Diagnosis of Inherited Platelet Disorders on a Blood Smear


{kind=link}
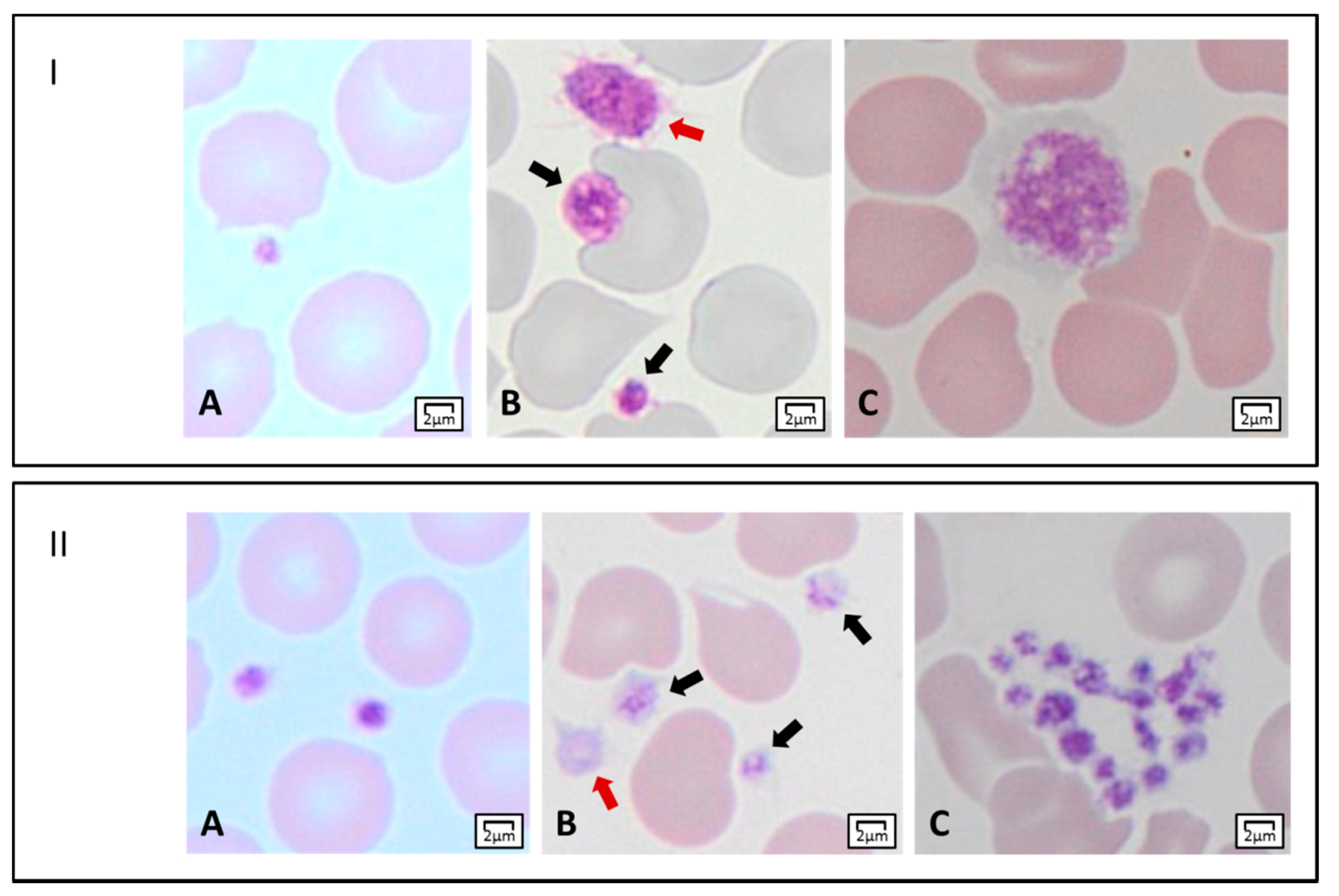{kind=link}
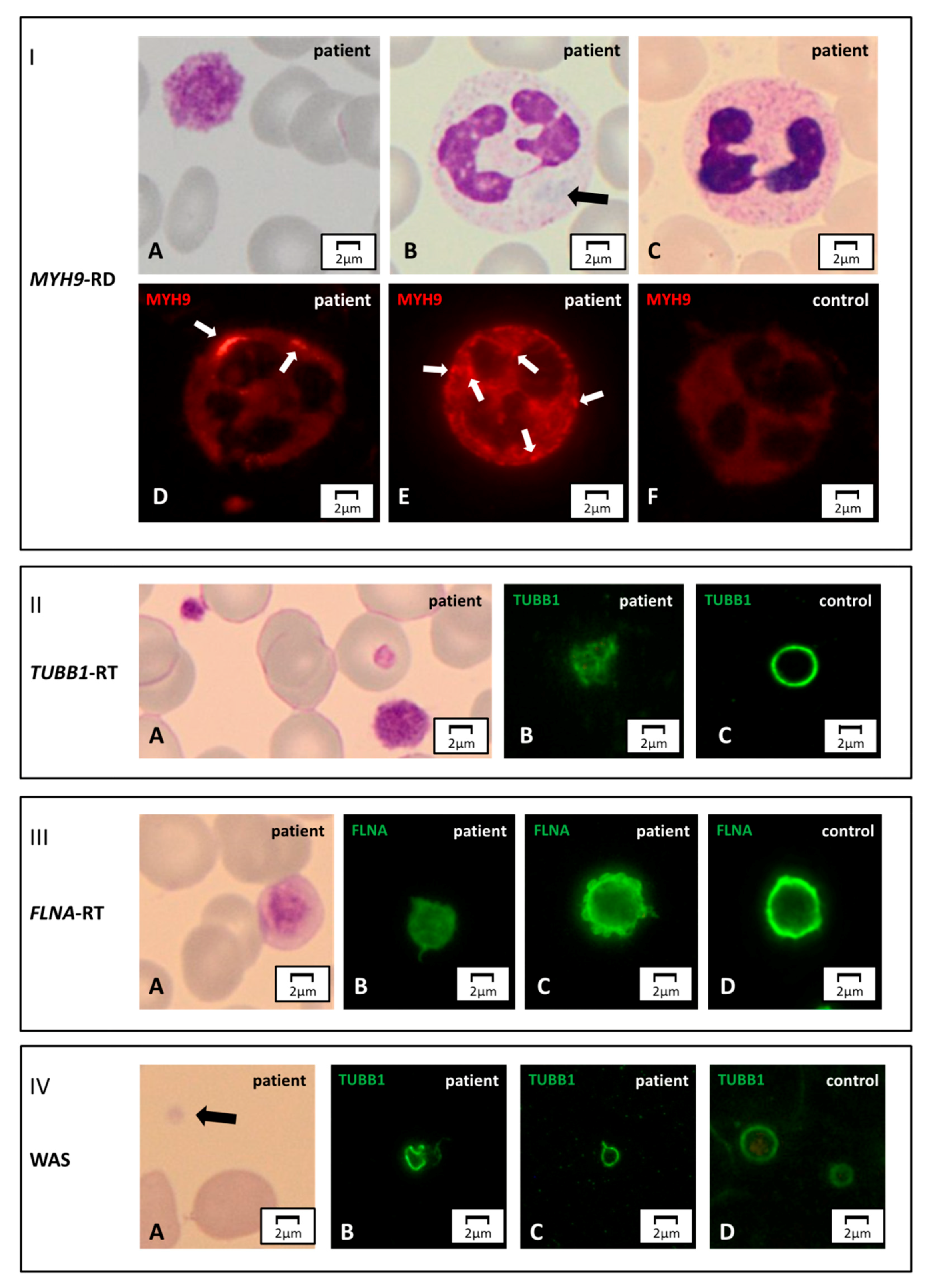{kind=link}
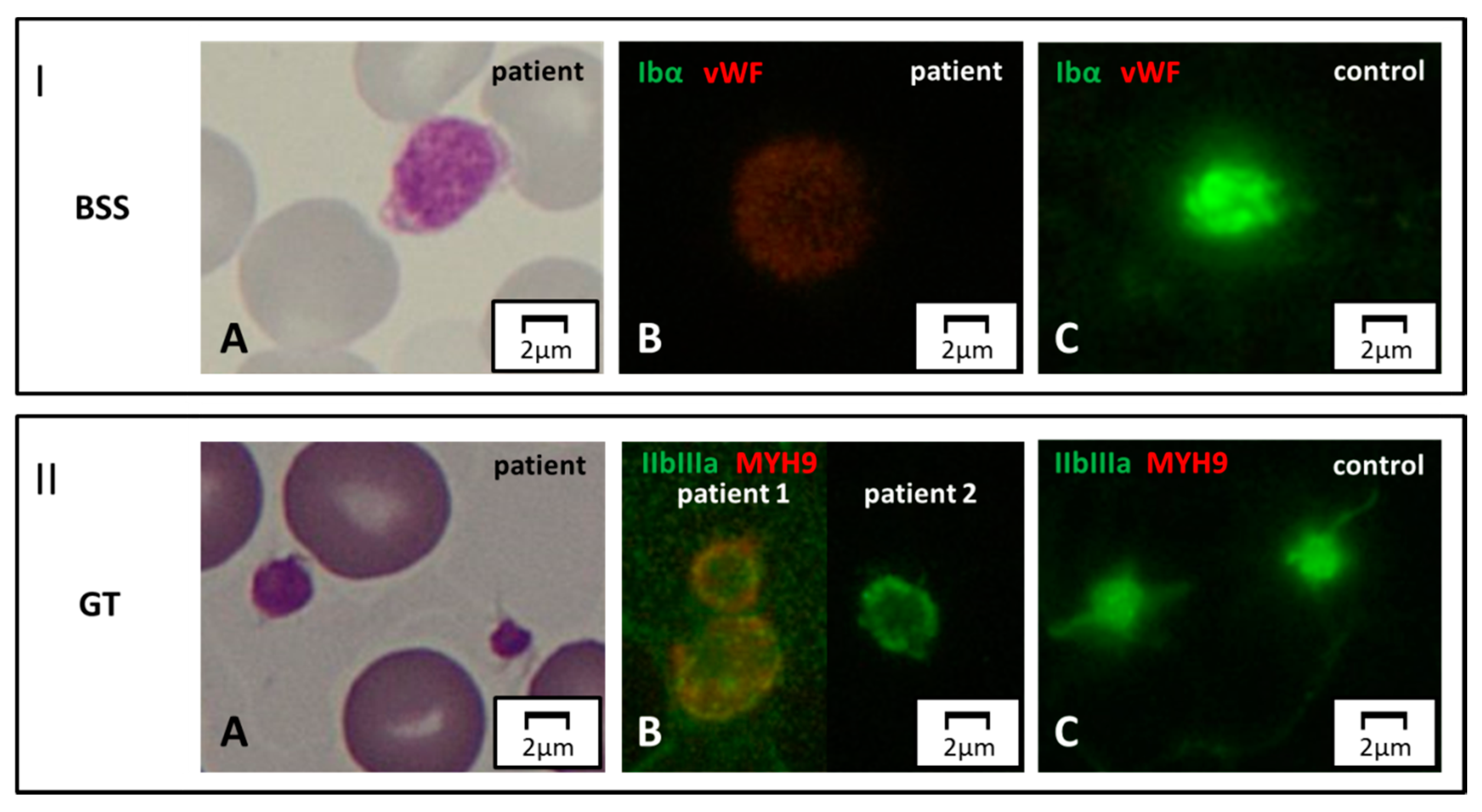{kind=link}
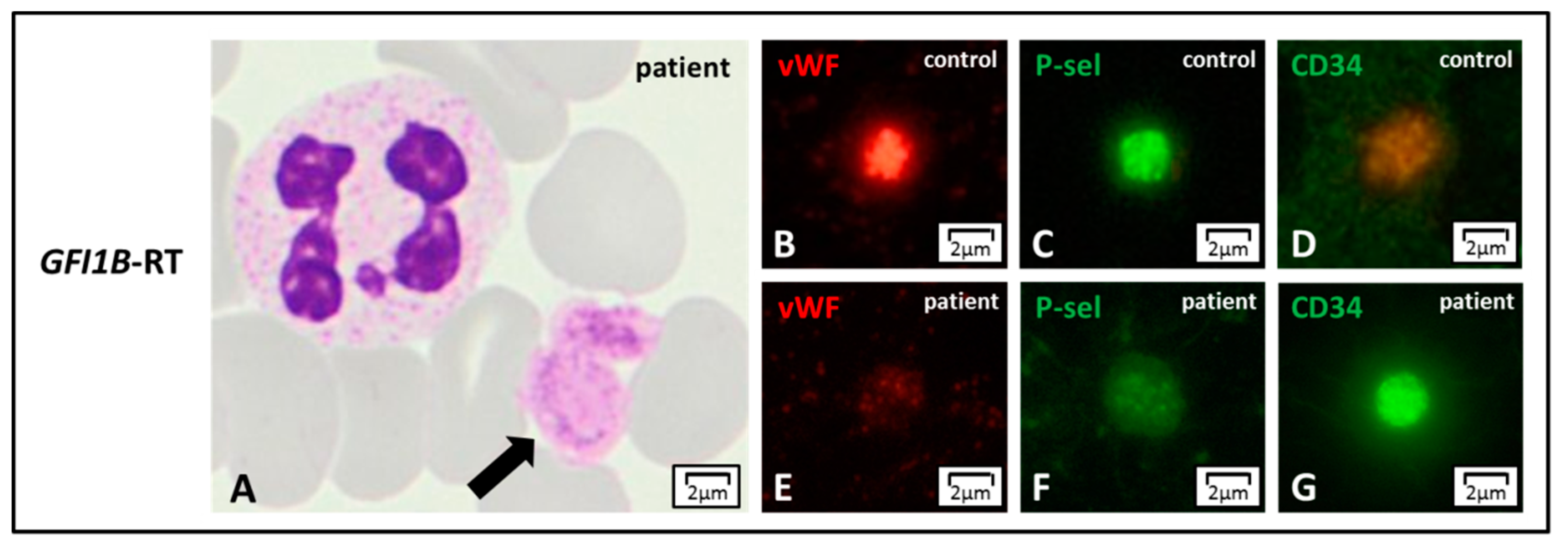{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Inherited Platelet Disorders
1.2. Bleeding Symptoms do not Allow Specific Diagnosis of an IPD
1.3. Non-Platelet Features Clinically Characterize some IPDs
1.4. Treatment of IPDs is Usually Symptomatic
1.5. Current Diagnostic Tools for IPDs
1.6. Main Advantages of Blood Smear Analysis
2. Technical Considerations
2.1. Preparation of Blood Smears
2.2. Immunofluorescence Labelling
2.3. Note of Caution
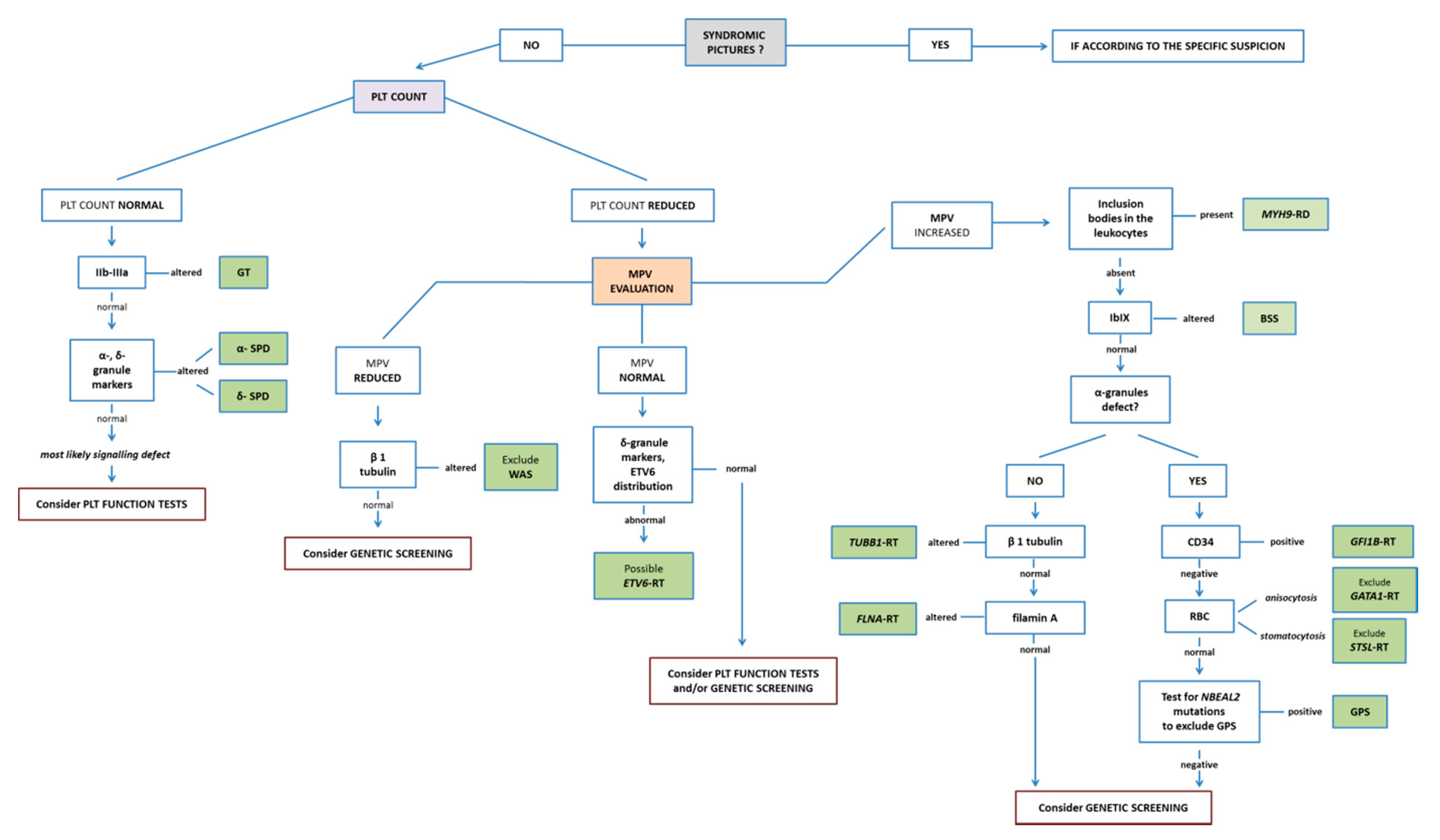
3. Main Diagnostic Patterns
3.1. Light Microscopy
3.2. Platelet Size
3.3. Platelet Staining
3.4. Other Morphological Alterations of Platelets
4. Morphological Alterations of Other Blood Cells
4.1. Immunofluorescence Microscopy
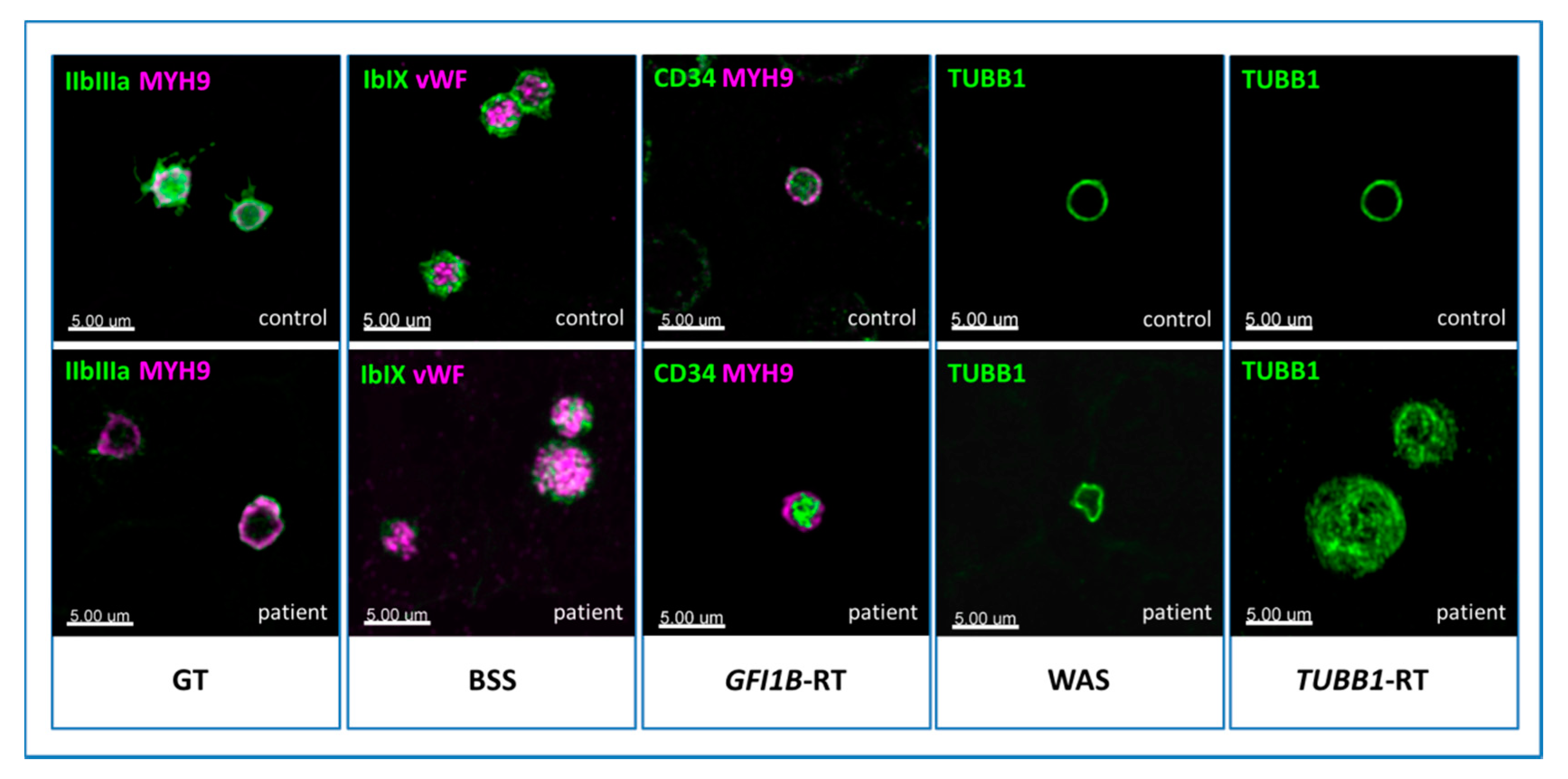
4.2. Cytoskeleton Proteins
4.3. Surface Receptors
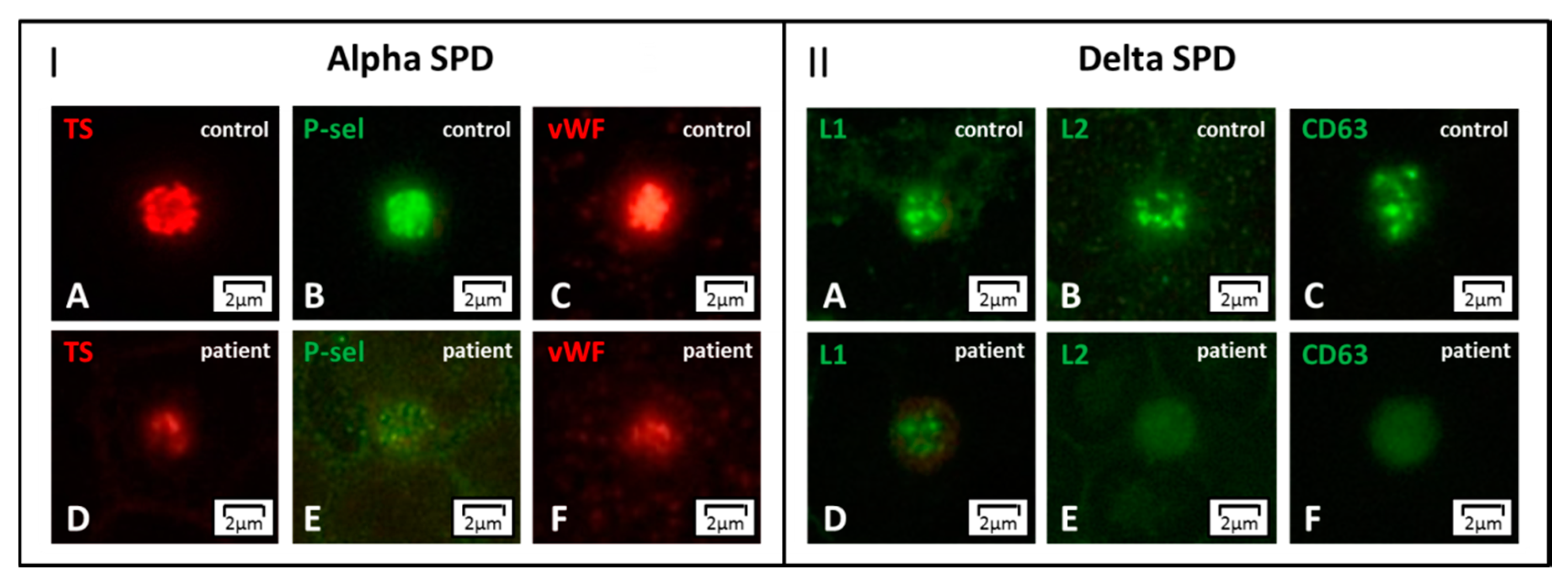
4.4. Alpha and Delta Granule Markers
5. Weaknesses and Ethical Considerations
5.1. Limitations Related to this Method and New Perspectives
5.2. Ethical Considerations
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Noris, P.; Pecci, A. Hereditary thrombocytopenias: A growing list of disorders. Hematology 2017, 2017, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Lentaigne, C.; Greene, D.; Sivapalaratnam, S.; Favier, R.; Seyres, D.; Thys, C.; Grassi, L.; Mangles, S.; Sibson, K.; Stubbs, M.J.; et al. Germline mutations in the transcription factor IKZF5 cause thrombocytopenia. Blood 2019, 134, 2070–2081. [Google Scholar] [CrossRef] [PubMed]
- Almazni, I.; Stapley, R.; Morgan, N.V. Inherited Thrombocytopenia: Update on Genes and Genetic Variants which may be Associated with Bleeding. Front. Cardiovasc. Med. 2019, 6, 80. [Google Scholar] [CrossRef]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Freson, K.; Turro, E. High-throughput sequencing approaches for diagnosing hereditary bleeding and platelet disorders. J. Thromb. Haemost. 2017, 15, 1262–1272. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leinøe, E.; Zetterberg, E.; Kinalis, S.; Østrup, O.; Kampmann, P.; Norström, E.; Andersson, N.; Klintman, J.; Qvortrup, K.; Nielsen, F.C.; et al. Application of whole-exome sequencing to direct the specific functional testing and diagnosis of rare inherited bleeding disorders in patients from the Öresund Region, Scandinavia. Br. J. Haematol. 2017, 179, 308–322. [Google Scholar] [CrossRef]
- Lentaigne, C.; Freson, K.; Laffan, M.A.; Turro, E.; Ouwehand, W.H. Inherited platelet disorders: Toward DNA-based diagnosis. Blood 2016, 127, 2814–2823. [Google Scholar] [CrossRef]
- Vannucchi, A.M. Pegasus causes inherited thrombocytopenia. Blood 2019, 134, 2000–2002. [Google Scholar] [CrossRef]
- Pecci, A.; Balduini, C.L. Lessons in platelet production from inherited thrombocytopenias. Br. J. Haematol. 2014, 165, 179–192. [Google Scholar] [CrossRef]
- Hirata, S.; Takayama, N.; Jono-Ohnishi, R.; Endo, H.; Nakamura, S.; Dohda, T.; Nishi, M.; Hamazaki, Y.; Ishii, E.I.; Kaneko, S.; et al. Congenital amegakaryocytic thrombocytopenia iPS cells exhibit defective MPL-mediated signaling. J. Clin. Investig. 2013, 123, 3802–3814. [Google Scholar] [CrossRef]
