Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research





 ,
, 
Abstract
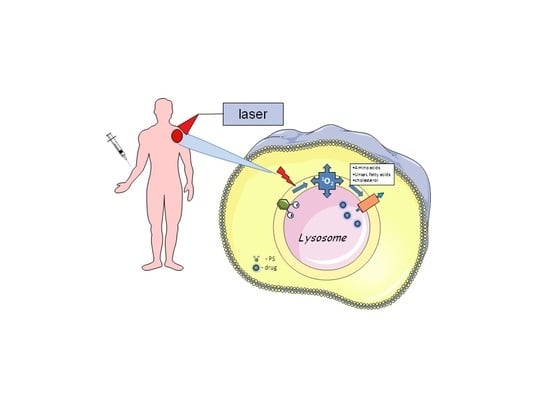
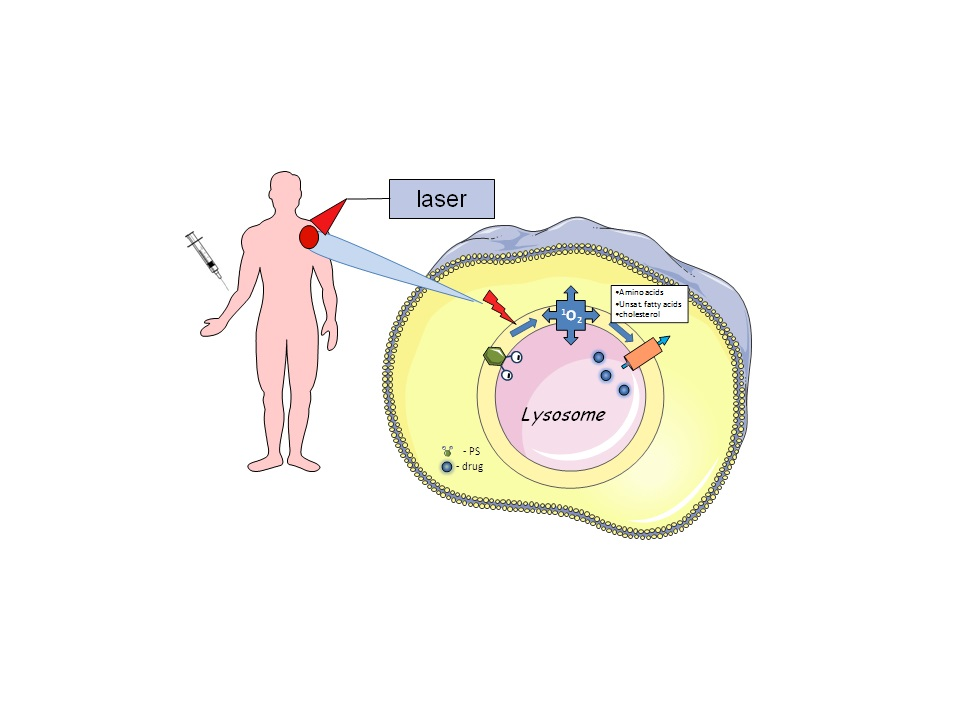{kind=link}
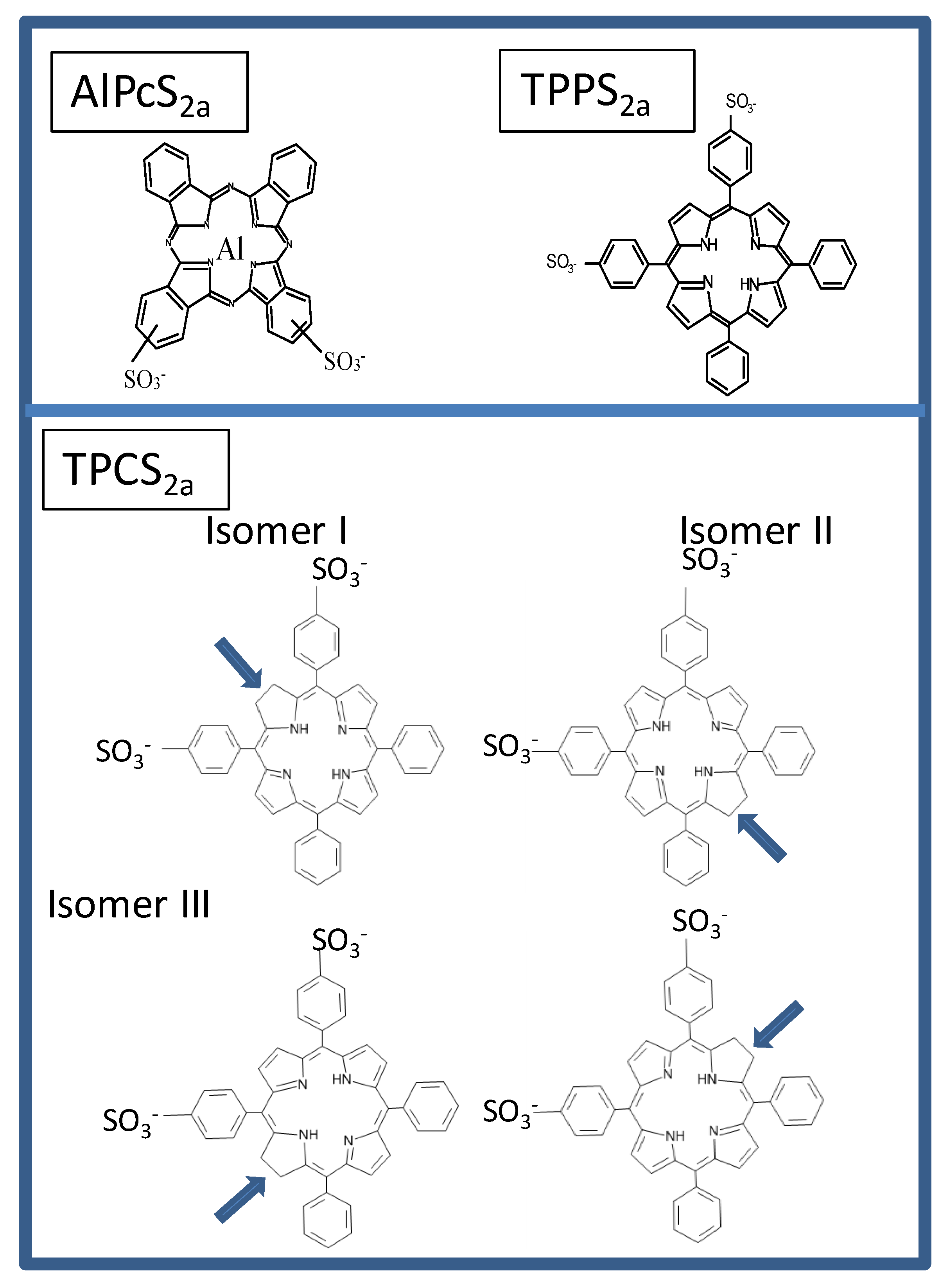{kind=link}
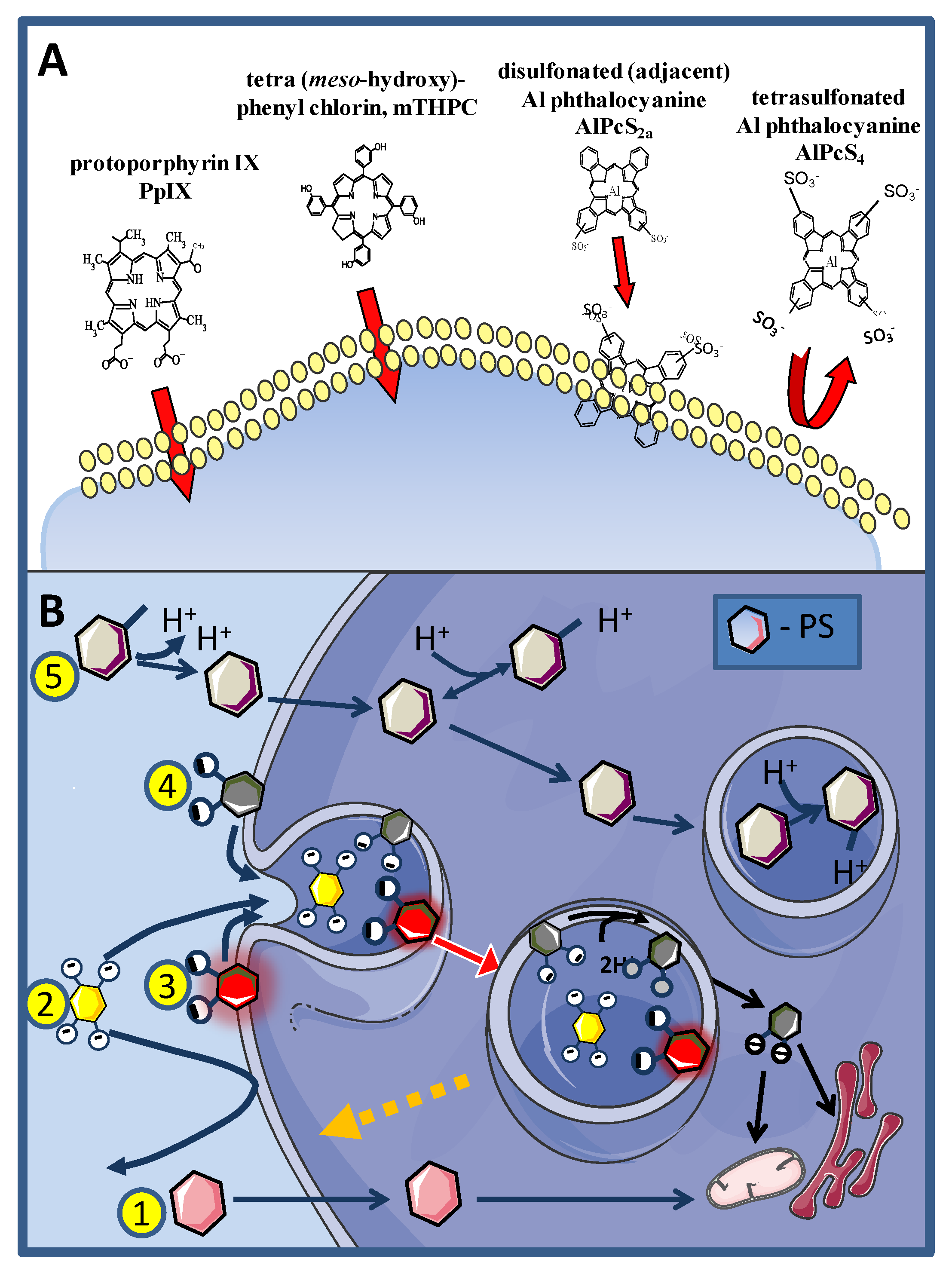{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Photodynamic Therapy
1.1.1. Higher Evidence-Based Studies Required
1.1.2. The Cancer Margin
1.2. Photochemical Internalisation (PCI)
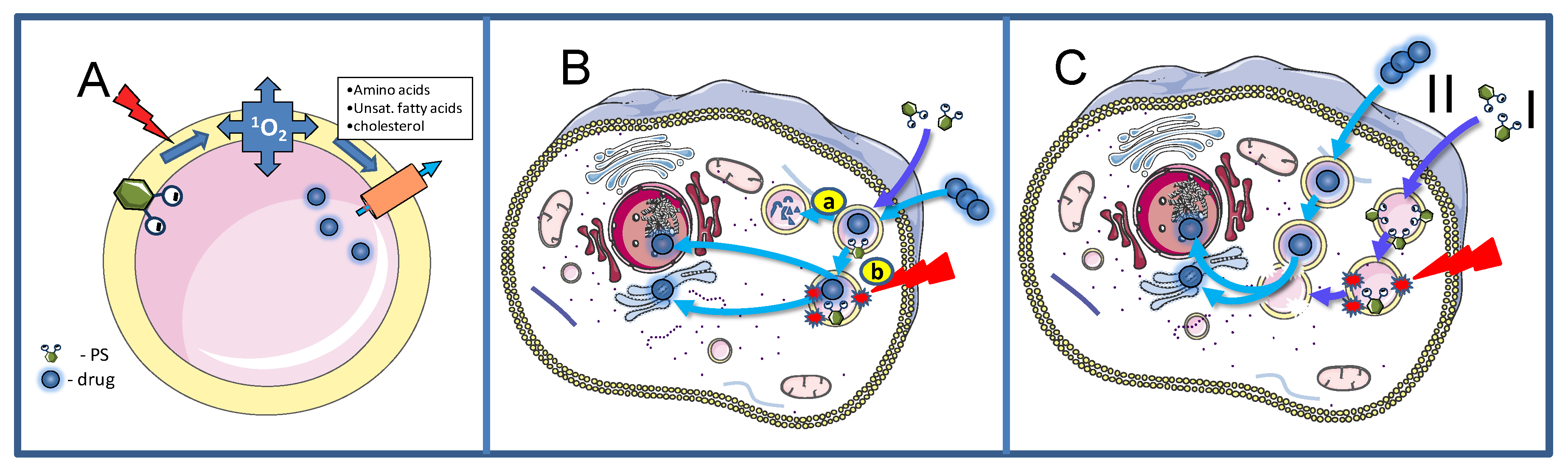
2. Basic Mechanisms
2.1. Cellular Uptake Mechanisms of PSs for PCI
2.2. Treatment Effects on Endocytic Vesicles
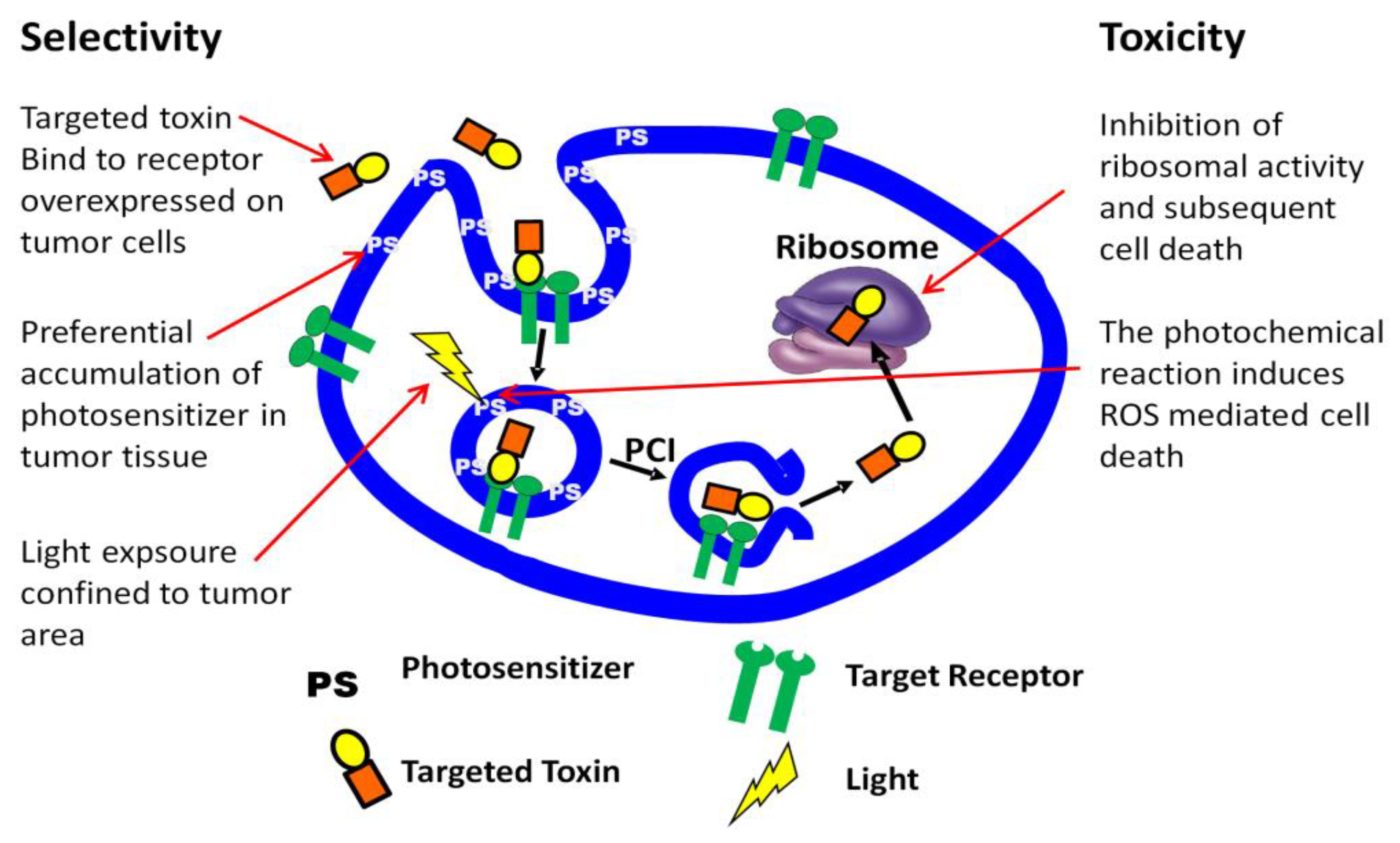
3. PCI Stimulated Activation of Targeted Toxins
3.1. Targeted Toxins
3.2. PCI Enhancement of Targeted Toxins
3.3. PCI of Recombinant Targeting Protein Toxins; Current Status
4. Nanoparticle-Mediated PCI as a Powerful Anticancer Drug Delivery System
5. PCI Delivery of Oligo- and Polynucleotides
5.1. PCI Mediated Oligonucleotide Delivery
5.2. PCI for the Delivery of mRNA
6. PCI for Gene Delivery
6.1. PCI for Plasmid Delivery
6.2. Viral Gene Delivery Systems
7. Considerations Regarding the Use of PCI to Enhance the Therapeutic Effect of Nucleic Acids
8. PCI in Antitumor Immunity and Cancer Vaccination
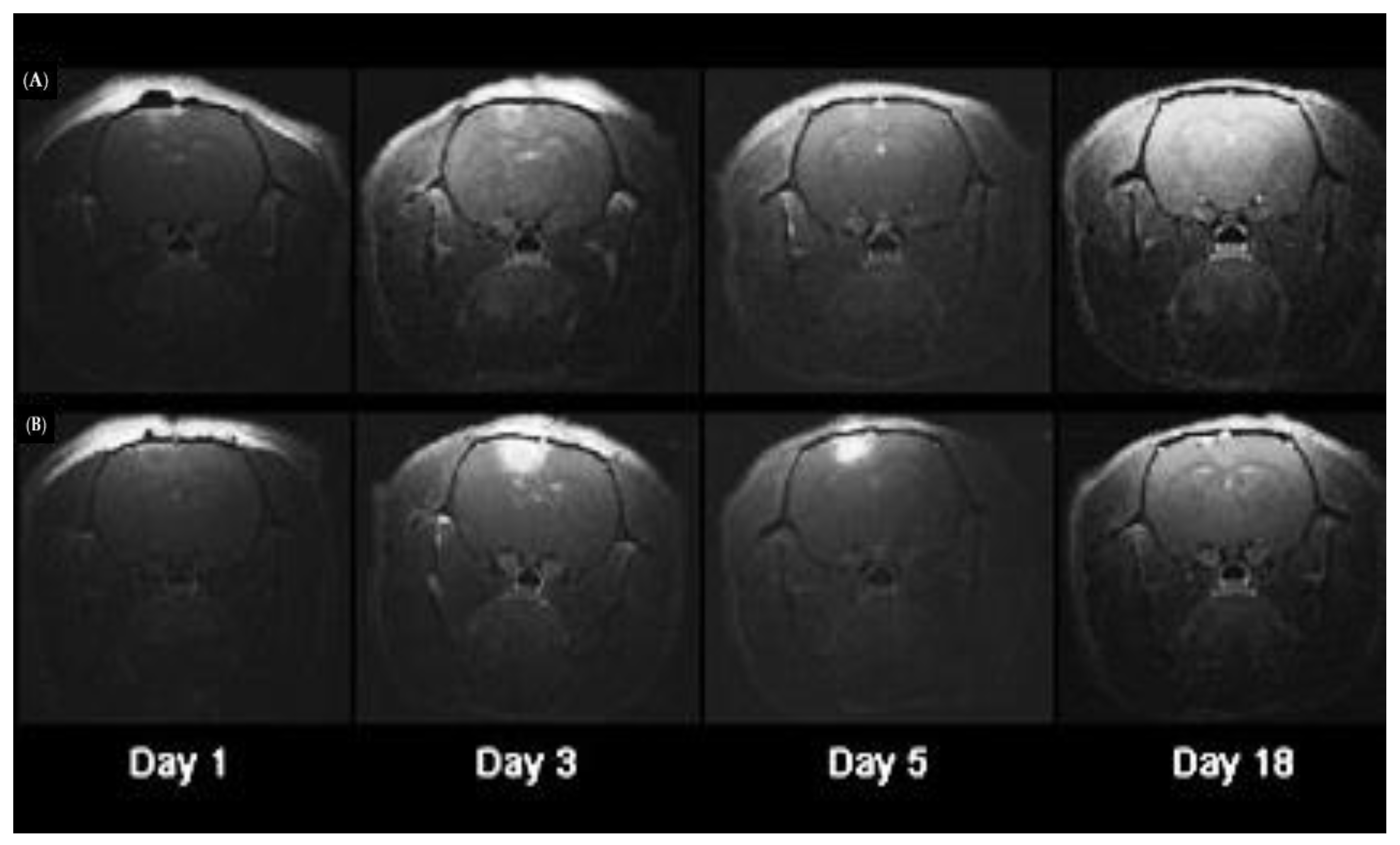
9. PCI Approaches to Treatment of Brain Tumors
9.1. Background
9.2. Potential Uses of PCI for the Treatment of Brain Tumors
9.2.1. Bypassing the BBB
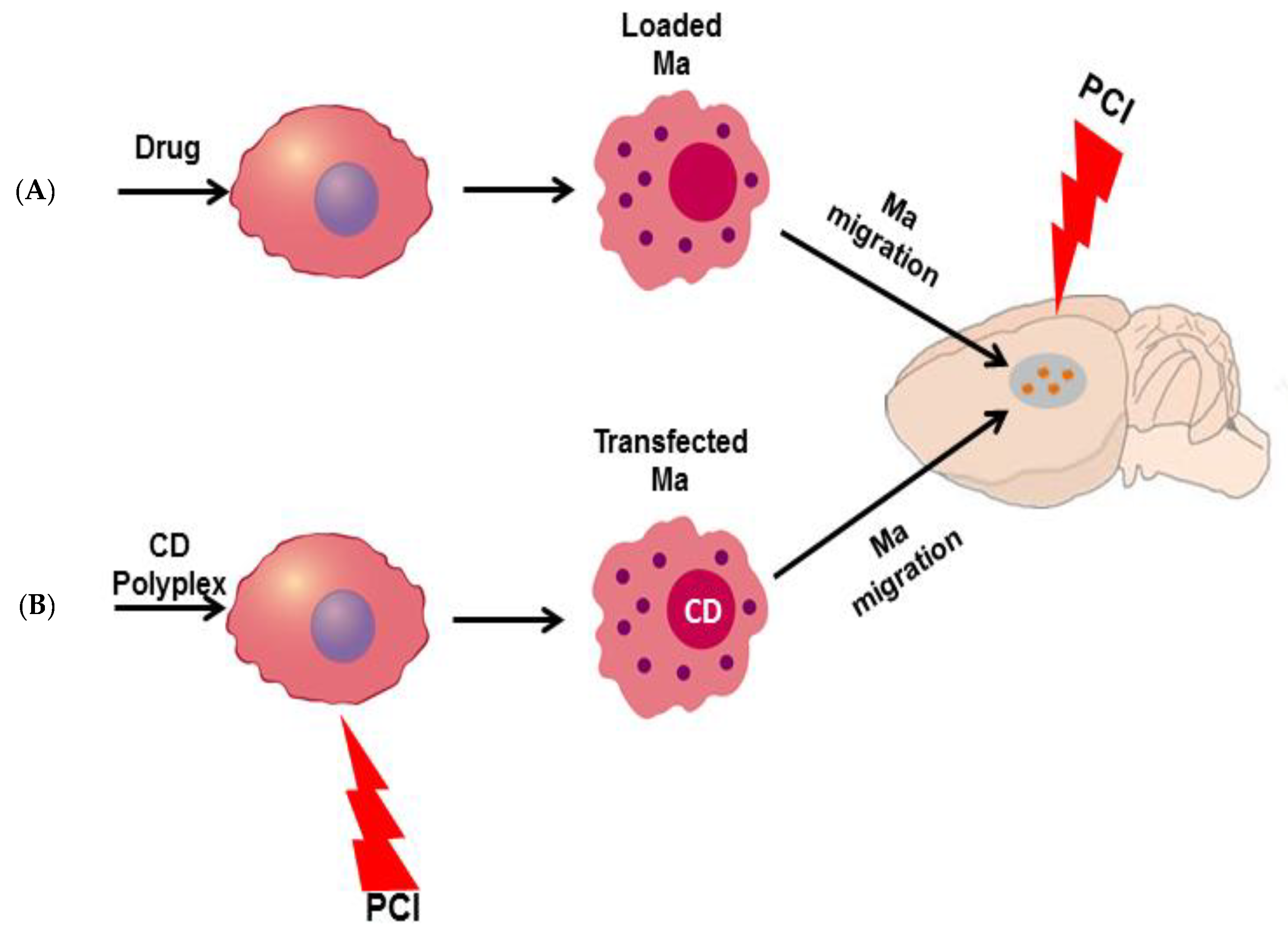
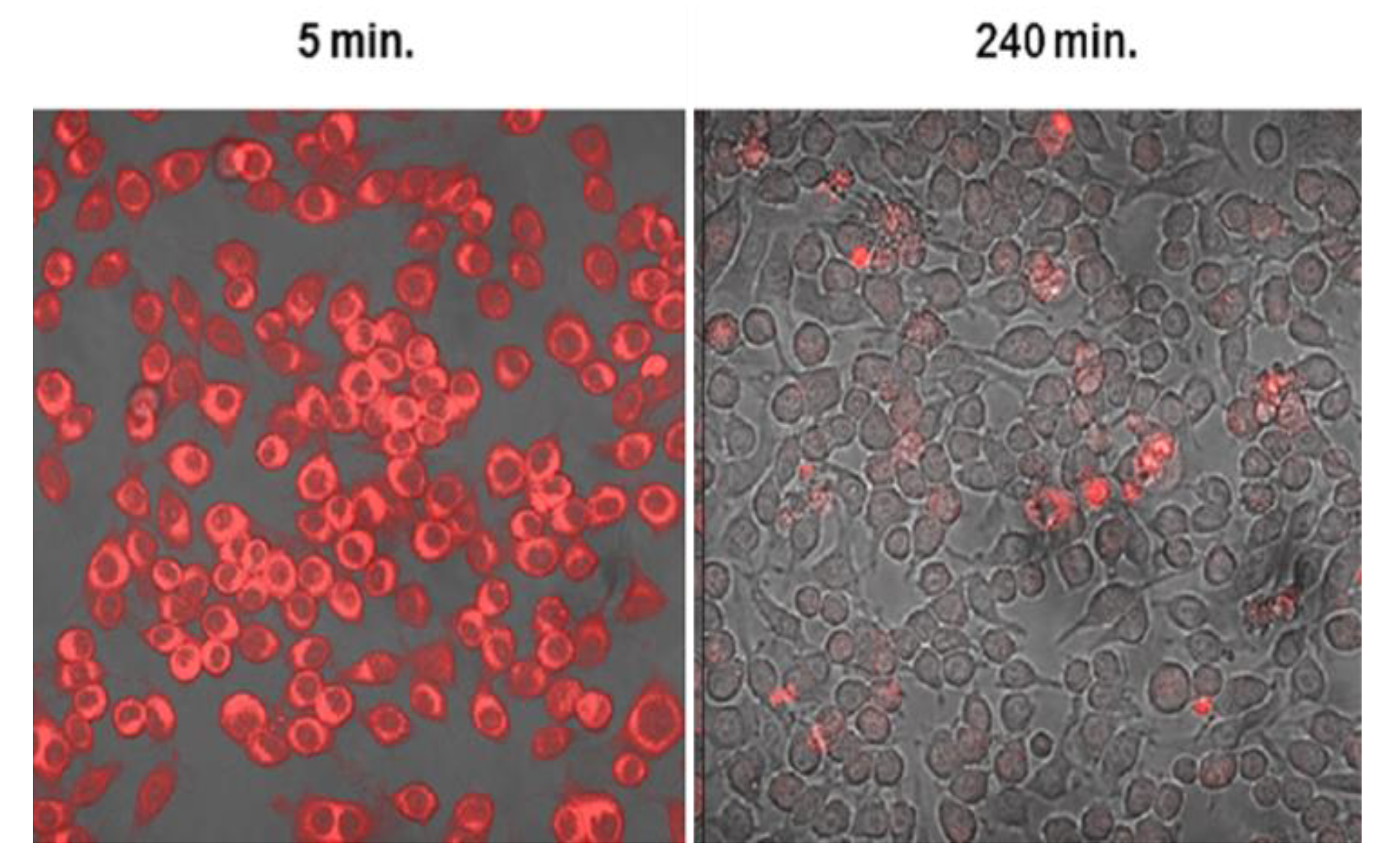
9.2.2. Macrophages as Delivery Vectors for PCI Mediated Chemotherapy
Drug Release from Loaded Ma
Ma-Mediated PCI-Enhanced Gene-Directed Enzyme Prodrug Therapy
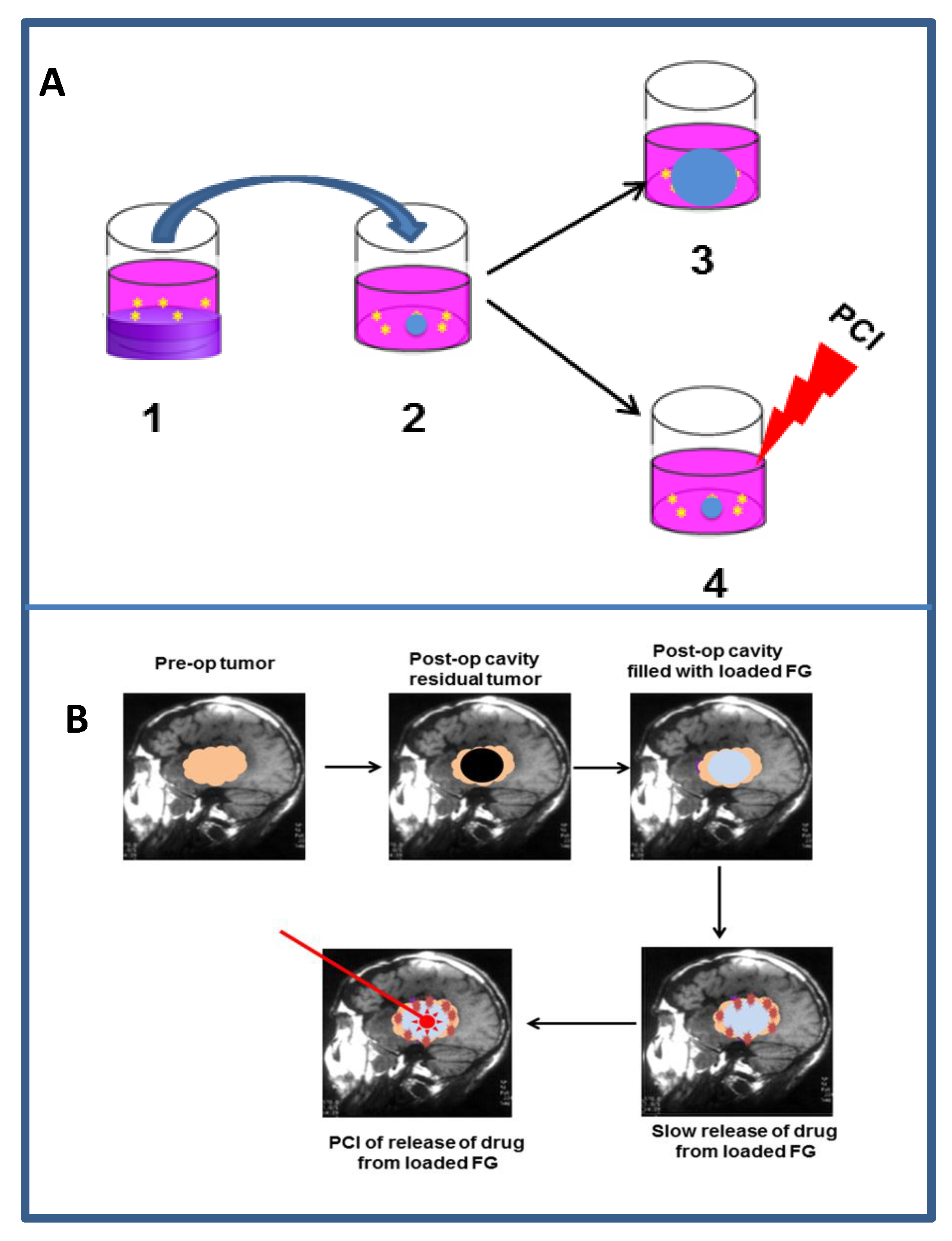
9.2.3. Localized Drug Delivery
9.2.4. Light Delivery to the Resection Cavity
9.2.5. Metronomic PCI
9.2.6. PCI for Treatment of Brain Tumors-Conclusion
10. Clinical Experience and Future Potential of PCI
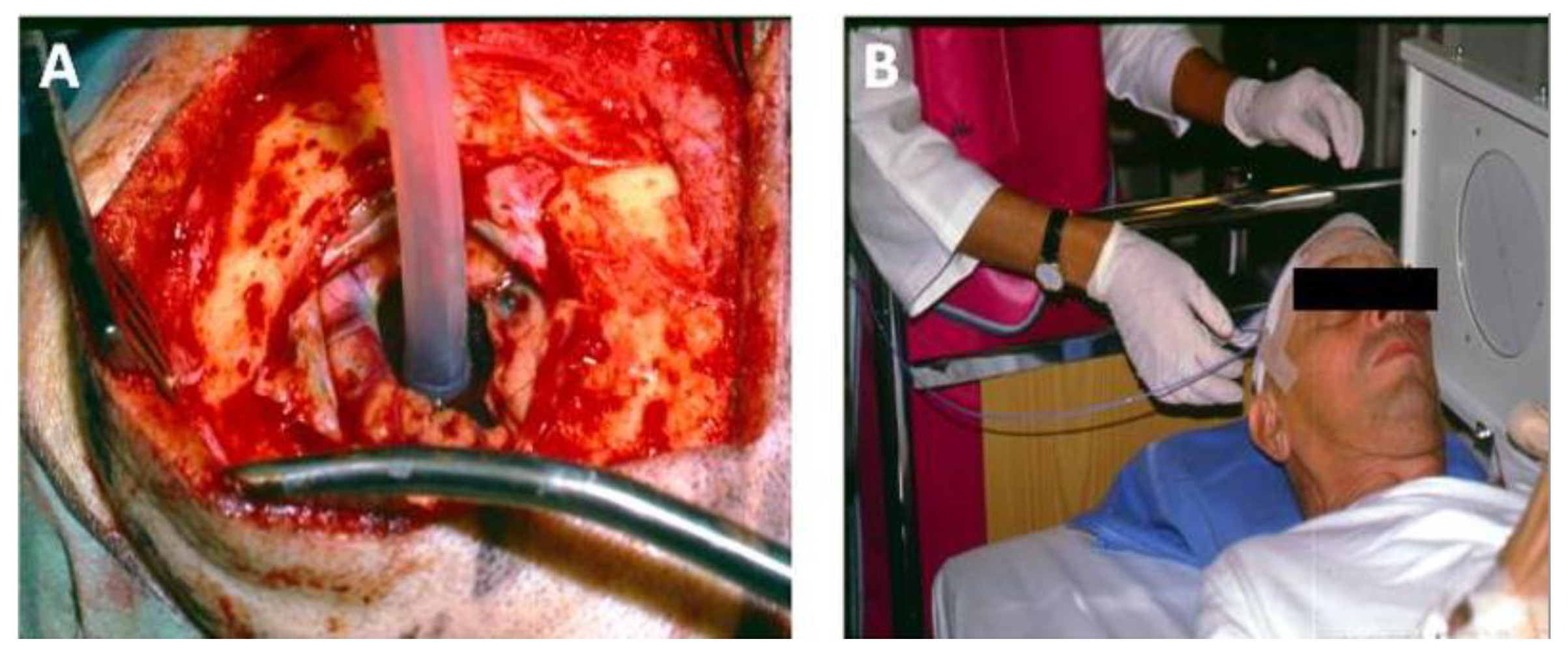
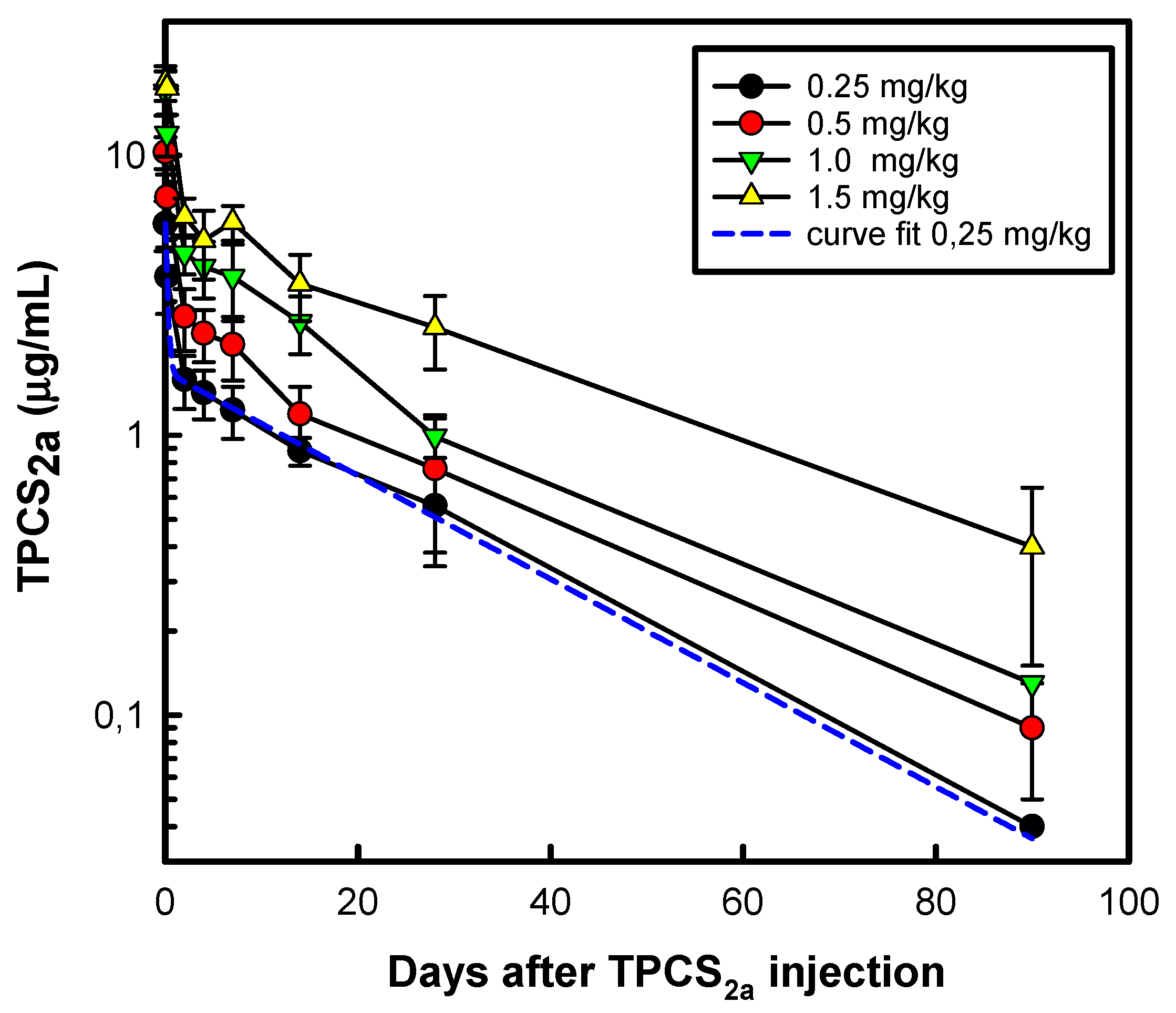
10.1. Summary of the First-in-Human PCI Trial
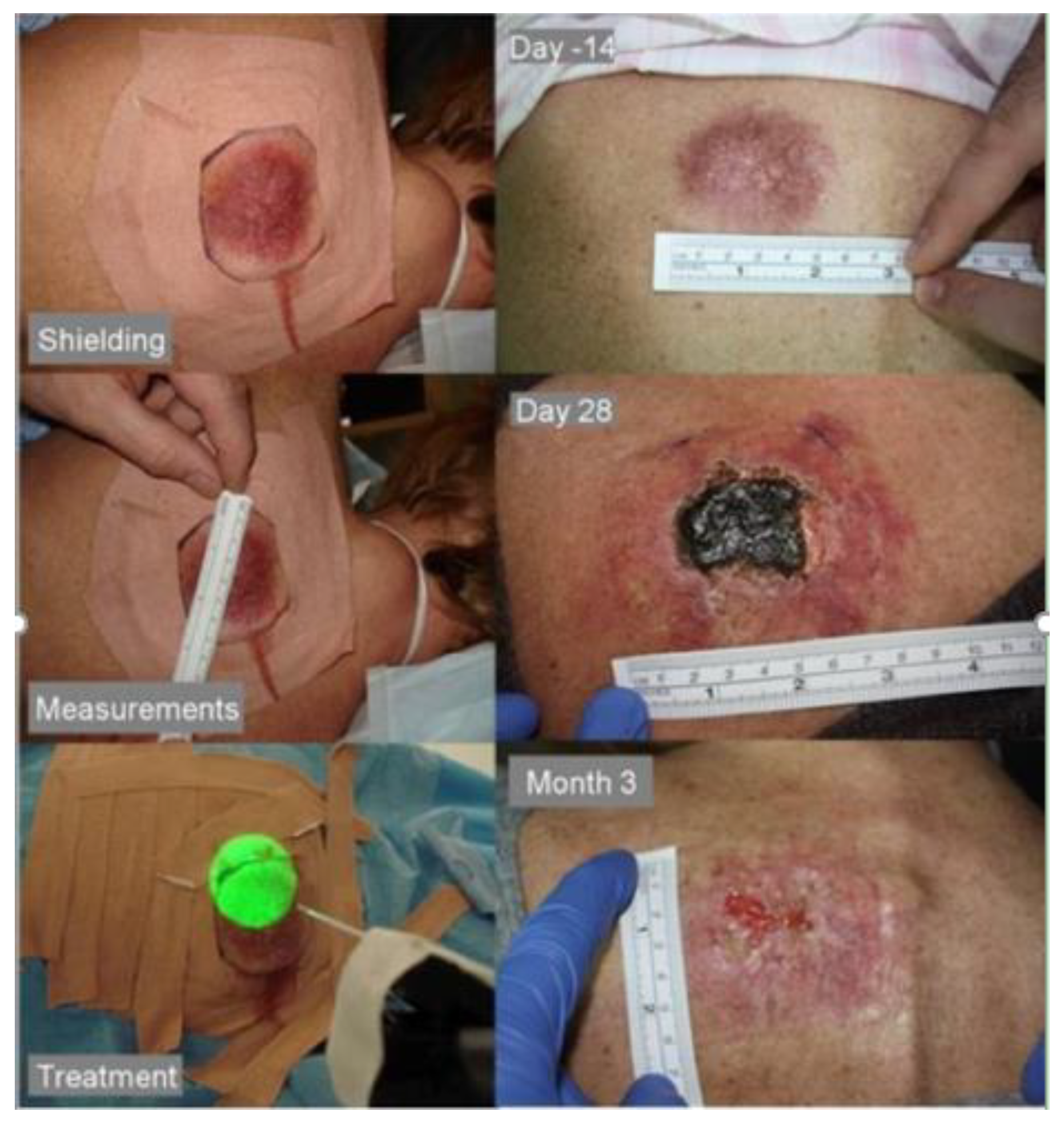
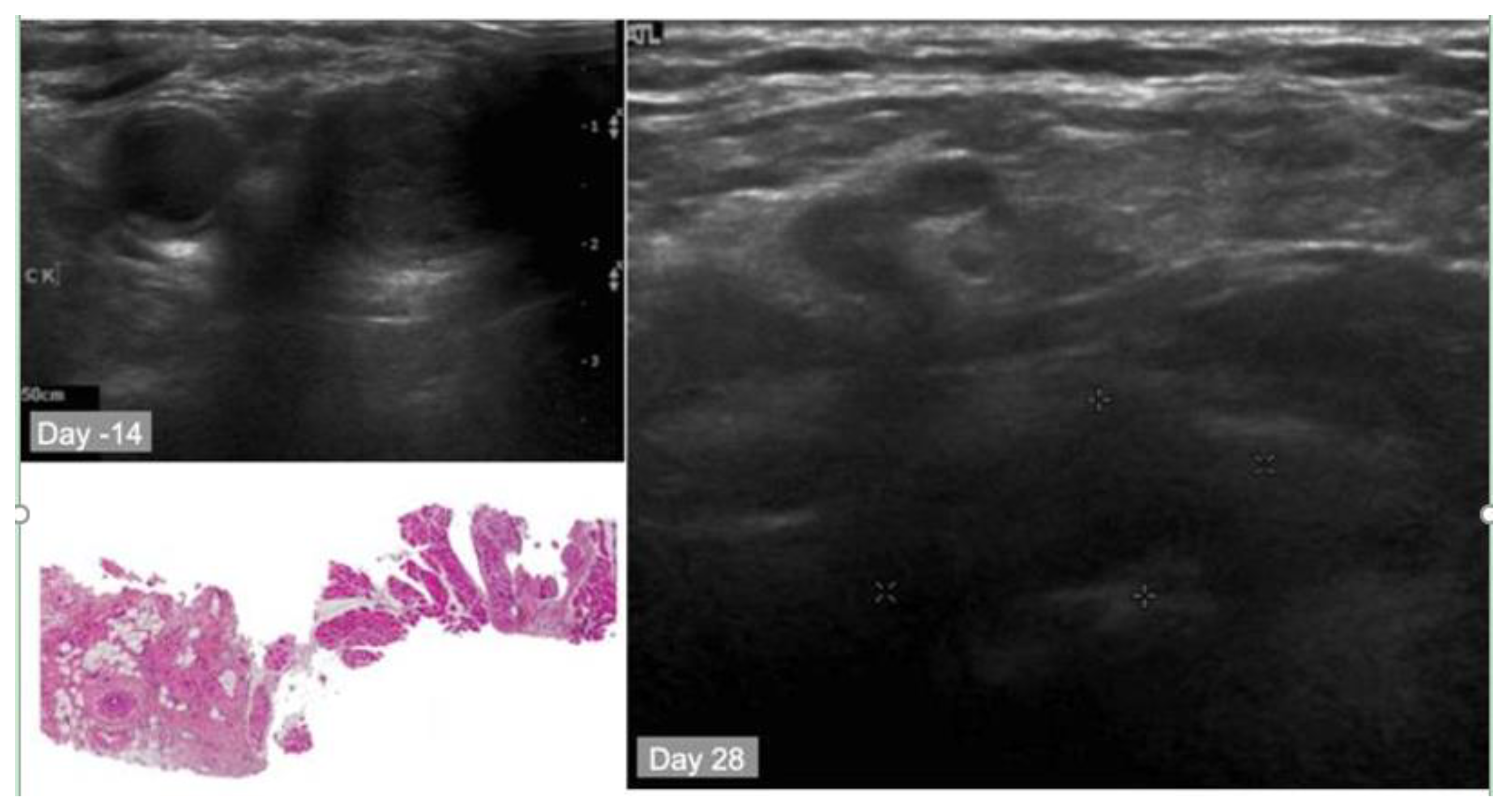
10.2. Case Study
11. Clinical Efficacy of PDT vs. PCI
12. Summary
13. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Ree, A.H.; Redalen, K.R. Personalized radiotherapy: Concepts, biomarkers and trial design. Br. J. Radiol. 2015, 88, 20150009. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Tannock, I.F. Repopulation of cancer cells during therapy: An important cause of treatment failure. Nat. Rev. Cancer 2005, 5, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, B.L.; Francis, P.A.; Parker, B.S.; Anderson, R.L. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat. Rev. Drug Discov. 2012, 11, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. 2013, 41, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.V.; Lienau, P.; Fricker, G.; Reichel, A. Quantitation of lysosomal trapping of basic lipophilic compounds using in vitro assays and in silico predictions based on the determination of the full pH profile of the endo-/lysosomal system in rat hepatocytes. Drug Metab. Dispos. 2019, 47, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Jerjes, W.; Upile, T.; Akram, S.; Hopper, C. The surgical palliation of advanced head and neck cancer using photodynamic therapy. Clin. Oncol. (R. Coll. Radiol.) 2010, 22, 785–791. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kedzierska, E.; Knap-Czop, K.; Kotlinska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Lan, M.; Zhao, S.; Liu, W.; Lee, C.S.; Zhang, W.; Wang, P. Photosensitizers for Photodynamic Therapy. Adv. Healthc. Mater. 2019, 8, e1900132. [Google Scholar] [CrossRef]
- Baskaran, R.; Lee, J.; Yang, S.G. Clinical development of photodynamic agents and therapeutic applications. Biomater. Res. 2018, 22, 25. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, C.; Figueiro Longo, J.P.; Azevedo, R.B.; Zhang, H.; Muehlmann, L.A. An updated overview on the development of new photosensitizers for anticancer photodynamic therapy. Acta Pharm. Sin. B 2018, 8, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Lucky, S.S.; Soo, K.C.; Zhang, Y. Nanoparticles in photodynamic therapy. Chem. Rev. 2015, 115, 1990–2042. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R.; Newman, E.L. On the mechanism of the tumour-localising effect in photodynamic therapy. J. Photochem. Photobiol. B 1994, 23, 3–8. [Google Scholar] [CrossRef]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and photodynamic therapy: Mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef]
- Jerjes, W.; Upile, T.; Hamdoon, Z.; Abbas, S.; Akram, S.; Mosse, C.A.; Morley, S.; Hopper, C. Photodynamic therapy: The minimally invasive surgical intervention for advanced and/or recurrent tongue base carcinoma. Lasers Surg. Med. 2011, 43, 283–292. [Google Scholar] [CrossRef]
- Jerjes, W.; Upile, T.; Hamdoon, Z.; Alexander Mosse, C.; Morcos, M.; Hopper, C. Photodynamic therapy outcome for T1/T2 N0 oral squamous cell carcinoma. Lasers Surg. Med. 2011, 43, 463–469. [Google Scholar] [CrossRef]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef]
- Abbas, S.; Jerjes, W.; Upile, T.; Vaz, F.; Hopper, C. The palliative role of PDT in recurrent advanced nasopharyngeal carcinoma: Case series. Photodiagnosis Photodyn. Ther. 2012, 9, 142–147. [Google Scholar] [CrossRef]
- de Visscher, S.A.; Melchers, L.J.; Dijkstra, P.U.; Karakullukcu, B.; Tan, I.B.; Hopper, C.; Roodenburg, J.L.; Witjes, M.J. mTHPC-mediated photodynamic therapy of early stage oral squamous cell carcinoma: A comparison to surgical treatment. Ann. Surg. Oncol. 2013, 20, 3076–3082. [Google Scholar] [CrossRef]
- Peng, W.; de Bruijn, H.S.; Farrell, E.; Sioud, M.; Mashayekhi, V.; Oliveira, S.; van Dam, G.M.; Roodenburg, J.L.N.; Witjes, M.J.H.; Robinson, D.J. Epidermal growth factor receptor (EGFR) density may not be the only determinant for the efficacy of EGFR-targeted photoimmunotherapy in human head and neck cancer cell lines. Lasers Surg. Med. 2018, 50, 513–522. [Google Scholar] [CrossRef]
- Fernandes, S.R.G.; Fernandes, R.; Sarmento, B.; Pereira, P.M.R.; Tome, J.P.C. Photoimmunoconjugates: Novel synthetic strategies to target and treat cancer by photodynamic therapy. Org. Biomol. Chem. 2019, 17, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, C.; Kruger, C.A.; Abrahamse, H. Photodynamic therapy for metastatic melanoma treatment: A review. Technol. Cancer Res. Treat. 2018, 17, 1533033818791795. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.E.; Hopper, C.; Speight, P.M.; MacRobert, A.J.; Bown, S.G. Photodynamic therapy of malignant and premalignant lesions in patients with ‘field cancerization’ of the oral cavity. J. Laryngol. Otol. 1993, 107, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Jerjes, W.; Hamdoon, Z.; Hopper, C. Photodynamic therapy in the management of basal cell carcinoma: Retrospective evaluation of outcome. Photodiagnosis Photodyn. Ther. 2017, 19, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Jerjes, W.; Hamdoon, Z.; Abdulkareem, A.A.; Hopper, C. Photodynamic therapy in the management of actinic keratosis: Retrospective evaluation of outcome. Photodiagnosis Photodyn. Ther. 2017, 17, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Kaalhus, O.; Sivam, G.; Berg, K. 5-Aminolevulinic acid-based photochemical internalization of the immunotoxin MOC31-gelonin generates synergistic cytotoxic effects in vitro. Photochem. Photobiol. 2001, 74, 303–310. [Google Scholar] [CrossRef]
- Wilson, B.C.; Olivo, M.; Singh, G. Subcellular localization of Photofrin(R) and aminolevulinic acid and photodynamic cross-resistance in vitro in radiation-induced fibrosarcoma cells sensitive or resistant to photofrin-mediated photodynamic therapy. Photochem. Photobiol. 1997, 65, 166–176. [Google Scholar] [CrossRef]
- Ji, Z.; Yang, G.; Vasovic, V.; Cunderlikova, B.; Suo, Z.; Nesland, J.M.; Peng, Q. Subcellular localization pattern of protoporphyrin IX is an important determinant for its photodynamic efficiency of human carcinoma and normal cell lines. J. Photochem. Photobiol. B 2006, 84, 213–220. [Google Scholar] [CrossRef]
- Story, W.; Sultan, A.A.; Bottini, G.; Vaz, F.; Lee, G.; Hopper, C. Strategies of airway management for head and neck photo-dynamic therapy. Lasers Surg. Med. 2013, 45, 370–376. [Google Scholar] [CrossRef]
- Green, B.; Cobb, A.R.; Hopper, C. Photodynamic therapy in the management of lesions of the head and neck. Br. J. Oral Maxillofac. Surg. 2013, 51, 283–287. [Google Scholar] [CrossRef]
- Betz, C.S.; Rauschning, W.; Stranadko, E.P.; Riabov, M.V.; Volgin, V.N.; Albrecht, V.; Nifantiev, N.E.; Hopper, C. Long-term outcomes following Foscan(R)-PDT of basal cell carcinomas. Lasers Surg. Med. 2012, 44, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Jerjes, W.; Upile, T.; Hamdoon, Z.; Mosse, C.A.; Akram, S.; Morley, S.; Hopper, C. Interstitial PDT for vascular anomalies. Lasers Surg. Med. 2011, 43, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D. Photodynamic therapy: A brief history. J. Clin. Med. 2019, 8, 1581. [Google Scholar] [CrossRef] [PubMed]
- Yanovsky, R.L.; Bartenstein, D.W.; Rogers, G.S.; Isakoff, S.J.; Chen, S.T. Photodynamic therapy for solid tumors: A review of the literature. Photodermatol. Photoimmunol. Photomed. 2019, 35, 295–303. [Google Scholar] [CrossRef]
- Nhembe, F.; Jerjes, W.; Upile, T.; Hamdoon, Z.; Hopper, C. Chondrosarcoma of the hyoid treated with interstitial photodynamic therapy: Case study. Photodiagnosis Photodyn. Ther. 2009, 6, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Jerjes, W.; Upile, T.; Hamdoon, Z.; Nhembe, F.; Bhandari, R.; Mackay, S.; Shah, P.; Mosse, C.A.; Brookes, J.A.; Morley, S.; et al. Ultrasound-guided photodynamic therapy for deep seated pathologies: Prospective study. Lasers Surg. Med. 2009, 41, 612–621. [Google Scholar] [CrossRef]
- Borgia, F.; Giuffrida, R.; Caradonna, E.; Vaccaro, M.; Guarneri, F.; Cannavo, S.P. Early and Late Onset Side Effects of Photodynamic Therapy. Biomedicines 2018, 6, 12. [Google Scholar] [CrossRef]
- Mayor, P.C.; Lele, S. Photodynamic therapy in gynecologic malignancies: A review of the Roswell Park Cancer Institute experience. Cancers 2016, 8, 88. [Google Scholar] [CrossRef]
- Fayter, D.; Corbett, M.; Heirs, M.; Fox, D.; Eastwood, A. A systematic review of photodynamic therapy in the treatment of pre-cancerous skin conditions, Barrett’s oesophagus and cancers of the biliary tract, brain, head and neck, lung, oesophagus and skin. Health Technol. Assess. 2010, 14, 1–288. [Google Scholar] [CrossRef]
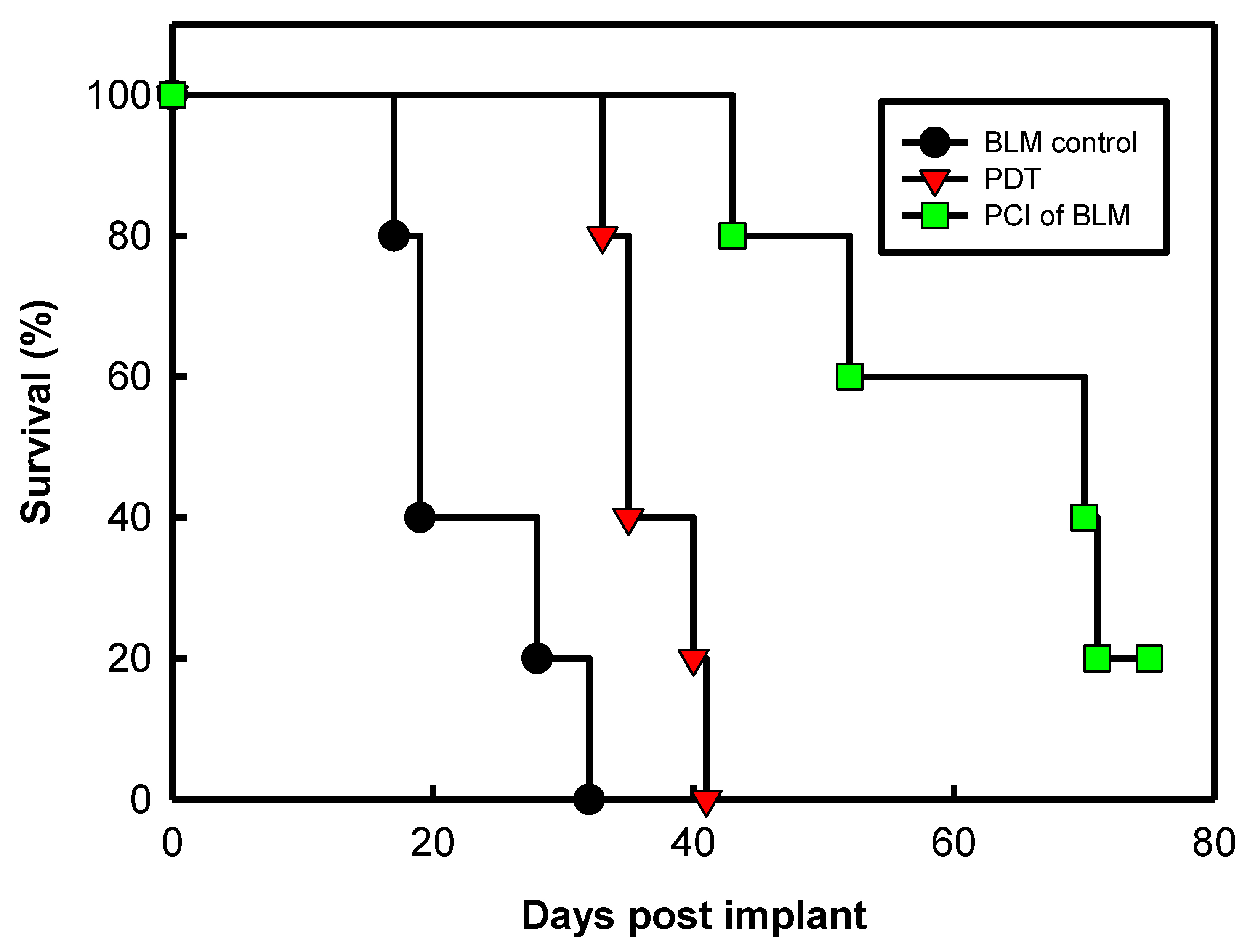
- Norum, O.J.; Gaustad, J.V.; Angell-Petersen, E.; Rofstad, E.K.; Peng, Q.; Giercksky, K.E.; Berg, K. Photochemical internalization of bleomycin is superior to photodynamic therapy due to the therapeutic effect in the tumor periphery. Photochem. Photobiol. 2009, 85, 740–749. [Google Scholar] [CrossRef]
- Norum, O.J.; Giercksky, K.E.; Berg, K. Photochemical internalization as an adjunct to marginal surgery in a human sarcoma model. Photochem. Photobiol. Sci. 2009, 8, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Fremstedal, A.S.; Skarpen, E.; Peng, Q.; Mohamedali, K.A.; Eng, M.S.; Cheung, L.H.; Rosenblum, M.G.; Waltenberger, J.; Berg, K. Light-enhanced VEGF121/rGel: A tumor targeted modality with vascular and immune-mediated efficacy. J. Control. Release 2018, 288, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Berstad, M.B.; Cheung, L.H.; Berg, K.; Peng, Q.; Fremstedal, A.S.; Patzke, S.; Rosenblum, M.G.; Weyergang, A. Design of an EGFR-targeting toxin for photochemical delivery: In vitro and in vivo selectivity and efficacy. Oncogene 2015, 34, 5582–5592. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Cheung, L.H.; Rosenblum, M.G.; Mohamedali, K.A.; Peng, Q.; Waltenberger, J.; Berg, K. Photochemical internalization augments tumor vascular cytotoxicity and specificity of VEGF(121)/rGel fusion toxin. J. Control. Release 2014, 180, 1–9. [Google Scholar] [CrossRef]
- Jerjes, W.; Upile, T.; Radhi, H.; Hopper, C. Photodynamic therapy vs. photochemical internalization: The surgical margin. Head Neck Oncol. 2011, 3, 53. [Google Scholar] [CrossRef]
- Selbo, P.K.; Weyergang, A.; Hogset, A.; Norum, O.J.; Berstad, M.B.; Vikdal, M.; Berg, K. Photochemical internalization provides time- and space-controlled endolysosomal escape of therapeutic molecules. J. Control. Release 2010, 148, 2–12. [Google Scholar] [CrossRef]
- Berg, K.; Berstad, M.; Prasmickaite, L.; Weyergang, A.; Selbo, P.K.; Hedfors, I.; Hogset, A. Photochemical internalization: A new tool for gene and oligonucleotide delivery. Top. Curr. Chem. 2010, 296, 251–281. [Google Scholar]
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; Tjelle, T.E.; Sandvig, K.; Moan, D.; Gaudernack, G.; Fodstad, Ø.; Kjolsrud, S.; Anholt, H.; et al. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183. [Google Scholar]
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. In vivo documentation of photochemical internalization, a novel approach to site specific cancer therapy. Int. J. Cancer 2001, 92, 761–766. [Google Scholar] [CrossRef]
- Berg, K.; Dietze, A.; Kaalhus, O.; Hogset, A. Site-specific drug delivery by photochemical internalization enhances the antitumor effect of bleomycin. Clin. Cancer Res. 2005, 11, 8476–8485. [Google Scholar] [CrossRef]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.; Angell-Petersen, E.; Hogset, A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem. Photobiol. Sci. 2011, 10, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Lilletvedt, M.; Smistad, G.; Tonnesen, H.H.; Hogset, A.; Kristensen, S. Solubilization of the novel anionic amphiphilic photosensitizer TPCS2a by nonionic Pluronic block copolymers. Eur. J. Pharm. Sci. 2011, 43, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Lilletvedt, M.; Tonnesen, H.H.; Hogset, A.; Nardo, L.; Kristensen, S. Physicochemical characterization of the photosensitizers TPCS2a and TPPS2a 1. Spectroscopic evaluation of drug--solvent interactions. Pharmazie 2010, 65, 588–595. [Google Scholar] [PubMed]
- Lilletvedt, M.; Tonnesen, H.H.; Hogset, A.; Sande, S.A.; Kristensen, S. Evaluation of physicochemical properties and aggregation of the photosensitizers TPCS2a and TPPS2a in aqueous media. Pharmazie 2011, 66, 325–333. [Google Scholar]
- Tovsen, M.L.; Tho, I.; Tonnesen, H.H. Viscosity reduction of isotonic solutions of the photosensitizer TPCS2a by cyclodextrin complexation. Drug Dev. Ind. Pharm. 2018, 44, 261–265. [Google Scholar] [CrossRef]
- Wang, J.T.; Berg, K.; Hogset, A.; Bown, S.G.; MacRobert, A.J. Photophysical and photobiological properties of a sulfonated chlorin photosensitiser TPCS(2a) for photochemical internalisation (PCI). Photochem. Photobiol. Sci. 2013, 12, 519–526. [Google Scholar] [CrossRef]
- Cuccato, N.; Nardo, L.; Kristensen, S.; Hjorth Tonnesen, H.; Lilletvedt Tovsen, M. Solubilization of the chlorin TPCS2a in the presence of Pluronic((R)) F127/Tween 80 mixtures. Pharm. Dev. Technol. 2019, 24, 513–520. [Google Scholar] [CrossRef]
- Zhang, X.; de Boer, L.; Heiliegers, L.; Man-Bovenkerk, S.; Selbo, P.K.; Drijfhout, J.W.; Hogset, A.; Zaat, S.A.J. Photochemical internalization enhances cytosolic release of antibiotic and increases its efficacy against staphylococcal infection. J. Control. Release 2018, 283, 214–222. [Google Scholar] [CrossRef]
- Dietze, A.; Engesaeter, B.; Berg, K. Transgene delivery and gelonin cytotoxicity enhanced by photochemical internalization in fibroblast-like synoviocytes (FLS) from rheumatoid arthritis patients. Photochem. Photobiol. Sci. 2005, 4, 341–347. [Google Scholar] [CrossRef]
- Rud, E.; Gederaas, O.; Hogset, A.; Berg, K. 5-aminolevulinic acid, but not 5-aminolevulinic acid esters, is transported into adenocarcinoma cells by system BETA transporters. Photochem. Photobiol. 2000, 71, 640–647. [Google Scholar] [CrossRef]
- Bohley, P.; Seglen, P.O. Proteases and proteolysis in the lysosome. Experientia 1992, 48, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.S. Photochemical internalization (PCI)—A technology for intracellular drug delivery. In Handbook in Porphyrin Science; Kadish, K.M., Smith, K.M., Eds.; World Scientific Publishing Co.: Singapore, 2016; Volume 43, pp. 245–300. [Google Scholar]
- Berg, K.; Moan, J. Lysosomes as photochemical targets. Int. J. Cancer 1994, 59, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Friberg, E.G.; Cunderlikova, B.; Pettersen, E.O.; Moan, J. pH effects on the cellular uptake of four photosensitizing drugs evaluated for use in photodynamic therapy of cancer. Cancer Lett. 2003, 195, 73–80. [Google Scholar] [CrossRef]
- Engesaeter, B.O.; Bonsted, A.; Berg, K.; Hogset, A.; Engebraten, O.; Fodstad, O.; Curiel, D.T.; Maelandsmo, G.M. PCI-enhanced adenoviral transduction employs the known uptake mechanism of adenoviral particles. Cancer Gene Ther. 2005, 12, 439–448. [Google Scholar] [CrossRef]
- Garaiova, Z.; Strand, S.P.; Reitan, N.K.; Lelu, S.; Storset, S.O.; Berg, K.; Malmo, J.; Folasire, O.; Bjorkoy, A.; Davies Cde, L. Cellular uptake of DNA-chitosan nanoparticles: The role of clathrin- and caveolae-mediated pathways. Int. J. Biol. Macromol. 2012, 51, 1043–1051. [Google Scholar] [CrossRef]
- Lamaze, C.; Tardif, N.; Dewulf, M.; Vassilopoulos, S.; Blouin, C.M. The caveolae dress code: Structure and signaling. Curr. Opin. Cell Biol. 2017, 47, 117–125. [Google Scholar] [CrossRef]
- Caruso, J.A.; Mathieu, P.A.; Reiners, J.J., Jr. Sphingomyelins suppress the targeted disruption of lysosomes/endosomes by the photosensitizer NPe6 during photodynamic therapy. Biochem. J. 2005, 392, 325–334. [Google Scholar] [CrossRef]
- Kerdous, R.; Heuvingh, J.; Bonneau, S. Photo-dynamic induction of oxidative stress within cholesterol-containing membranes: Shape transitions and permeabilization. Biochim. Biophys. Acta 2011, 1808, 2965–2972. [Google Scholar] [CrossRef]
- Moan, J.; Berg, K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem. Photobiol. 1991, 53, 549–553. [Google Scholar] [CrossRef]
- Lindig, B.A.; Rodgers, M.A.J.; Schaap, A.P. Determination of the lifetime of singlet oxygen in water-d2 using 9,10-anthracenedipropionic acid, a water-soluble probe. J. Am. Chem. Soc. 1980, 102, 5590–5593. [Google Scholar] [CrossRef]
- Rodgers, M.A. Time resolved studies of 1.27 micron luminescence from singlet oxygen generated in homogeneous and microheterogeneous fluids. Photochem. Photobiol. 1983, 37, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberg, B.; Anderson, J.L.; Foote, C.S. Kinetics and yield of singlet oxygen photosensitized by hypericin in organic and biological media. Photochem. Photobiol. 1998, 68, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Bronshtein, I.; Afri, M.; Weitman, H.; Frimer, A.A.; Smith, K.M.; Ehrenberg, B. Porphyrin depth in lipid bilayers as determined by iodide and parallax fluorescence quenching methods and its effect on photosensitizing efficiency. Biophys. J. 2004, 87, 1155–1164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berg, K.; Madslien, K.; Bommer, J.C.; Oftebro, R.; Winkelman, J.W.; Moan, J. Light induced relocalization of sulfonated meso- tetraphenylporphines in NHIK 3025 cells and effects of dose fractionation. Photochem. Photobiol. 1991, 53, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Moan, J.; Berg, K.; Anholt, H.; Madslien, K. Sulfonated aluminium phthalocyanines as sensitizers for photochemotherapy. Effects of small light doses on localization, dye fluorescence and photosensitivity in V79 cells. Int. J. Cancer 1994, 58, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Miki, S.; Kobayashi, S.; Haraguchi, T.; Nakata, E.; Hirakawa, K.; Sumita, K.; Watanabe, K.; Okazaki, S. The molecular mechanism of photochemical internalization of cell penetrating peptide-cargo-photosensitizer conjugates. Sci. Rep. 2015, 5, 18577. [Google Scholar] [CrossRef] [PubMed]
- Sakharov, D.V.; Elstak, E.D.; Chernyak, B.; Wirtz, K.W. Prolonged lipid oxidation after photodynamic treatment. Study with oxidation-sensitive probe C11-BODIPY581/591. FEBS Lett. 2005, 579, 1255–1260. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- de Bruin, K.G.; Fella, C.; Ogris, M.; Wagner, E.; Ruthardt, N.; Brauchle, C. Dynamics of photoinduced endosomal release of polyplexes. J. Control. Release 2008, 130, 175–182. [Google Scholar] [CrossRef]
- Yao, J.L.; Zhang, G.J. Loss of lysosomal integrity caused by the decrease of proton translocation in Methylene blue-mediated photosensitization. Biochim. Biophys. Acta Biomembr. 1996, 1284, 35–40. [Google Scholar] [CrossRef]
- Yao, J.L.; Zhang, G.J. Lysosomal destabilization via increased potassium ion permeability following photodamage. Biochim. Biophys. Acta Biomembr. 1997, 1323, 334–342. [Google Scholar] [CrossRef][Green Version]
- Wan, F.Y.; Zhang, G.J. Enhancement of lysosomal proton permeability induced by photooxidation of membrane thiol groups. Arch. Biochem. Biophys. 2002, 402, 268–274. [Google Scholar] [CrossRef]
- Wan, F.Y.; Yang, L.; Zhong, Y.G.; Zhu, W.; Wang, Y.N.; Zhang, G.J. Enhancement of lysosomal osmotic sensitivity induced by the photooxidation of membrane thiol groups. Photochem. Photobiol. 2002, 75, 134–139. [Google Scholar] [CrossRef]
- Prasmickaite, L.; Hogset, A.; Tjelle, T.E.; Olsen, V.M.; Berg, K. Role of endosomes in gene transfection mediated by photochemical internalisation (PCI). J. Gene Med. 2000, 2, 477–488. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.Y.; Pellois, J.P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Soe, T.H.; Nanjo, T.; Watanabe, K.; Ohtsuki, T. Relation of Photochemical internalization to heat, pH and Ca(2+) ions. Photochem. Photobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dondi, R.; Yaghini, E.; Tewari, K.M.; Wang, L.; Giuntini, F.; Loizidou, M.; MacRobert, A.J.; Eggleston, I.M. Flexible synthesis of cationic peptide-porphyrin derivatives for light-triggered drug delivery and photodynamic therapy. Org. Biomol. Chem. 2016, 14, 11488–11501. [Google Scholar] [CrossRef] [PubMed]
- Meerovich, I.; Muthukrishnan, N.; Johnson, G.A.; Erazo-Oliveras, A.; Pellois, J.P. Photodamage of lipid bilayers by irradiation of a fluorescently labeled cell-penetrating peptide. Biochim. Biophys. Acta 2014, 1840, 507–515. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, C.Y.; Wu, J.F.; Huang, Y.B.; Liu, C.B. Enhancement of TAT cell membrane penetration efficiency by dimethyl sulphoxide. J. Control. Release 2010, 143, 64–70. [Google Scholar] [CrossRef]
- Gramlich, P.A.; Remington, M.P.; Amin, J.; Betenbaugh, M.J.; Fishman, P.S. Tat-tetanus toxin fragment C: A novel protein delivery vector and its use with photochemical internalization. J. Drug Target. 2013, 21, 662–674. [Google Scholar] [CrossRef]
- Folini, M.; Berg, K.; Millo, E.; Villa, R.; Prasmickaite, L.; Daidone, M.G.; Benatti, U.; Zaffaroni, N. Photochemical internalization of a peptide nucleic acid targeting the catalytic subunit of human telomerase. Cancer Res. 2003, 63, 3490–3494. [Google Scholar] [PubMed]
- Watanabe, K.; Fujiwara, H.; Kitamatsu, M.; Ohtsuki, T. Photoinduced apoptosis using a peptide carrying a photosensitizer. Bioorg. Med. Chem. Lett. 2016, 26, 3115–3118. [Google Scholar] [CrossRef] [PubMed]
- Mora-Espi, I.; Barrios, L.; Ibanez, E.; Soriano, J.; Nogues, C. Membrane reorganization after photochemical internalization to release transferrin-biofunctionalized polystyrene microparticles. Sci. Rep. 2018, 8, 17617. [Google Scholar] [CrossRef]
- Prasmickaite, L.; Hogset, A.; Selbo, P.K.; Engesaeter, B.O.; Hellum, M.; Berg, K. Photochemical disruption of endocytic vesicles before delivery of drugs: A new strategy for cancer therapy. Br. J. Cancer 2002, 86, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Samplonius, D.F.; de Visscher, S.; Roodenburg, J.L.; Helfrich, W.; Witjes, M.J. Photochemical internalization (PCI)-mediated enhancement of bleomycin cytotoxicity by liposomal mTHPC formulations in human head and neck cancer cells. Lasers Surg. Med. 2014, 46, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Berg, K. Photochemical Internalization (PCI)—A technology for intracellular drug delivery. The bleomycin case. In Photodynamic Medicine. From Bench to Clinic; Kostron, H., Hasan, T., Eds.; RSC Publishing: Cambridge, UK, 2016; pp. 181–196. [Google Scholar]
- Kessel, D. Apoptosis, Paraptosis and Autophagy: Death and survival pathways associated with photodynamic therapy. Photochem. Photobiol. 2019, 95, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Domagala, A.; Fidyt, K.; Bobrowicz, M.; Stachura, J.; Szczygiel, K.; Firczuk, M. Typical and atypical inducers of lysosomal cell death: A promising anticancer strategy. Int. J. Mol. Sci. 2018, 19, 2256. [Google Scholar] [CrossRef]
- Caruso, J.A.; Mathieu, P.A.; Joiakim, A.; Leeson, B.; Kessel, D.; Sloane, B.F.; Reiners, J.J., Jr. Differential susceptibilities of murine hepatoma 1c1c7 and Tao cells to the lysosomal photosensitizer NPe6: Influence of aryl hydrocarbon receptor on lysosomal fragility and protease contents. Mol. Pharmacol. 2004, 65, 1016–1028. [Google Scholar] [CrossRef]
- Cesen, M.H.; Pegan, K.; Spes, A.; Turk, B. Lysosomal pathways to cell death and their therapeutic applications. Exp. Cell Res. 2012, 318, 1245–1251. [Google Scholar] [CrossRef]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 2012, 1824, 22–33. [Google Scholar] [CrossRef]
- Muthukrishnan, N.; Johnson, G.A.; Lim, J.; Simanek, E.E.; Pellois, J.P. TAT-mediated photochemical internalization results in cell killing by causing the release of calcium into the cytosol of cells. Biochim. Biophys. Acta 2012, 1820, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Vikdal, M.; Weyergang, A.; Selbo, P.K.; Berg, K. Vascular endothelial cells as targets for photochemical internalization (PCI). Photochem. Photobiol. 2013, 89, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, C.; Hopper, C.; MacRobert, A.J.; Phillips, J.B.; Woodhams, J.H. Could clinical photochemical internalisation be optimised to avoid neuronal toxicity? Int. J. Pharm. 2017, 528, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.A.; Jerjes, W.; Berg, K.; Hogset, A.; Mosse, C.A.; Hamoudi, R.; Hamdoon, Z.; Simeon, C.; Carnell, D.; Forster, M.; et al. Disulfonated tetraphenyl chlorin (TPCS2a)-induced photochemical internalisation of bleomycin in patients with solid malignancies: A phase 1, dose-escalation, first-in-man trial. Lancet Oncol. 2016, 17, 1217–1229. [Google Scholar] [CrossRef]
- Antignani, A.; Fitzgerald, D. Immunotoxins: The role of the toxin. Toxins 2013, 5, 1486–1502. [Google Scholar] [CrossRef]
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef]
- Dhillon, S. Moxetumomab Pasudotox: First global approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta 1993, 1154, 237–282. [Google Scholar] [CrossRef]
- Pirie, C.M.; Hackel, B.J.; Rosenblum, M.G.; Wittrup, K.D. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J. Biol. Chem. 2011, 286, 4165–4172. [Google Scholar] [CrossRef]
- Weyergang, A.; Selbo, P.K.; Berstad, M.E.; Bostad, M.; Berg, K. Photochemical internalization of tumor-targeted protein toxins. Lasers Surg. Med. 2011, 43, 721–733. [Google Scholar] [CrossRef]
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. Photochemical internalisation increases the cytotoxic effect of the immunotoxin MOC31-gelonin. Int. J. Cancer 2000, 87, 853–859. [Google Scholar] [CrossRef]
- Weyergang, A.; Selbo, P.K.; Berg, K. Photochemically stimulated drug delivery increases the cytotoxicity and specificity of EGF-saporin. J. Control. Release 2006, 111, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Berstad, M.B.; Weyergang, A.; Berg, K. Photochemical internalization (PCI) of HER2-targeted toxins: Synergy is dependent on the treatment sequence. Biochim. Biophys. Acta 2012, 1820, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Yip, W.L.; Weyergang, A.; Berg, K.; Tonnesen, H.H.; Selbo, P.K. Targeted delivery and enhanced cytotoxicity of cetuximab-saporin by photochemical internalization in EGFR-positive cancer cells. Mol. Pharm. 2007, 4, 241–251. [Google Scholar] [CrossRef]
- Bostad, M.; Kausberg, M.; Weyergang, A.; Olsen, C.E.; Berg, K.; Hogset, A.; Selbo, P.K. Light-triggered, efficient cytosolic release of IM7-saporin targeting the putative cancer stem cell marker CD44 by photochemical internalization. Mol. Pharm. 2014, 11, 2764–2776. [Google Scholar] [CrossRef] [PubMed]
- Eng, M.S.; Kaur, J.; Prasmickaite, L.; Engesaeter, B.O.; Weyergang, A.; Skarpen, E.; Berg, K.; Rosenblum, M.G.; Maelandsmo, G.M.; Hogset, A.; et al. Enhanced targeting of triple-negative breast carcinoma and malignant melanoma by photochemical internalization of CSPG4-targeting immunotoxins. Photochem. Photobiol. Sci. 2018, 17, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Stratford, E.W.; Bostad, M.; Castro, R.; Skarpen, E.; Berg, K.; Hogset, A.; Myklebost, O.; Selbo, P.K. Photochemical internalization of CD133-targeting immunotoxins efficiently depletes sarcoma cells with stem-like properties and reduces tumorigenicity. Biochim. Biophys. Acta 2013, 1830, 4235–4243. [Google Scholar] [CrossRef]
- Brenner, B.M.; Hostetter, T.H.; Humes, H.D. Glomerular permselectivity: Barrier function based on discrimination of molecular size and charge. Am. J. Physiol. 1978, 234, F455–F460. [Google Scholar] [CrossRef]
- Maack, T.; Johnson, V.; Kau, S.T.; Figueiredo, J.; Sigulem, D. Renal filtration, transport, and metabolism of low-molecular-weight proteins: A review. Kidney Int. 1979, 16, 251–270. [Google Scholar] [CrossRef]
- Jain, R.K.; Baxter, L.T. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: Significance of elevated interstitial pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar]
- Jain, R.K. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990, 50 (Suppl. 3), 814s–819s. [Google Scholar] [PubMed]
- Selbo, P.K.; Rosenblum, M.G.; Cheung, L.H.; Zhang, W.; Berg, K. Multi-modality therapeutics with potent anti-tumor effects: Photochemical internalization enhances delivery of the fusion toxin scFvMEL/rGel. PLoS ONE 2009, 4, e6691. [Google Scholar] [CrossRef] [PubMed]
- Bull-Hansen, B.; Cao, Y.; Berg, K.; Skarpen, E.; Rosenblum, M.G.; Weyergang, A. Photochemical activation of the recombinant HER2-targeted fusion toxin MH3-B1/rGel; Impact of HER2 expression on treatment outcome. J. Control. Release 2014, 182, 58–66. [Google Scholar] [CrossRef]
- Bull-Hansen, B.; Berstad, M.B.; Berg, K.; Cao, Y.; Skarpen, E.; Fremstedal, A.S.; Rosenblum, M.G.; Peng, Q.; Weyergang, A. Photochemical activation of MH3-B1/rGel: A HER2-targeted treatment approach for ovarian cancer. Oncotarget 2015, 6, 12436–12451. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Stern, D.F. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim. Biophys. Acta 1994, 1198, 165–184. [Google Scholar] [CrossRef]
- Veenendaal, L.M.; Jin, H.; Ran, S.; Cheung, L.; Navone, N.; Marks, J.W.; Waltenberger, J.; Thorpe, P.; Rosenblum, M.G. In vitro and in vivo studies of a VEGF121/rGelonin chimeric fusion toxin targeting the neovasculature of solid tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7866–7871. [Google Scholar] [CrossRef]
- Selbo, P.K.; Bostad, M.; Olsen, C.E.; Edwards, V.T.; Hogset, A.; Weyergang, A.; Berg, K. Photochemical internalisation, a minimally invasive strategy for light-controlled endosomal escape of cancer stem cell-targeting therapeutics. Photochem. Photobiol. Sci. 2015, 14, 1433–1450. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Sauer, A.M.; Schlossbauer, A.; Ruthardt, N.; Cauda, V.; Bein, T.; Brauchle, C. Role of endosomal escape for disulfide-based drug delivery from colloidal mesoporous silica evaluated by live-cell imaging. Nano Lett. 2010, 10, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.C.; Cabral, H.; Mi, P.; Toh, K.; Matsumoto, Y.; Liu, X.; Koori, H.; Kim, A.; Miyazaki, K.; Miura, Y.; et al. Light-induced cytosolic activation of reduction-sensitive camptothecin-loaded polymeric micelles for spatiotemporally controlled in vivo chemotherapy. ACS Nano 2014, 8, 11591–11602. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, J.; Zhao, J.; Shan, S.; Chu, C.C. A light-facilitated drug delivery system from a pseudo-protein/hyaluronic acid nanocomplex with improved anti-tumor effects. Nanoscale 2019, 11, 9987–10003. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Liu, G.; Hu, J.; Liu, S. Near-Infrared Light-Activated Photochemical internalization of reduction-responsive polyprodrug vesicles for synergistic photodynamic therapy and chemotherapy. Biomacromolecules 2017, 18, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.L.; Syu, W.J.; Nishiyama, N.; Kataoka, K.; Lai, P.S. Dendrimer phthalocyanine-encapsulated polymeric micelle-mediated photochemical internalization extends the efficacy of photodynamic therapy and overcomes drug-resistance in vivo. J. Control. Release 2011, 155, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Grabski, S.; Hoff, E.; Simon, S.M. Defective pH regulation of acidic compartments in human breast cancer cells (MCF-7) is normalized in adriamycin-resistant cells (MCF-7adr). Biochemistry 1996, 35, 2811–2817. [Google Scholar] [CrossRef]
- Altan, N.; Chen, Y.; Schindler, M.; Simon, S.M. Defective acidification in human breast tumor cells and implications for chemotherapy. J. Exp. Med. 1998, 187, 1583–1598. [Google Scholar] [CrossRef]
- Rajagopal, A.; Simon, S.M. Subcellular localization and activity of multidrug resistance proteins. Mol. Biol. Cell 2003, 14, 3389–3399. [Google Scholar] [CrossRef]
- Lou, P.J.; Lai, P.S.; Shieh, M.J.; Macrobert, A.J.; Berg, K.; Bown, S.G. Reversal of doxorubicin resistance in breast cancer cells by photochemical internalization. Int. J. Cancer 2006, 119, 2692–2698. [Google Scholar] [CrossRef]
- Chen, H.; Xiao, L.; Anraku, Y.; Mi, P.; Liu, X.; Cabral, H.; Inoue, A.; Nomoto, T.; Kishimura, A.; Nishiyama, N.; et al. Polyion complex vesicles for photoinduced intracellular delivery of amphiphilic photosensitizer. J. Am. Chem. Soc. 2014, 136, 157–163. [Google Scholar] [CrossRef]
- Park, H.; Park, W.; Na, K. Doxorubicin loaded singlet-oxygen producible polymeric micelle based on chlorine e6 conjugated pluronic F127 for overcoming drug resistance in cancer. Biomaterials 2014, 35, 7963–7969. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, G.; Manouras, T.; Vamvakaki, M.; Argitis, P. Harnessing photochemical internalization with dual degradable nanoparticles for combinatorial photo-chemotherapy. Nat. Commun. 2014, 5, 3623. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Hu, J.; Zhang, G.; Liu, S. Rationally engineering phototherapy modules of eosin-conjugated responsive polymeric nanocarriers via intracellular endocytic pH gradients. Bioconjug. Chem. 2015, 26, 1328–1338. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Z.; Wang, Z.; Li, W.; Ju, E.; Yan, Z.; Liu, Z.; Ren, J.; Qu, X. A graphitic hollow carbon nitride nanosphere as a novel photochemical internalization agent for targeted and stimuli-responsive cancer therapy. Nanoscale 2016, 8, 12570–12578. [Google Scholar] [CrossRef] [PubMed]
- McAtee, C.O.; Berkebile, A.R.; Elowsky, C.G.; Fangman, T.; Barycki, J.J.; Wahl, J.K., 3rd; Khalimonchuk, O.; Naslavsky, N.; Caplan, S.; Simpson, M.A. Hyaluronidase Hyal1 increases tumor cell proliferation and motility through accelerated vesicle trafficking. J. Biol. Chem. 2015, 290, 13144–13156. [Google Scholar] [CrossRef]
- Yaghini, E.; Dondi, R.; Edler, K.J.; Loizidou, M.; MacRobert, A.J.; Eggleston, I.M. Codelivery of a cytotoxin and photosensitiser via a liposomal nanocarrier: A novel strategy for light-triggered cytosolic release. Nanoscale 2018, 10, 20366–20376. [Google Scholar] [CrossRef]
- Galanou, M.C.; Theodossiou, T.A.; Tsiourvas, D.; Sideratou, Z.; Paleos, C.M. Interactive transport, subcellular relocation and enhanced phototoxicity of hypericin encapsulated in guanidinylated liposomes via molecular recognition. Photochem. Photobiol. 2008, 84, 1073–1083. [Google Scholar] [CrossRef]
- Martinez-Jothar, L.; Beztsinna, N.; van Nostrum, C.F.; Hennink, W.E.; Oliveira, S. Selective Cytotoxicity to HER2 positive breast cancer cells by saporin-loaded nanobody-targeted polymeric nanoparticles in combination with photochemical internalization. Mol. Pharm. 2019, 16, 1633–1647. [Google Scholar] [CrossRef]
- Cheng, H.; Fan, J.H.; Zhao, L.P.; Fan, G.L.; Zheng, R.R.; Qiu, X.Z.; Yu, X.Y.; Li, S.Y.; Zhang, X.Z. Chimeric peptide engineered exosomes for dual-stage light guided plasma membrane and nucleus targeted photodynamic therapy. Biomaterials 2019, 211, 14–24. [Google Scholar] [CrossRef]
- Luo, Z.; Li, M.; Zhou, M.; Li, H.; Chen, Y.; Ren, X.; Dai, Y. O2-evolving and ROS-activable nanoparticles for treatment of multi-drug resistant cancer by combination of photodynamic therapy and chemotherapy. Nanomedicine 2019, 19, 49–57. [Google Scholar] [CrossRef]
- Tian, J.; Xu, L.; Xue, Y.; Jiang, X.; Zhang, W. Enhancing Photochemical Internalization of DOX through a porphyrin-based amphiphilic block copolymer. Biomacromolecules 2017, 18, 3992–4001. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Lovell, J.F.; Chen, J.; Ng, K.; Cao, W.; Ding, L.; Zhang, Z.; Zheng, G. Cytosolic delivery of LDL nanoparticle cargo using photochemical internalization. Photochem. Photobiol. Sci. 2011, 10, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Theodossiou, T.A.; Goncalves, A.R.; Yannakopoulou, K.; Skarpen, E.; Berg, K. Photochemical internalization of tamoxifens transported by a “Trojan-horse” nanoconjugate into breast-cancer cell lines. Angew. Chem. Int. Ed. Engl. 2015, 54, 4885–4889. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Park, W.; Na, K. Gadolinium-chelate nanoparticle entrapped human mesenchymal stem cell via photochemical internalization for cancer diagnosis. Biomaterials 2015, 36, 90–97. [Google Scholar] [CrossRef]
- Sonawane, N.D.; Szoka, F.C., Jr.; Verkman, A.S. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [Google Scholar] [CrossRef]
- Turell, L.; Radi, R.; Alvarez, B. The thiol pool in human plasma: The central contribution of albumin to redox processes. Free Radic. Biol. Med. 2013, 65, 244–253. [Google Scholar] [CrossRef]
- Høgset, A.; Prasmickaite, L.; Tjelle, T.E.; Berg, K. Photochemical transfection: A new technology for light-induced, site-directed gene delivery. Hum. Gene Ther. 2000, 11, 869–880. [Google Scholar] [CrossRef]
- Shiraishi, T.; Nielsen, P.E. Photochemically enhanced cellular delivery of cell penetrating peptide-PNA conjugates. FEBS Lett. 2006, 580, 1451–1456. [Google Scholar] [CrossRef]
- Boe, S.; Hovig, E. Photochemically induced gene silencing using PNA-peptide conjugates. Oligonucleotides 2006, 16, 145–157. [Google Scholar] [CrossRef]
- Yarani, R.; Shiraishi, T.; Nielsen, P.E. Effective photo-enhancement of cellular activity of fluorophore-octaarginine antisense PNA conjugates correlates with singlet oxygen formation, endosomal escape and chromophore lipophilicity. Sci. Rep. 2018, 8, 638. [Google Scholar] [CrossRef]
- Boe, S.L.; Longva, A.S.; Hovig, E. A novel photosensitizer for light-controlled gene silencing. Nucleic Acid Ther. 2011, 21, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Boe, S.; Longva, A.S.; Hovig, E. Photochemically induced gene silencing using small interfering RNA molecules in combination with lipid carriers. Oligonucleotides 2007, 17, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Boe, S.L.; Longva, A.S.; Hovig, E. Cyclodextrin-containing polymer delivery system for light-directed siRNA gene silencing. Oligonucleotides 2010, 20, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, J.A.; Longva, A.S.; Hovig, E.; Boe, S.L. Evaluation of biodegradable peptide carriers for light-directed targeting. Nucleic Acid Ther. 2013, 23, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Boe, S.L.; Jorgensen, J.A.; Longva, A.S.; Lavelle, T.; Saeboe-Larssen, S.; Hovig, E. Light-controlled modulation of gene expression using polyamidoamine formulations. Nucleic Acid Ther. 2013, 23, 160–165. [Google Scholar] [CrossRef]
- Yuan, A.; Hu, Y.; Ming, X. Dendrimer conjugates for light-activated delivery of antisense oligonucleotides. RSC Adv. 2015, 5, 35195–35200. [Google Scholar] [CrossRef]
- Oliveira, S.; Fretz, M.M.; Hogset, A.; Storm, G.; Schiffelers, R.M. Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA. Biochim. Biophys. Acta 2007, 1768, 1211–1217. [Google Scholar] [CrossRef]
- Oliveira, S.; Hogset, A.; Storm, G.; Schiffelers, R.M. Delivery of siRNA to the target cell cytoplasm: Photochemical internalization facilitates endosomal escape and improves silencing efficiency, in vitro and in vivo. Curr. Pharm. Des. 2008, 14, 3686–3697. [Google Scholar] [CrossRef]
- Raemdonck, K.; Naeye, B.; Hogset, A.; Demeester, J.; De Smedt, S.C. Prolonged gene silencing by combining siRNA nanogels and photochemical internalization. J. Control. Release 2010, 145, 281–288. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Lammers, T.; Schiffelers, R.M.; van Steenbergen, M.J.; Hennink, W.E.; Storm, G. Gene silencing activity of siRNA polyplexes based on biodegradable polymers. Eur. J. Pharm. Biopharm. 2011, 77, 450–457. [Google Scholar] [CrossRef]
- Zhang, Z.; Jayakumar, M.K.G.; Zheng, X.; Shikha, S.; Zhang, Y.; Bansal, A.; Poon, D.J.J.; Chu, P.L.; Yeo, E.L.L.; Chua, M.L.K.; et al. Upconversion superballs for programmable photoactivation of therapeutics. Nat. Commun. 2019, 10, 4586. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, M.K.; Bansal, A.; Huang, K.; Yao, R.; Li, B.N.; Zhang, Y. Near-infrared-light-based nano-platform boosts endosomal escape and controls gene knockdown in vivo. ACS Nano 2014, 8, 4848–4858. [Google Scholar] [CrossRef] [PubMed]
- Ekineker, G.; Nguyen, C.; Bayir, S.; Dominguez Gil, S.; Isci, U.; Daurat, M.; Godefroy, A.; Raehm, L.; Charnay, C.; Oliviero, E.; et al. Phthalocyanine-based mesoporous organosilica nanoparticles: NIR photodynamic efficiency and siRNA photochemical internalization. Chem. Commun. (Camb.) 2019, 55, 11619–11622. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics--developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Kariko, K. Nucleoside modifications in RNA limit activation of 2’-5’-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338. [Google Scholar] [CrossRef]
- Lorenz, C.; Fotin-Mleczek, M.; Roth, G.; Becker, C.; Dam, T.C.; Verdurmen, W.P.; Brock, R.; Probst, J.; Schlake, T. Protein expression from exogenous mRNA: Uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011, 8, 627–636. [Google Scholar] [CrossRef]
- Boe, S.; Saeboe-Larssen, S.; Hovig, E. Light-induced gene expression using messenger RNA molecules. Oligonucleotides 2010, 20, 1–6. [Google Scholar] [CrossRef]
- Hobernik, D.; Bros, M. DNA Vaccines-How far from clinical use? Int. J. Mol. Sci. 2018, 19, 605. [Google Scholar] [CrossRef]
- Liu, M.A. A Comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef]
- Maurice-Duelli, A.; Ndoye, A.; Bouali, S.; Leroux, A.; Merlin, J.L. Enhanced cell growth inhibition following PTEN nonviral gene transfer using polyethylenimine and photochemical internalization in endometrial cancer cells. Technol. Cancer Res. Treat. 2004, 3, 459–465. [Google Scholar] [CrossRef]
- Shieh, M.J.; Peng, C.L.; Lou, P.J.; Chiu, C.H.; Tsai, T.Y.; Hsu, C.Y.; Yeh, C.Y.; Lai, P.S. Non-toxic phototriggered gene transfection by PAMAM-porphyrin conjugates. J. Control. Release 2008, 129, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Gaware, V.S.; Hakerud, M.; Leosson, K.; Jonsdottir, S.; Hogset, A.; Berg, K.; Masson, M. Tetraphenylporphyrin tethered chitosan based carriers for photochemical transfection. J. Med. Chem. 2013, 56, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, N.; Iriyama, A.; Jang, W.D.; Miyata, K.; Itaka, K.; Inoue, Y.; Takahashi, H.; Yanagi, Y.; Tamaki, Y.; Koyama, H.; et al. Light-induced gene transfer from packaged DNA enveloped in a dendrimeric photosensitizer. Nat. Mater. 2005, 4, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Arnida; Nishiyama, N.; Kanayama, N.; Jang, W.D.; Yamasaki, Y.; Kataoka, K. PEGylated gene nanocarriers based on block catiomers bearing ethylenediamine repeating units directed to remarkable enhancement of photochemical transfection. J. Control. Release 2006, 115, 208–215. [Google Scholar] [CrossRef]
- Xu, X.; Li, Y.; Liang, Q.; Song, Z.; Li, F.; He, H.; Wang, J.; Zhu, L.; Lin, Z.; Yin, L. Efficient gene delivery mediated by a helical polypeptide: Controlling the membrane activity via multivalency and light-assisted photochemical internalization (PCI). ACS Appl. Mater. Interfaces 2018, 10, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Lin, Q.M.; Zhang, L.M.; Liang, Y.Y.; Xue, W. A star-shaped porphyrin-arginine functionalized poly(l-lysine) copolymer for photo-enhanced drug and gene co-delivery. Biomaterials 2014, 35, 4357–4367. [Google Scholar] [CrossRef]
- Kloeckner, J.; Prasmickaite, L.; Hogset, A.; Berg, K.; Wagner, E. Photochemically enhanced gene delivery of EGF receptor-targeted DNA polyplexes. J. Drug Target. 2004, 12, 205–213. [Google Scholar] [CrossRef]
- Kloeckner, J.; Boeckle, S.; Persson, D.; Roedl, W.; Ogris, M.; Berg, K.; Wagner, E. DNA polyplexes based on degradable oligoethylenimine-derivatives: Combination with EGF receptor targeting and endosomal release functions. J. Control. Release 2006, 116, 115–122. [Google Scholar] [CrossRef]
- Lu, X.; Liu, L. Asymmetric polyplex-nanocapsules loaded with photosentisizer for light-assisted gene transfer. J. Photochem. Photobiol. B 2017, 174, 269–275. [Google Scholar] [CrossRef]
- Cho, H.; Cho, Y.W.; Kang, S.W.; Kwak, M.K.; Huh, K.M.; Bae, Y.H.; Kang, H.C. Tempo-spatial Activation of sequential quadruple stimuli for high gene expression of polymeric gene nanocomplexes. Mol. Pharm. 2017, 14, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hwang, H.S.; Na, K. TRAIL-secreting human mesenchymal stem cells engineered by a non-viral vector and photochemical internalization for pancreatic cancer gene therapy. Biomaterials 2018, 182, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, H.; Xu, X.; Wang, X.; Chen, Y.; Yin, L. Far-red light-mediated programmable anti-cancer gene delivery in cooperation with photodynamic therapy. Biomaterials 2018, 171, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Hellum, M.; Hogset, A.; Engesaeter, B.O.; Prasmickaite, L.; Stokke, T.; Wheeler, C.; Berg, K. Photochemically enhanced gene delivery with cationic lipid formulations. Photochem. Photobiol. Sci. 2003, 2, 407–411. [Google Scholar] [CrossRef]
- Prasmickaite, L.; Hogset, A.; Olsen, V.M.; Kaalhus, O.; Mikalsen, S.O.; Berg, K. Photochemically enhanced gene transfection increases the cytotoxicity of the herpes simplex virus thymidine kinase gene combined with ganciclovir. Cancer Gene Ther. 2004, 11, 514–523. [Google Scholar] [CrossRef][Green Version]
- Wang, F.; Zamora, G.; Sun, C.H.; Trinidad, A.; Chun, C.; Kwon, Y.J.; Berg, K.; Madsen, S.J.; Hirschberg, H. Increased sensitivity of glioma cells to 5-fluorocytosine following photo-chemical internalization enhanced nonviral transfection of the cytosine deaminase suicide gene. J. Neurooncol. 2014, 118, 29–37. [Google Scholar] [CrossRef]
- Ndoye, A.; Merlin, J.L.; Leroux, A.; Dolivet, G.; Erbacher, P.; Behr, J.P.; Berg, K.; Guillemin, F. Enhanced gene transfer and cell death following p53 gene transfer using photochemical internalisation of glucosylated PEI-DNA complexes. J. Gene Med. 2004, 6, 884–894. [Google Scholar] [CrossRef]
- Ndoye, A.; Bouali, S.; Dolivet, G.; Leroux, A.; Erbacher, P.; Behr, J.P.; Berg, K.; Guillemin, F.; Merlin, J.L. Sustained gene transfer and enhanced cell death following glucosylated-PEI-mediated p53 gene transfer with photochemical internalisation in p53-mutated head and neck carcinoma cells. Int. J. Oncol. 2004, 25, 1575–1581. [Google Scholar] [CrossRef]
- Ndoye, A.; Dolivet, G.; Hogset, A.; Leroux, A.; Fifre, A.; Erbacher, P.; Berg, K.; Behr, J.P.; Guillemin, F.; Merlin, J.L. Eradication of p53-mutated head and neck squamous cell carcinoma xenografts using nonviral p53 gene therapy and photochemical internalization. Mol. Ther. 2006, 13, 1156–1162. [Google Scholar] [CrossRef]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef]
- Leopold, P.L.; Ferris, B.; Grinberg, I.; Worgall, S.; Hackett, N.R.; Crystal, R.G. Fluorescent virions: Dynamic tracking of the pathway of adenoviral gene transfer vectors in living cells. Hum. Gene Ther. 1998, 9, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Hogset, A.; Engesaeter, B.O.; Prasmickaite, L.; Berg, K.; Fodstad, O.; Maelandsmo, G.M. Light-induced adenovirus gene transfer, an efficient and specific gene delivery technology for cancer gene therapy. Cancer Gene Ther. 2002, 9, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Høgset, A.B.K.; Mælandsmo, G.; Engesæter, B.; Prasmickatie, L. Photochemical Internalization for Virus-Mediated Molecule Delivery into the Cytosol. Eur.Pat.Appl. KR20087031843, 29 December 2008. [Google Scholar]
- Fasbender, A.; Zabner, J.; Chillon, M.; Moninger, T.O.; Puga, A.P.; Davidson, B.L.; Welsh, M.J. Complexes of adenovirus with polycationic polymers and cationic lipids increase the efficiency of gene transfer in vitro and in vivo. J. Biol. Chem. 1997, 272, 6479–6489. [Google Scholar] [CrossRef] [PubMed]
- Arcasoy, S.M.; Latoche, J.D.; Gondor, M.; Pitt, B.R.; Pilewski, J.M. Polycations increase the efficiency of adenovirus-mediated gene transfer to epithelial and endothelial cells in vitro. Gene Ther. 1997, 4, 32–38. [Google Scholar] [CrossRef]
- Bonsted, A.; Engesaeter, B.O.; Hogset, A.; Maelandsmo, G.M.; Prasmickaite, L.; Kaalhus, O.; Berg, K. Transgene expression is increased by photochemically mediated transduction of polycation-complexed adenoviruses. Gene Ther. 2004, 11, 152–160. [Google Scholar] [CrossRef][Green Version]
- Bonsted, A.; Engesaeter, B.O.; Hogset, A.; Berg, K. Photochemically enhanced adenoviral transduction in a multicellular environment. Photochem. Photobiol. Sci. 2006, 5, 411–421. [Google Scholar] [CrossRef]
- Bonsted, A.; Engesaeter, B.O.; Hogset, A.; Maelandsmo, G.M.; Prasmickaite, L.; D’Oliveira, C.; Hennink, W.E.; van Steenis, J.H.; Berg, K. Photochemically enhanced transduction of polymer-complexed adenovirus targeted to the epidermal growth factor receptor. J. Gene Med. 2006, 8, 286–297. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Hub, B.; Kartenbeck, J. Endocytosis of adeno-associated virus type 5 leads to accumulation of virus particles in the Golgi compartment. J. Virol. 2002, 76, 2340–2349. [Google Scholar] [CrossRef]
- Hansen, J.; Qing, K.; Srivastava, A. Adeno-associated virus type 2-mediated gene transfer: Altered endocytic processing enhances transduction efficiency in murine fibroblasts. J. Virol. 2001, 75, 4080–4090. [Google Scholar] [CrossRef]
- Bonsted, A.; Hogset, A.; Hoover, F.; Berg, K. Photochemical enhancement of gene delivery to glioblastoma cells is dependent on the vector applied. Anticancer Res. 2005, 25, 291–297. [Google Scholar]
- Hogset, A.; Prasmickaite, L.; Engesaeter, B.O.; Hellum, M.; Selbo, P.K.; Olsen, V.M.; Maelandsmo, G.M.; Berg, K. Light directed gene transfer by photochemical internalisation. Curr. Gene Ther. 2003, 3, 89–112. [Google Scholar] [CrossRef]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Boe, S.; Longva, A.S.; Hovig, E. Evaluation of various polyethylenimine formulations for light-controlled gene silencing using small interfering RNA molecules. Oligonucleotides 2008, 18, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Nielsen, P.E. Enhanced cellular delivery of cell-penetrating peptide-peptide nucleic acid conjugates by photochemical internalization. Methods Mol. Biol. 2011, 683, 391–397. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Håkerud, M.; Waeckerle-Men, Y.; Selbo, P.K.; Kundig, T.M.; Hogset, A.; Johansen, P. Intradermal photosensitisation facilitates stimulation of MHC class-I restricted CD8 T-cell responses of co-administered antigen. J. Control. Release 2014, 174, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Håkerud, M.; Selbo, P.K.; Waeckerle-Men, Y.; Contassot, E.; Dziunycz, P.; Kundig, T.M.; Hogset, A.; Johansen, P. Photosensitisation facilitates cross-priming of adjuvant-free protein vaccines and stimulation of tumour-suppressing CD8 T cells. J. Control. Release 2015, 198, 10–17. [Google Scholar] [CrossRef]
- Varypataki, E.M.; Hasler, F.; Waeckerle-Men, Y.; Vogel-Kindgen, S.; Hogset, A.; Kundig, T.M.; Gander, B.; Halin, C.; Johansen, P. Combined photosensitization and vaccination enable CD8 T-cell immunity and tumor suppression independent of CD4 T-cell help. Front. Immunol. 2019, 10, 1548. [Google Scholar] [CrossRef]
- Haug, M.; Brede, G.; Hakerud, M.; Nedberg, A.G.; Gederaas, O.A.; Flo, T.H.; Edwards, V.T.; Selbo, P.K.; Hogset, A.; Halaas, O. Photochemical internalization of peptide antigens provides a novel strategy to realize therapeutic cancer vaccination. Front. Immunol. 2018, 9, 650. [Google Scholar] [CrossRef]
- Norum, O.J.; Fremstedal, A.S.V.; Weyergang, A.; Golab, J.; Berg, K. Photochemical delivery of bleomycin induces T-cell activation of importance for curative effect and systemic anti-tumor immunity. J. Control. Release 2017, 268, 120–127. [Google Scholar] [CrossRef]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, H.; Samset, E.; Hol, P.K.; Tillung, T.; Lote, K. Impact of intraoperative MRI on the surgical results for high-grade gliomas. Minim. Invasive Neurosurg. 2005, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Radiographic patterns of relapse in glioblastoma. J. Neurooncol. 2011, 101, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Dobelbower, M.C.; Burnett Iii, O.L.; Nordal, R.A.; Nabors, L.B.; Markert, J.M.; Hyatt, M.D.; Fiveash, J.B. Patterns of failure for glioblastoma multiforme following concurrent radiation and temozolomide. J. Med. Imaging Radiat. Oncol. 2011, 55, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, H.; Uzal, F.A.; Chighvinadze, D.; Zhang, M.J.; