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef]
- Tian, L.; Goldstein, A.; Wang, H.; Lo, H.C.; Kim, I.S.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, A.E.M.; Oude Egbrink, M.G.A.; Castermans, K.; Van Der Schaft, D.W.J.; Thijssen, V.L.J.L.; Dings, R.P.M.; Kwee, L.; Mayo, K.H.; Wagstaff, J.; Bouma-ter Steege, J.C.A.; et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006, 20, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Taylor, J.L.; Alber, S.; Watkins, S.C.; Garcia, J.A.; Rini, B.I.; Ko, J.S.; Cohen, P.A.; Finke, J.H.; Storkus, W.J. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int. J. Cancer 2011, 129, 2158–2170. [Google Scholar] [CrossRef] [PubMed]
- Ozao-Choy, J.; Ge, M.; Kao, J.; Wang, G.X.; Meseck, M.; Sung, M.; Schwartz, M.; Divino, C.M.; Pan, P.Y.; Chen, S.H. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009, 69, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Santoni, M.; Ciccarese, C.; Santini, D.; Alfieri, S.; Martignoni, G.; Brunelli, M.; Piva, F.; Berardi, R.; Montironi, R.; et al. PD-1 blockade therapy in renal cell carcinoma: Current studies and future promises. Cancer Treat. Rev. 2015, 41, 114–121. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Shi, S.; Chen, L. Bevacizumab improves the antitumor efficacy of adoptive cytokine-induced killer cells therapy in non-small cell lung cancer models. Med. Oncol. 2014, 31, 777. [Google Scholar] [CrossRef]
- Pircher, A.; Wolf, D.; Heidenreich, A.; Hilbe, W.; Pichler, R.; Heidegger, I. Synergies of targeting tumor angiogenesis and immune checkpoints in non-small cell lung cancer and renal cell cancer: From basic concepts to clinical reality. Int. J. Mol. Sci. 2017, 18, 2291. [Google Scholar] [CrossRef]
- Zhao, S.; Jiang, T.; Li, X.; Zhou, C. OA11.07 Combining Anti-Angiogenesis and Immunotherapy Enhances Antitumor Effect by Promoting Immune Response in Lung Cancer. J. Thorac. Oncol. 2017, 12, S288. [Google Scholar] [CrossRef][Green Version]
- Meder, L.; Schuldt, P.; Thelen, M.; Schmitt, A.; Dietlein, F.; Klein, S.; Borchmann, S.; Wennhold, K.; Vlasic, I.; Oberbeck, S.; et al. Combined VEGF and PD-L1 blockade displays synergistic treatment effects in an autochthonous mouse model of small cell lung cancer. Cancer Res. 2018, 78, 4270–4281. [Google Scholar] [CrossRef] [PubMed]
- Finke, J.H.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Lambrechts, D. It’s T Time for Normal Blood Vessels. Dev. Cell 2017, 41, 125–126. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; De Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro. Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef]
- Passardi, A.; Scarpi, E.; Cavanna, L.; Agata, M.D.; Tassinari, D.; Leo, S.; Bernardini, I.; Gelsomino, F.; Tamberi, S.; Brandes, A.A.; et al. Inflammatory indexes as predictors of prognosis and bevacizumab efficacy in patients with metastatic colorectal cancer. Oncotarget 2016, 7, 33210–33219. [Google Scholar] [CrossRef]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; De Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage migration inhibitory factor downregulation: A novel mechanism of resistance to anti-angiogenic therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef]
- Liu, X.D.; Hoang, A.; Zhou, L.; Kalra, S.; Yetil, A.; Sun, M.; Ding, Z.; Zhang, X.; Bai, S.; German, P.; et al. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic Renal cell carcinoma. Cancer Immunol. Res. 2015, 3, 1017–1029. [Google Scholar] [CrossRef]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Rmili, C.W.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, 385. [Google Scholar] [CrossRef]
- Farolfi, A.; Schepisi, G.; Conteduca, V.; Burgio, S.L.; Lolli, C.; De Giorgi, U. Pharmacokinetics, pharmacodynamics and clinical efficacy of nivolumab in the treatment of metastatic renal cell carcinoma. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1089–1096. [Google Scholar] [CrossRef]
- Escudier, B.; Motzer, R.J.; Sharma, P.; Wagstaff, J.; Plimack, E.R.; Hammers, H.J.; Donskov, F.; Gurney, H.; Sosman, J.A.; Zalewski, P.G.; et al. Treatment Beyond Progression in Patients with Advanced Renal Cell Carcinoma Treated with Nivolumab in CheckMate 025. Eur. Urol. 2017, 72, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Eruslanov, E.; Kübler, H.; Tseng, T.; Sakai, Y.; Su, Z.; Kaliberov, S.; Heiser, A.; Rosser, C.; Dahm, P.; et al. Oxidative Stress Regulates Expression of VEGFR1 in Myeloid Cells: Link to Tumor-Induced Immune Suppression in Renal Cell Carcinoma. J. Immunol. 2008, 181, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, O.; Pere, H.; Ravel, P.; Haicheur, N.; Badoual, C.; Merillon, N.; Medioni, J.; Peyrard, S.; Roncelin, S.; Verkarre, V.; et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J. Immunother. 2010, 33, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wu, D.; Li, L.; Chai, Y.; Huang, J. PD-L1 and survival in solid tumors: A meta-analysis. PLoS ONE 2015, 10, e0131403. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, F.; Liu, L. Prognostic significance of PD-L1 in solid tumor: An updated meta-analysis. Medical 2017, 96, e6369. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csöszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1279–1290. [Google Scholar] [CrossRef]
- Li, D.; Chen, R.; Wang, Y.W.; Fornace, A.J.; Li, H.H. Prior irradiation results in elevated programmed cell death protein 1 (PD-1) in T cells. Int. J. Radiat. Biol. 2018, 94, 488–494. [Google Scholar] [CrossRef]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in Melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef]
- Munari, E.; Zamboni, G.; Lunardi, G.; Marchionni, L.; Marconi, M.; Sommaggio, M.; Brunelli, M.; Martignoni, G.; Netto, G.J.; Hoque, M.O.; et al. PD-L1 Expression Heterogeneity in Non–Small Cell Lung Cancer: Defining Criteria for Harmonization between Biopsy Specimens and Whole Sections. J. Thorac. Oncol. 2018, 13, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Gradecki, S.E.; Grange, J.S.; Stelow, E.B. Concordance of PD-L1 expression between core biopsy and resection specimens of non-small cell lung cancer. Am. J. Surg. Pathol. 2018, 42, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Figueroa, D.J.; Fay, A.P.; Signoretti, S.; Liu, Y.; Gagnon, R.; Deen, K.; Carpenter, C.; Benson, P.; Ho, T.H.; et al. Correlation of PD-L1 tumor expression and treatment outcomes in patients with renal cell carcinoma receiving sunitinib or pazopanib: Results from COMPARZ, a randomized controlled trial. Clin. Cancer Res. 2015, 21, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-J.; Jeon, Y.K.; Cho, Y.M.; Lee, J.-L.; Chung, D.H.; Park, J.Y.; Go, H. The Association Between PD-L1 Expression and the Clinical Outcomes to Vascular Endothelial Growth Factor-Targeted Therapy in Patients with Metastatic Clear Cell Renal Cell Carcinoma. Oncologist 2015, 20, 1253–1260. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. Proc. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Amin, A.; Plimack, E.R.; Ernstoff, M.S.; Lewis, L.D.; Bauer, T.M.; McDermott, D.F.; Carducci, M.; Kollmannsberger, C.; Rini, B.I.; Heng, D.Y.C.; et al. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: The CheckMate 016 study. J. Immunother. Cancer 2018, 6, 109. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Keeler, M.E.; Kessler, E.R.; Bernard, B.; Weisdack, S.; Breaker, K.M.; Wold, M.; Ertz, D.; Weitzenkamp, D.; Flaig, T.W.; Lam, E.T. Pembrolizumab (pembro) and cabozantinib (cabo) in patients (pts) with metastatic renal cell carcinoma (mRCC): Phase I results. J. Clin. Oncol. 2019, 7, 600. [Google Scholar] [CrossRef]
- Nadal, R.M.; Mortazavi, A.; Stein, M.; Pal, S.K.; Davarpanah, N.N.; Parnes, H.L.; Ning, Y.-M.; Cordes, L.M.; Bagheri, M.H.; Lindenberg, L.; et al. Results of phase I plus expansion cohorts of cabozantinib (Cabo) plus nivolumab (Nivo) and CaboNivo plus ipilimumab (Ipi) in patients (pts) with with metastatic urothelial carcinoma (mUC) and other genitourinary (GU) malignancies. J. Clin. Oncol. 2018, (Suppl. 6), 515. [Google Scholar] [CrossRef]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef]
- Reck, M.; Von Pawel, J.; Zatloukal, P.; Ramlau, R.; Gorbounova, V.; Hirsh, V.; Leighl, N.; Mezger, J.; Archer, V.; Moore, N.; et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAiL. J. Clin. Oncol. 2009, 27, 1227–1234. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Kowanetz, M.; Zou, W.; Mccleland, M.; Gandara, D.R.; Gadgeel, S.; Rittmeyer, A.; Barlesi, F.; Park, K.; Shames, D.; Koeppen, H.; et al. MA 05.09 Pre-Existing Immunity Measured by Teff Gene Expression in Tumor Tissue is Associated with Atezolizumad Efficacy in NSCLC. J. Thorac. Oncol. 2017, 12, S1817–S1818. [Google Scholar] [CrossRef][Green Version]
- Jotte, R.M.; Cappuzzo, F.; Vynnychenko, I.; Stroyakovskiy, D.; Rodriguez Abreu, D.; Hussein, M.A.; Soo, R.A.; Conter, H.J.; Kozuki, T.; Silva, C.; et al. IMpower131: Primary PFS and safety analysis of a randomized phase III study of atezolizumab + carboplatin + paclitaxel or nab-paclitaxel vs. carboplatin + nab-paclitaxel as 1L therapy in advanced squamous NSCLC. J. Clin. Oncol. 2018, 36 (Suppl. 18). [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 204–2051. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Langer, C.J.; Gadgeel, S.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. 24-Month Overall Survival from KEYNOTE-021 Cohort G: Pemetrexed and Carboplatin with or without Pembrolizumab as First-Line Therapy for Advanced Nonsquamous Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Cassidy, J.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Rittweger, K.; Gilberg, F.; Saltz, L. XELOX vs. FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br. J. Cancer 2011, 105, 58–64. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Bennouna, J.; Sastre, J.; Arnold, D.; Österlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Viéitez, J.M.; Bouché, O.; Borg, C.; et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef]
- Simkens, L.H.J.; Van Tinteren, H.; May, A.; Ten Tije, A.J.; Creemers, G.J.M.; Loosveld, O.J.L.; De Jongh, F.E.; Erdkamp, F.L.G.; Erjavec, Z.; Van Der Torren, A.M.E.; et al. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): A phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 2015, 385, 843–852. [Google Scholar] [CrossRef]
- Aparicio, T.; Ghiringhelli, F.; Boige, V.; Le Malicot, K.; Taieb, J.; Bouche, O.; Phelip, J.M.; François, E.; Borel, C.; Faroux, R.; et al. Bevacizumab maintenance versus no maintenance during chemotherapy-free intervals in metastatic colorectal cancer: A randomized phase III trial (PRODIGE 9). J. Clin. Oncol. 2018, 36, 674–681. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; Van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef]
- Tabernero, J.; Van Cutsem, E.; Lakomý, R.; Prausová, J.; Ruff, P.; Van Hazel, G.A.; Moiseyenko, V.M.; Ferry, D.R.; McKendrick, J.J.; Soussan-Lazard, K.; et al. Aflibercept versus placebo in combination with fluorouracil, leucovorin and irinotecan in the treatment of previously treated metastatic colorectal cancer: Prespecified subgroup analyses from the VELOUR trial. Eur. J. Cancer 2014, 50, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blin. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef]
- Arnold, D.; Fuchs, C.S.; Tabernero, J.; Ohtsu, A.; Zhu, A.X.; Garon, E.B.; Mackey, J.R.; Paz-Ares, L.; Baron, A.D.; Okusaka, T.; et al. Meta-analysis of individual patient safety data from six randomized, placebo-controlled trials with the antiangiogenic VEGFR2-binding monoclonal antibody ramucirumab. Ann. Oncol. 2017, 28, 2932–2942. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L.; et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 619–629. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Hochster, H.S.; Bendell, J.C.; Cleary, J.M.; Foster, P.; Zhang, W.; He, X.; Hernandez, G.; Iizuka, K.; Eckhardt, S.G. Efficacy and safety of atezolizumab (atezo) and bevacizumab (bev) in a phase Ib study of microsatellite instability (MSI)-high metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, (Suppl. 4), 673. [Google Scholar] [CrossRef]
- Grothey, A.; Tabernero, J.; Arnold, D.; De Gramont, A.; Ducreux, M.P.; O’Dwyer, P.J.; Van Cutsem, E.; Bosanac, I.; Srock, S.; Mancao, C.; et al. LBA19Fluoropyrimidine (FP) + bevacizumab (BEV) + atezolizumab vs. FP/BEV in BRAFwt metastatic colorectal cancer (mCRC): Findings from Cohort 2 of MODUL—A multicentre, randomized trial of biomarker-driven maintenance treatment following first-line inducti. Ann. Oncol. 2018, 29 (Suppl. 8), viii714–viii715. [Google Scholar] [CrossRef]
- Lee, J.J.; Yothers, G.; Jacobs, S.A.; Sanoff, H.K.; Cohen, D.J.; Guthrie, K.A.; Henry, N.L.; Ganz, P.A.; Kopetz, S.; Lucas, P.C.; et al. Colorectal Cancer Metastatic dMMR Immuno-Therapy (COMMIT) study (NRG- GI004/SWOG-S1610): A randomized phase III study of mFOLFOX6/bevacizumab combination chemotherapy with or without atezolizumab or atezolizumab monotherapy in the first-line treatment of patients (pts) with deficient DNA mismatch repair (dMMR) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2019, 37, TPS728. [Google Scholar]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Chau, I.; Penel, N.; Arkenau, H.-T.; Santana-Davila, R.; Calvo, E.; Soriano, A.O.; Mi, G.; Jin, J.; Ferry, D.; Herbst, R.S.; et al. Safety and antitumor activity of ramucirumab plus pembrolizumab in treatment naïve advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma: Preliminary results from a multi-disease phase I study (JVDF). J. Clin. Oncol. 2018, 36, 101. [Google Scholar] [CrossRef]
- Michielin, O.; van Akkooi, A.; Ascierto, P.; Dummer, R.; Keilholz, U. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2019, 26, 126–132. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Corrie, P.G.; Marshall, A.; Nathan, P.D.; Lorigan, P.; Gore, M.; Tahir, S.; Faust, G.; Kelly, C.G.; Marples, M.; Danson, S.J.; et al. Adjuvant bevacizumab for melanoma patients at high risk of recurrence: Survival analysis of the AVAST-M trial. Ann. Oncol. 2018, 29, 1843–1852. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Diallo, A.; Etienne-Grimaldi, M.-C.; Bidard, F.-C.; Rodrigues, M.; Plancher, C.; Mariani, P.; Cassoux, N.; Decaudin, D.; Asselain, B.; et al. Phase II Trial of Bevacizumab in Combination With Temozolomide as First-Line Treatment in Patients With Metastatic Uveal Melanoma. Oncologist 2016, 21, 281–282. [Google Scholar] [CrossRef][Green Version]
- Glitza, I.C.; Guha-Thakurta, N.; D’Souza, N.M.; Amaria, R.N.; McGovern, S.L.; Rao, G.; Li, J. Bevacizumab as an effective treatment for radiation necrosis after radiotherapy for melanoma brain metastases. Melanoma Res. 2017, 27, 580–584. [Google Scholar] [CrossRef]
- Grignol, V.P.; Olencki, T.; Relekar, K.; Taylor, C.; Kibler, A.; Kefauver, C.; Wei, L.; Walker, M.J.; Chen, H.X.; Kendra, K.; et al. A phase 2 trial of bevacizumab and high-dose interferon alpha 2b in metastatic melanoma. J. Immunother. 2011, 34, 509–515. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A. Breast cancer immunobiology driving immunotherapy: Vaccines and immune checkpoint blockade. Expert Rev. Anticancer Ther. 2012, 12, 1597–1611. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Bergh, J.C.S.; Pusztai, L.; Denkert, C.; Verma, S.; McArthur, H.L.; Kummel, S.; Ding, Y.; Karantza, V.; et al. KEYNOTE-522: Phase III study of pembrolizumab (pembro) + chemotherapy (chemo) vs. placebo + chemo as neoadjuvant therapy followed by pembro vs. placebo as adjuvant therapy for triple-negative breast cancer (TNBC). J. Clin. Oncol. 2018, 36, TPS602. [Google Scholar] [CrossRef]
- Quintela-Fandino, M.; Manso Sànchez, L.M.; Holgado Martín, E.; Moreno, M.C.; Morales Murillo, S.; Bermejo De Las Heras, B.; Malon Gimenez, D.; Colomer Bosch, R.; Gonzalez Cortijo, L.; Hornedo, J.; et al. 72OAddition of durvalumab (Dur) upon progression to bevacizumab (Bev) maintenance in advanced HER2-negative (HERNEG) breast cancer (BC): Safety, efficacy and biomarkers. Ann. Oncol. 2018, 29 (Suppl. 9), ix23–ix27. [Google Scholar] [CrossRef]
- Lamm, D.; Thomas, L.; Flaig, W.; Philippe, E.S. NCCN Clinical Practice Guidelines in Oncology Bladder Cancer. Semin. Surg. Oncol. 2019, 15, 1240–1267. [Google Scholar]
- Gallagher, D.J.; Milowsky, M.I.; Gerst, S.R.; Ishill, N.; Riches, J.; Regazzi, A.; Boyle, M.G.; Trout, A.; Flaherty, A.M.; Bajorin, D.F. Phase II study of sunitinib in patients with metastatic urothelial cancer. J. Clin. Oncol. 2010, 28, 1373–1379. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Ballman, K.V.; Halabi, S.; Watt, C.; Hahn, O.M.; Steen, P.D.; Dreicer, R.; Flaig, T.W.; Stadler, W.M.; Sweeney, C.; et al. CALGB 90601 (Alliance): Randomized, double-blind, placebo-controlled phase III trial comparing gemcitabine and cisplatin with bevacizumab or placebo in patients with metastatic urothelial carcinoma. J. Clin. Oncol. 2019, 37, 4503. [Google Scholar] [CrossRef]
- McConkey, D.J.; Choi, W.; Shen, Y.; Lee, I.L.; Porten, S.; Matin, S.F.; Kamat, A.M.; Corn, P.; Millikan, R.E.; Dinney, C.; et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naïve Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisp. Eur. Urol. 2016, 69, 855–862. [Google Scholar] [CrossRef]
- Grivas, P.D.; Daignault, S.; Tagawa, S.T.; Nanus, D.M.; Stadler, W.M.; Dreicer, R.; Kohli, M.; Petrylak, D.P.; Vaughn, D.J.; Bylow, K.A.; et al. Double-blind, randomized, phase 2 trial of maintenance sunitinib versus placebo after response to chemotherapy in patients with advanced urothelial carcinoma. Cancer 2014, 120, 692–701. [Google Scholar] [CrossRef]
- Necchi, A.; Mariani, L.; Zaffaroni, N.; Schwartz, L.H.; Giannatempo, P.; Crippa, F.; Morosi, C.; Lanocita, R.; Sava, T.; Ortega, C.; et al. Pazopanib in advanced and platinum-resistant urothelial cancer: An open-label, single group, phase 2 trial. Lancet Oncol. 2012, 13, 810–816. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Ross, R.W.; Jacobus, S.; Vaishampayan, U.; Yu, E.Y.; Quinn, D.I.; Hahn, N.M.; Hutson, T.E.; Sonpavde, G.; Morrissey, S.C.; et al. Double-blind, randomized trial of docetaxel plus vandetanib versus docetaxel plus placebo in platinum-pretreated metastatic urothelial cancer. J. Clin. Oncol. 2012, 30, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.; de Wit, R.; Chi, K.N.; Drakaki, A.; Sternberg, C.N.; Nishiyama, H.; Castellano, D.; Hussain, S.; Fléchon, A.; Bamias, A.; et al. Ramucirumab plus docetaxel versus placebo plus docetaxel in patients with locally advanced or metastatic urothelial carcinoma after platinum-based therapy (RANGE): A randomised, double-blind, phase 3 trial. Lancet 2017, 390, 2266–2277. [Google Scholar] [CrossRef]


{kind=link}
| Authors, Year | Disease/Setting | Clinical Trial/Phase | Study Design | Total No. of Patients, mFU | Primary End Points | Results |
|---|---|---|---|---|---|---|
| Brian I. Rini et al. The Lancet 2019 [128] | LA or mRCC (clear cell, sarcomatoid), first line | IMmotion 151, phase III, NCT02420821 | R (1:1), OL, atezolizumab + bevacizumab vs. sunitinib | 951 pts, mFU: 15 m | IA PFS in PD-L1+ pts and OS in ITT population | IA mPFS in PD-L1+: 11.2 vs. 7.7 months (HR 0.74, p = 0.0217) mOS in ITT: 33.6 vs. 34.9 (HR 0.93, p = 0.4751) |
| Motzer et al. NEJM 2019 [129] | mRCC (clear cell), first line | Javeline Renal 101, phase III, NCT02684006 | R (1:1), OL, avelumab + axitinib vs. sunitinib | 866 pts, mFU: 11.6 m and 10.7 m | PFS and OS in PD-L1 + pts | mPFS in PD-L1+: 13.8 vs. 7.2 months (HR 0.61, p < 0.001) mOS in PD-L1+: data immature |
| Mark A. Socinski et al. NEJM 2018 [130] | Non-squamous NSCLC, First line | IMpower 150, phase III, NCT02366143 | R (1:1:1), OL, ACP vs. BCP vs. ABCP x4-6 cycles (induction), followed by A or B or A+B (maintenance) | 1202 pts, mFU: 15.4 months (ABCP) and 15.5 months (BCP) | IA PFS (both in ITT-WT and in Teff- high WT populations) and OS in ITT- WT population | mPFS in ITT-WT: 8.3 vs. 6.8 months (HR 0.62, p < 0.001); mPFS in Teff-high WT: 11.3 vs. 6.8 months (HR 0.51, p < 0.001); mOS in ITT-WT: 19.2 vs. 14.7 months (HR 0.78, p = 0.02) |
| Amin A. et al. J Immunother Cancer 2018 [131] | mRCC (clear cell or non-clear cell), first and subsequent lines | Checkmate 016, phase I, NCT01472081 | OL, nivolumab + sunitib (N+S), nivolumab + pazopanib (N+P) | N+S: 33 pts mFU: 50 m N+P: 20 pts mFU: 27.1 m (closed early) | Safety and tolerability | AEs in N+S: 100% G3-4 AEs in N+S: 82% AEs N+P: 100% G3-4 AEs in N+P: 70% |
| Brian I. Rini et al. NEJM 2019 [132] | mRCC (clear cell), first line | KEYNOTE-426, phase III, NCT02853331 | R (1:1), OL, pembrolizumab + axitinib vs. sunitinib | 861 pts, mFU:12.8m | PFS and OS in ITT population | mPFS in ITT: 15.1 vs. 11.1 months (HR 0.69, p < 0.001) mOS in ITT: data immature |
| A.Grothey et al. Ann of Oncol. 2018 [156] | mCRC, first line, Results from Cohort 2 available (BRAFwt) | MODUL, Phase II, NCT02291289 | Umbrella, FOLFOX+ B x 16 weeks (induction), then R (2:1) to FP/B vs. FP/B +A (maintenance in Cohort 2). | 696 pts (445 pts in cohort 2) mFU Cohort 2: 10.5 months | PFS per investigator | mPFS: 7.39 vs. 7.20 months (HR 0.96, p = 0.727) |
| F. S. Hodi et al. Cancer Immunol Res. 2014 [173] | Metastatic melanoma, First and subsequent lines | Phase I, NCT00790010 | OL, non-R, 4 cohorts, Ipilimumab (induction and maintenance), bevacizumab (continuous) | 46 pts, mFU: 17.3 months | Primary end points: Safety and tolerability, Secondary end points: BORR, DCR, TTP, mOS | MTD: cohort 2 (ipilimumab 10 mg/kg + bevacizumab 15 mg/kg) Overall G3-4: 28.3% BORR: 19.6% DCR: 67.4% TTP: 9 months mOS: 25.1 months |
| M. Quintela-Fandino et al. JCO 2018 [177] | mBC, subsequent lines | Phase Ib, NCT02802098 | OL, single arm, durvalumab + bevacizumab after PD to taxane+ bevacizumab regimen | 24 pts mFU: NA | PFS | mPFS: 76 days |
| Combination Drugs | Disease Condition | Study Phase | Status at Time of Search | Clinical Trial ID |
|---|---|---|---|---|
| Nivolumab as maintenance therapy after sunitinib/pazopanib | LA/mRCC Maintenance therapy | Phase II | Active, not recruiting | NCT02959554 |
| Cabozantinib + nivolumab +/− ipilimumab | LA/m urothelial carcinoma, mRCC, other genitourinary tumors subsequent lines | Phase I | recruiting | NCT02496208 |
| Pembrolizumab + cabozantinib | mRCC subsequent lines | Phase I/II | recruiting | NCT03149822 |
| Pembrolizumab + CT 1 + bevacizumab/ipilimumab/antiEGFR | LA/m NSCLC First line | Phase I/II | Active, not recruiting | NCT02039674 |
| Pembrolizumab + bevacizumab | untreated brain metastases from melanoma or NSCLC | Phase II | recruiting | NCT02681549 |
| Nintedanib + ipilimumab + nivolumab | mNSCLC First and subsequent lines | Phase I/II | recruiting | NCT03377023 |
| Atezolizumab + bevacizumab +/− CT 2 Atezolizumab + CT 3 | Advanced solid tumors Subsequent lines | Phase I | Active, not recruiting | NCT01633970 |
| Atezolizumab + bevacizumab + mFOLFOX6 | mCRC first line | Phase III | recruiting | NCT02997228 |
| Ramucirumab + Pembrolizumab | LA/m G or GEJ adenocarcinoma NSCLC BTC Urothelial carcinoma First and Subsequent lines | Phase I | Active, not recruiting | NCT02443324 |
| Ramucirumab + pembrolizumab + paclitaxel | LA/m G or GEJ adenocarcinoma Subsequent lines | Phase II | Active Not yet recruiting | NCT04069273 |
| Ipilimumab + bevacizumab | LA/m melanoma First and subsequent lines | Phase II | Active, not recruiting | NCT01950390 |
| Lenvatinib + pembrolizumab | Advanced solid tumors Subsequent lines | Phase I/II | Active, not recruiting | NCT02501096 |
| Lenvatinib + pembrolizumab | LA/m urothelial carcinoma First line | Phase III | recruiting | NCT03898180 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciciola, P.; Cascetta, P.; Bianco, C.; Formisano, L.; Bianco, R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J. Clin. Med. 2020, 9, 675. https://doi.org/10.3390/jcm9030675
Ciciola P, Cascetta P, Bianco C, Formisano L, Bianco R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. Journal of Clinical Medicine. 2020; 9(3):675. https://doi.org/10.3390/jcm9030675
Chicago/Turabian StyleCiciola, Paola, Priscilla Cascetta, Cataldo Bianco, Luigi Formisano, and Roberto Bianco. 2020. "Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents" Journal of Clinical Medicine 9, no. 3: 675. https://doi.org/10.3390/jcm9030675
APA StyleCiciola, P., Cascetta, P., Bianco, C., Formisano, L., & Bianco, R. (2020). Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. Journal of Clinical Medicine, 9(3), 675. https://doi.org/10.3390/jcm9030675