- Songdej, N.; Rao, A.K. Hematopoietic transcription factor mutations: Important players in inherited platelet defects. Blood 2017, 129, 2873–2881. [Google Scholar] [CrossRef]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.L.; et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat. Genet. 2015, 47, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Kasahara, H.; Yoshida, K.; Yoshimi, A.; Kunimoto, H.; Watanabe, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Harada, Y.; et al. Genetic basis of myeloid transformation in familial platelet disorder/acute myeloid leukemia patients with haploinsufficient RUNX1 allele. Blood Cancer J. 2016, 6, e392. [Google Scholar] [CrossRef] [PubMed]
- Karastaneva, A.; Nebral, K.; Schlagenhauf, A.; Baschin, M.; Palankar, R.; Juch, H.; Heitzer, E.; Speicher, M.R.; Höfler, G.; Grigorow, I.; et al. Novel phenotypes observed in patients with ETV6-linked leukaemia/familial thrombocytopenia syndrome and a biallelic ARID5B risk allele as leukaemogenic cofactor. J. Med. Genet. 2019. [Google Scholar] [CrossRef]
- Pecci, A.; Malara, A.; Badalucco, S.; Bozzi, V.; Torti, M.; Balduini, C.L.; Balduini, A. Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb. Haemost. 2009, 102, 90–96. [Google Scholar]
- Kunishima, S.; Kobayashi, R.; Itoh, T.J.; Hamaguchi, M.; Saito, H. Mutation of the β1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood 2009, 113, 458–461. [Google Scholar] [CrossRef]
- Nurden, P.; Debili, N.; Coupry, I.; Bryckaert, M.; Youlyouz-Marfak, I.; Solé, G.; Pons, A.C.; Berrou, E.; Adam, F.; Kauskot, A.; et al. Thrombocytopenia resulting from mutations in filamin A can be expressed as an isolated syndrome. Blood 2011, 118, 5928–5937. [Google Scholar] [CrossRef]
- Kunishima, S.; Okuno, Y.; Yoshida, K.; Shiraishi, Y.; Sanada, M.; Muramatsu, H.; Chiba, K.; Tanaka, H.; Miyazaki, K.; Sakai, M.; et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am. J. Hum. Genet. 2013, 92, 431–438. [Google Scholar] [CrossRef]
- Futterer, J.; Dalby, A.; Lowe, G.C.; Johnson, B.; Simpson, M.A.; Motwani, J.; Williams, M.; Watson, S.P.; Morgan, N.V. Mutation in GNE is associated with severe congenital thrombocytopenia. Blood 2018, 132, 1855–1858. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Shai, E.; Turro, E.; Jahshan, N.; Hi-Am, E.; Spectre, G.; Daum, H.; Kalish, Y.; Althaus, K.; Greinacher, A.; et al. GNE variants causing autosomal recessive macrothrombocytopenia without associated muscle wasting. Blood 2018, 132, 1851–1854. [Google Scholar] [CrossRef]
- Kraemer, B.F.; Weyrich, A.S. Insight Increased formation of PaCSs in megakaryocytes and platelets from patients with autosomal-dominant ANKRD26-related thrombocytopenia: Polyubiquinated protein depots in platelets and megakaryocytes from patients with ANKRD26-RT. Thromb. Haemost. 2012, 109, 180. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Balduini, A.; Noris, P.; Barozzi, S.; Sommi, P.; di Buduo, C.; Balduini, C.L.; Solcia, E.; Pecci, A. Ubiquitin/proteasome-rich particulate cytoplasmic structures (PaCSs) in the platelets and megakaryocytes of ANKRD26-related thrombocytopenia. Thromb. Haemost. 2013, 109, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Study investigators. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: A communication from the Platelet Physiology SSC. J. Thromb. Haemost. 2019. [Google Scholar] [CrossRef] [PubMed]
- Orsini, S.; Noris, P.; Bury, L.; Heller, P.G.; Santoro, C.; Kadir, R.A.; Butta, N.C.; Falcinelli, E.; Cid, A.R.; Fabris, F.; et al. Bleeding risk of surgery and its prevention in patients with inherited platelet disorders. Haematologica 2017, 102, 1192–1203. [Google Scholar] [CrossRef]
- Toriello, H.V. Thrombocytopenia-absent radius syndrome. Semin. Thromb. Hemost. 2011, 37, 707–712. [Google Scholar] [CrossRef]
- Niihori, T.; Ouchi-Uchiyama, M.; Sasahara, Y.; Kaneko, T.; Hashii, Y.; Irie, M.; Sato, A.; Saito-Nanjo, Y.; Funayama, R.; Nagashima, T.; et al. Mutations in MECOM, Encoding Oncoprotein EVI1, Cause Radioulnar Synostosis with Amegakaryocytic Thrombocytopenia. Am. J. Hum. Genet. 2015, 97, 848–854. [Google Scholar] [CrossRef]
- Candotti, F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome. J. Clin. Immunol. 2018, 38, 13–27. [Google Scholar] [CrossRef]
- Albert, M.H.; Bittner, T.C.; Nonoyama, S.; Notarangelo, L.D.; Burns, S.; Imai, K.; Espanol, T.; Fasth, A.; Pellier, I.; Strauss, G.; et al. X-linked thrombocytopenia (XLT) due to WAS mutations: Clinical characteristics, long-term outcome, and treatment options. Blood 2010, 115, 3231–3238. [Google Scholar] [CrossRef]
- Freson, K.; Wijgaerts, A.; Van Geet, C. GATA1 gene variants associated with thrombocytopenia and anemia. Platelets 2017, 28, 731–734. [Google Scholar] [CrossRef]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef]
- Markello, T.; Chen, D.; Kwan, J.Y.; Horkayne-Szakaly, I.; Morrison, A.; Simakova, O.; Maric, I.; Lozier, J.; Cullinane, A.R.; Kilo, T.; et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol. Genet. Metab. 2015, 114, 474–482. [Google Scholar] [CrossRef]
- Pecci, A.; Klersy, C.; Gresele, P.; Lee, K.J.D.; De Rocco, D.; Bozzi, V.; Russo, G.; Heller, P.G.; Loffredo, G.; Ballmaier, M.; et al. MYH9-related disease: A novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations. Hum. Mutat. 2014, 35, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Stritt, S.; Nurden, P.; Turro, E.; Greene, D.; Jansen, S.B.; Westbury, S.K.; Petersen, R.; Astle, W.J.; Marlin, S.; Bariana, T.K.; et al. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood 2016, 127, 2903–2914. [Google Scholar] [CrossRef] [PubMed]
- Ballmaier, M.; Germeshausen, M. Congenital amegakaryocytic thrombocytopenia: Clinical presentation, diagnosis, and treatment. Semin. Thromb. Hemost. 2011, 37, 673–681. [Google Scholar] [CrossRef]
- Arber, D.A. The 2016 WHO classification of acute myeloid leukemia: What the practicing clinician needs to know. Semin. Hematol. 2019, 56, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Galera, P.; Dulau-Florea, A.; Calvo, K.R. Inherited thrombocytopenia and platelet disorders with germline predisposition to myeloid neoplasia. Int. J. Lab. Hematol. 2019, 41, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: Analysis of 78 patients from 21 families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef]
- Melazzini, F.; Palombo, F.; Balduini, A.; De Rocco, D.; Marconi, C.; Noris, P.; Gnan, C.; Pippucci, T.; Bozzi, V.; Faleschini, M.; et al. Clinical and pathogenic features of ETV6-related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica 2016, 101, 1333–1342. [Google Scholar] [CrossRef]
- Moriyama, T.; Metzger, M.L.; Wu, G.; Nishii, R.; Qian, M.; Devidas, M.; Yang, W.; Cheng, C.; Cao, X.; Quinn, E.; et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: A systematic genetic study. Lancet Oncol. 2015, 16, 1659–1666. [Google Scholar] [CrossRef]
- Owen, C.J.; Toze, C.L.; Koochin, A.; Forrest, D.L.; Smith, C.A.; Stevens, J.M.; Jackson, S.C.; Poon, M.C.; Sinclair, G.D.; Leber, B.; et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood 2008, 112, 4639–4645. [Google Scholar] [CrossRef]
- Turro, E.; Greene, D.; Wijgaerts, A.; Thys, C.; Lentaigne, C.; Bariana, T.K.; Westbury, S.K.; Kelly, A.M.; Selleslag, D.; Stephens, J.C.; et al. A dominant gain-of-function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci. Transl. Med. 2016, 8, 328ra30. [Google Scholar] [CrossRef] [PubMed]
- Saultier, P.; Alessi, M.C.; Michel, G.; Chambost, H. Hematopoietic stem cell transplantation for the treatment of leukocyte adhesion deficiency type III. Pediatr. Neonatol. 2017, 58, 560–561. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elfeky, R.A.; Furtado-Silva, J.M.; Chiesa, R.; Rao, K.; Amrolia, P.; Lucchini, G.; Gilmour, K.; Adams, S.; Bibi, S.; Worth, A.; et al. One hundred percent survival after transplantation of 34 patients with Wiskott-Aldrich syndrome over 20 years. J. Allergy Clin. Immunol. 2018, 142, 1654–1656. [Google Scholar] [CrossRef] [PubMed]
- Winikoff, R.; Scully, M.F.; Robinson, K.S. Women and inherited bleeding disorders—A review with a focus on key challenges for 2019. Transfus. Apher. Sci. 2019, 58, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Colucci, G.; Stutz, M.; Rochat, S.; Conte, T.; Pavicic, M.; Reusser, M.; Giabbani, E.; Huynh, A.; Thürlemann, C.; Keller, P.; et al. The effect of desmopressin on platelet function: A selective enhancement of procoagulant COAT platelets in patients with primary platelet function defects. Blood 2014, 123, 1905–1916. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B.; Chalmers, E.A.; Collins, P.W.; Harrison, P.; Kitchen, S.; Liesner, R.J.; Minford, A.; Mumford, A.D.; Parapia, L.A.; Perry, D.J.; et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br. J. Haematol. 2006, 135, 603–633. [Google Scholar] [CrossRef]
- Estcourt, L.J.; Birchall, J.; Allard, S.; Bassey, S.J.; Hersey, P.; Kerr, J.P.; Mumford, A.D.; Stanworth, S.J.; Tinegate, H. Guidelines for the use of platelet transfusions. Br. J. Haematol. 2017, 176, 365–394. [Google Scholar] [CrossRef]
- Kaufman, R.M.; Djulbegovic, B.; Gernsheimer, T.; Kleinman, S.; Tinmouth, A.T.; Capocelli, K.E.; Cipolle, M.D.; Cohn, C.S.; Fung, M.K.; Grossman, B.J.; et al. Platelet transfusion: A clinical practice guideline from the AABB. Ann. Intern. Med. 2015, 162, 205–213. [Google Scholar] [CrossRef]
- Noris, P.; Schlegel, N.; Klersy, C.; Heller, P.G.; Civaschi, E.; Pujol-Moix, N.; Fabris, F.; Favier, R.; Gresele, P.; Latger-Cannard, V.; et al. Analysis of 339 pregnancies in 181 women with 13 different forms of inherited thrombocytopenia. Haematologica 2014, 99, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Zaninetti, C.; Gresele, P.; Bertomoro, A.; Klersy, C.; De Candia, E.; Veneri, D.; Barozzi, S.; Fierro, T.; Alberelli, M.A.; Musella, V.; et al. Eltrombopag for the treatment of inherited thrombocytopenias: A phase 2 clinical trial. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Zaninetti, C.; Barozzi, S.; Bozzi, V.; Gresele, P.; Balduini, C.L.; Pecci, A. Eltrombopag in preparation for surgery in patients with severe MYH9-related thrombocytopenia. Am. J. Hematol. 2019, 94, E199–E201. [Google Scholar] [CrossRef] [PubMed]
- Ozsahin, H.; Cavazzana-Calvo, M.; Notarangelo, L.D.; Schulz, A.; Thrasher, A.J.; Mazzolari, E.; Slatter, M.A.; Le Deist, F.; Blanche, S.; Veys, P.; et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: Collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood 2008, 111, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-aldrich syndrome. Science 2013, 341, 123351. [Google Scholar] [CrossRef]
- Gerrits, A.J.; Leven, E.A.; Frelinger, A.L.; Brigstocke, S.L.; Berny-Lang, M.A.; Mitchell, W.B.; Revel-Vilk, S.; Tamary, H.; Carmichael, S.L.; Barnard, M.R.; et al. Effects of eltrombopag on platelet count and platelet activation in Wiskott-Aldrich syndrome/X-linked thrombocytopenia. Blood 2015, 126, 1367–1378. [Google Scholar] [CrossRef]
- Pecci, A.; Granata, A.; Fiore, C.E.; Balduini, C.L. Renin-angiotensin system blockade is effective in reducing proteinuria of patients with progressive nephropathy caused by MYH9 mutations (Fechtner-Epstein syndrome). Nephrol. Dial. Transplant. 2008, 23, 2690–2692. [Google Scholar] [CrossRef]
- Pecci, A.; Verver, E.J.; Schlegel, N.; Canzi, P.; Boccio, C.M.; Platokouki, H.; Krause, E.; Benazzo, M.; Topsakal, V.; Greinacher, A. Cochlear implantation is safe and effective in patients with MYH9-related disease. Orphanet J. Rare Dis. 2014, 9, 100. [Google Scholar] [CrossRef]
- Balduini, C.L.; Pecci, A.; Savoia, A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br. J. Haematol. 2011, 154, 161–174. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.; Arnold, D.M.; Coller, B.; James, P.; Neunert, C.; Lillicrap, D. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef]
- Noris, P.; Biino, G.; Pecci, A.; Civaschi, E.; Savoia, A.; Seri, M.; Melazzini, F.; Loffredo, G.; Russo, G.; Bozzi, V.; et al. Platelet diameters in inherited thrombocytopenias: Analysis of 376 patients with all known disorders. Blood 2014, 124, e4–e10. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Bury, L.; Falcinelli, E. Inherited platelet function disorders: Algorithms for phenotypic and genetic investigation. Semin. Thromb. Hemost. 2016, 42, 292–305. [Google Scholar] [PubMed]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.M.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, S.P.; et al. Recommendations for the standardization of light transmission aggregometry: A consensus of the working party from the platelet physiology subcommittee of SSC/ISTH. J. Thromb. Haemost. 2013, 11, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.; Mackie, I.; Mumford, A.; Briggs, C.; Liesner, R.; Winter, M.; Machin, S. Guidelines for the laboratory investigation of heritable disorders of platelet function. Br. J. Haematol. 2011, 155, 30–44. [Google Scholar] [CrossRef]
- Hechler, B.; Dupuis, A.; Mangin, P.H.; Gachet, C. Platelet preparation for function testing in the laboratory and clinic: Historical and practical aspects. Res. Pract. Thromb. Haemost. 2019, 3, 615–625. [Google Scholar] [CrossRef]
- Bastida, J.M.; Benito, R.; Lozano, M.L.; Marín-Quilez, A.; Janusz, K.; Martín-Izquierdo, M.; Hernández-Sánchez, J.; Palma-Barqueros, V.; Hernández-Rivas, J.M.; Rivera, J.; et al. Molecular Diagnosis of Inherited Coagulation and Bleeding Disorders. Semin. Thromb. Hemost. 2019, 45, 695–707. [Google Scholar] [CrossRef]
- Andres, O.; König, E.-M.; Althaus, K.; Bakchoul, T.; Bugert, P.; Eber, S.; Knöfler, R.; Kunstmann, E.; Manukjan, G.; Meyer, O.; et al. Use of Targeted High-Throughput Sequencing for Genetic Classification of Patients with Bleeding Diathesis and Suspected Platelet Disorder. TH Open 2018, 2, e445–e454. [Google Scholar] [CrossRef]
- Heremans, J.; Freson, K. High-throughput sequencing for diagnosing platelet disorders: Lessons learned from exploring the causes of bleeding disorders. Int. J. Lab. Hematol. 2018, 40, 89–96. [Google Scholar] [CrossRef]
- Johnson, B.; Doak, R.; Allsup, D.; Astwood, E.; Evans, G.; Grimley, C.; James, B.; Myers, B.; Stokley, S.; Thachil, J.; et al. A comprehensive targeted next-generation sequencing panel for genetic diagnosis of patients with suspected inherited thrombocytopenia. Res. Pract. Thromb. Haemost. 2018, 2, 640–652. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, L.; Sheng, G.; Shen, H.; Ling, J.; Xie, J.; Ma, Z.; Yin, J.; Wang, Z.; Yu, Z.; et al. Application of High-Throughput Sequencing in the Diagnosis of Inherited Thrombocytopenia. Clin. Appl. Thromb. 2018, 24, 94S–103S. [Google Scholar] [CrossRef]
- Maclachlan, A.; Watson, S.P.; Morgan, N.V. Inherited platelet disorders: Insight from platelet genomics using next-generation sequencing. Platelets 2017, 28, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, I.; Stephens, J.C.; Hu, F.; Deevi, S.V.V.; Megy, K.; Bariana, T.K.; Lentaigne, C.; Schulman, S.; Sivapalaratnam, S.; Vries, M.J.A.; et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood 2016, 127, 2791–2803. [Google Scholar] [CrossRef] [PubMed]
- Romasko, E.J.; Devkota, B.; Biswas, S.; Jayaraman, V.; Rajagopalan, R.; Dulik, M.C.; Thom, C.S.; Choi, J.; Jairam, S.; Scarano, M.I.; et al. Utility and limitations of exome sequencing in the molecular diagnosis of pediatric inherited platelet disorders. Am. J. Hematol. 2018, 93, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.P.; Lowe, G.C.; Lordkipanidzé, M.; Morgan, N.V. Genotyping and phenotyping of platelet function disorders. J. Thromb. Haemost. 2013, 11, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Ball, E.V.; Krawczak, M. The human gene mutation database. Nucleic Acids Res. 1998, 26, 285–287. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Howells, K.; Phillips, A.D.; Cooper, D.N.; Thomas, N.S.T. The human gene mutation database: 2008 update. Genome Med. 2009, 1, 13. [Google Scholar] [CrossRef]
- Downes, K.; Megy, K.; Duarte, D.; Vries, M.; Gebhart, J.; Hofer, S.; Shamardina, O.; Deevi, S.V.V.; Stephens, J.; Mapeta, R.; et al. Diagnostic high-throughput sequencing of 2,396 patients with bleeding, thrombotic and platelet disorders. Blood 2019, 134, 2082–2091. [Google Scholar] [CrossRef]
- Greinacher, A.; Pecci, A.; Kunishima, S.; Althaus, K.; Nurden, P.; Balduini, C.L.; Bakchoul, T. Diagnosis of inherited platelet disorders on a blood smear: A tool to facilitate worldwide diagnosis of platelet disorders. J. Thromb. Haemost. 2017, 15, 1511–1521. [Google Scholar] [CrossRef]
- Greinacher, A.; Eekels, J.J.M. Simplifying the diagnosis of inherited platelet disorders? The new tools do not make it any easier. Blood 2019, 133, 2478–2483. [Google Scholar] [CrossRef]
- Nurden, A.T.; Pillois, X. ITGA2B and ITGB3 gene mutations associated with Glanzmann thrombasthenia. Platelets 2018, 29, 98–101. [Google Scholar] [CrossRef]
- Bury, L.; Falcinelli, E.; Chiasserini, D.; Springer, T.A.; Italiano, J.E.; Gresele, P. Cytoskeletal perturbation leads to platelet dysfunction and thrombocytopenia in variant forms of Glanzmann thrombasthenia. Haematologica 2016, 101, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Savoia, A.; Kunishima, S.; De Rocco, D.; Zieger, B.; Rand, M.L.; Pujol-Moix, N.; Caliskan, U.; Tokgoz, H.; Pecci, A.; Noris, P.; et al. Spectrum of the mutations in bernard-soulier syndrome. Hum. Mutat. 2014, 35, 1033–1045. [Google Scholar] [CrossRef]
- Balduini, C.L.; Savoia, A.; Seri, M. Inherited thrombocytopenias frequently diagnosed in adults. J. Thromb. Haemost. 2013, 11, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Bottega, R.; Pecci, A.; De Candia, E.; Pujol-Moix, N.; Heller, P.G.; Noris, P.; De Rocco, D.; Podda, G.M.; Glembotsky, A.C.; Cattaneo, M.; et al. Correlation between platelet phenotype and NBEAL2 genotype in patients with congenital thrombocytopenia and α-granule deficiency. Haematologica 2013, 98, 868–874. [Google Scholar] [CrossRef][Green Version]
- Kunishima, S.; Nishimura, S.; Suzuki, H.; Imaizumi, M.; Saito, H. TUBB1 mutation disrupting microtubule assembly impairs proplatelet formation and results in congenital macrothrombocytopenia. Eur. J. Haematol. 2014, 92, 276–282. [Google Scholar] [CrossRef]
- Kitamura, K.; Okuno, Y.; Yoshida, K.; Sanada, M.; Shiraishi, Y.; Muramatsu, H.; Kobayashi, R.; Furukawa, K.; Miyano, S.; Kojima, S.; et al. Functional characterization of a novel GFI1B mutation causing congenital macrothrombocytopenia. J. Thromb. Haemost. 2016, 14, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Lex, L.; Wesche, J.; Baschin, M.; Greinacher, A.A.K. Immunofluorescence analysis on a blood smear—Validation of stability of blood smears during storage. Hamostaseologie 2018, 38. [Google Scholar] [CrossRef]
- Bender, M.; Stritt, S.; Nurden, P.; Van Eeuwijk, J.M.M.; Zieger, B.; Kentouche, K.; Schulze, H.; Morbach, H.; Stegner, D.; Heinze, K.; et al. Megakaryocyte-specific Profilin1-deficiency alters microtubule stability and causes a Wiskott-Aldrich syndrome-like platelet defect. Nat. Commun. 2014, 5, 4746. [Google Scholar] [CrossRef]
- Bain, B.J. Blood Cells: A Practical Guide, 4th ed.; Blackwell Science Ltd.: Oxford, UK, 2006. [Google Scholar]
- Noris, P.; Klersy, C.; Gresele, P.; Giona, F.; Giordano, P.; Minuz, P.; Loffredo, G.; Pecci, A.; Melazzini, F.; Civaschi, E.; et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: A multicentric, real life study. Br. J. Haematol. 2013, 162, 112–119. [Google Scholar] [CrossRef]
- Monteferrario, D.; Bolar, N.A.; Marneth, A.E.; Hebeda, K.M.; Bergevoet, S.M.; Veenstra, H.; Laros-van Gorkom, B.A.P.; MacKenzie, M.A.; Khandanpour, C.; Botezatu, L.; et al. A dominant-negative GFI1B mutation in the gray platelet syndrome. N. Engl. J. Med. 2014, 370, 245–253. [Google Scholar] [CrossRef]
- Balduini, C.L.; Pecci, A.; Loffredo, G.; Izzo, P.; Noris, P.; Grosso, M.; Bergamaschi, G.; Rosti, V.; Magrini, U.; Ceresa, I.F.; et al. Effects of the R216Q mutation of GATA-1 on erythropoiesis and megakaryocytopoiesis. Thromb. Haemost. 2004, 91, 129–140. [Google Scholar]
- Hatta, K.; Kunishima, S.; Suganuma, H.; Tanaka, N.; Ohkawa, N.; Shimizu, T. A family having type 2B von Willebrand disease with a novel VWF p.R1308S mutation: Detection of characteristic platelet aggregates on peripheral blood smears as the key aspect of diagnosis. Thromb. Res. 2015, 136, 813–817. [Google Scholar] [CrossRef]
- Federici, A.B.; Mannucci, P.M.; Castaman, G.; Baronciani, L.; Bucciarelli, P.; Canciani, M.T.; Pecci, A.; Lenting, P.J.; Groot, P.G.D. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: A cohort study of 67 patients. Blood 2009, 113, 526–534. [Google Scholar] [CrossRef]
- Bury, L.; Malara, A.; Momi, S.; Petito, E.; Balduini, A.; Gresele, P. Mechanisms of thrombocytopenia in platelet-type von Willebrand disease. Haematologica 2019, 104, 1473–1481. [Google Scholar] [CrossRef]
- Savoia, A.; De Rocco, D.; Panza, E.; Bozzi, V.; Scandellari, R.; Loffredo, G.; Mumford, A.; Heller, P.G.; Noris, P.; De Groot, M.R.; et al. Heavy chain myosin 9-related disease (MYH9-RD): Neutrophil inclusions of myosin-9 as a pathognomonic sign of the disorder. Thromb. Haemost. 2010, 103, 826–832. [Google Scholar]
- Millikan, P.D.; Balamohan, S.M.; Raskind, W.H.; Kacena, M.A. Inherited thrombocytopenia due to GATA-1 mutations. Semin. Thromb. Hemost. 2011, 37, 682–689. [Google Scholar] [CrossRef]
- Neff, A.T. Sitosterolemia’s stomatocytosis and macrothrombocytopenia. Blood 2012, 120, 4283. [Google Scholar] [CrossRef]
- Amoruso, M.; Alberio, L.; Nagy, M. EDTA-related degranulation mimicking Storage Pool Disease. Am. J. Hematol. 2018, 93, 1192–1193. [Google Scholar] [CrossRef]
- Kunishima, S.; Matsushita, T.; Kojima, T.; Sako, M.; Kimura, F.; Jo, E.K.; Inoue, C.; Kamiya, T.; Saito, H. Immunofluorescence analysis of neutrophil nonmuscle myosin heavy chain-A in MYH9 disorders: Association of subcellular localization with MYH9 mutations. Lab. Investig. 2003, 83, 115–122. [Google Scholar] [CrossRef]
- Kitamura, K.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Furukawa, K.; Miyano, S.; Ogawa, S.; Kunishima, S. Normal neutrophil myosin IIA localization in an immunofluorescence analysis can rule out MYH9 disorders. J. Thromb. Haemost. 2013, 11, 2071–2073. [Google Scholar] [CrossRef]
- Steinberg-Shemer, O.; Tamary, H. Gray platelet syndrome mimicking atypical autoimmune lymphoproliferative syndrome: The key is in the blood smear. Blood 2018, 131, 2737. [Google Scholar] [CrossRef]
- Kunishima, S.; Kitamura, K.; Yasutomi, M.; Kobayashi, R. Diagnostic biomarker for ACTN1 macrothrombocytopenia. Blood 2015, 126, 2525–2526. [Google Scholar] [CrossRef]
- Antony-Debré, I.; Bluteau, D.; Itzykson, R.; Baccini, V.; Renneville, A.; Boehlen, F.; Morabito, M.; Droin, N.; Deswarte, C.; Chang, Y.; et al. MYH10 protein expression in platelets as a biomarker of RUNX1 and FLI1 alterations. Blood 2012, 120, 2719–2722. [Google Scholar] [CrossRef]
- Greinacher, A.; Eekels, J.J.M. Diagnosis of hereditary platelet disorders in the era of next-generation sequencing: “primum non nocere”. J. Thromb. Haemost. 2019, 17, 551–554. [Google Scholar] [CrossRef]
- ACMG Board of Directors. Points to consider for informed consent for genome/exome sequencing. Genet. Med. 2013, 15, 748–749. [Google Scholar] [CrossRef]
- Baschin, M.; Blumentritt, C.; Holzhauer, S.; Karastaneva, A.; Seidel, M.; Greinacher, A. Diagnosis of ETV6-mutation Related Thrombocytopenia by Immunofluorescence Microscopy. Hämostaseologie 2019, 39, SY09-5. [Google Scholar]
- Zaninetti, C.; Santini, V.; Tiniakou, M.; Barozzi, S.; Savoia, A.; Pecci, A. Inherited thrombocytopenia caused by ANKRD26 mutations misdiagnosed and treated as myelodysplastic syndrome: Report on two cases. J. Thromb. Haemost. 2017, 15, 2388–2392. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaninetti, C.; Greinacher, A. Diagnosis of Inherited Platelet Disorders on a Blood Smear. J. Clin. Med. 2020, 9, 539. https://doi.org/10.3390/jcm9020539
Zaninetti C, Greinacher A. Diagnosis of Inherited Platelet Disorders on a Blood Smear. Journal of Clinical Medicine. 2020; 9(2):539. https://doi.org/10.3390/jcm9020539
Chicago/Turabian StyleZaninetti, Carlo, and Andreas Greinacher. 2020. "Diagnosis of Inherited Platelet Disorders on a Blood Smear" Journal of Clinical Medicine 9, no. 2: 539. https://doi.org/10.3390/jcm9020539
APA StyleZaninetti, C., & Greinacher, A. (2020). Diagnosis of Inherited Platelet Disorders on a Blood Smear. Journal of Clinical Medicine, 9(2), 539. https://doi.org/10.3390/jcm9020539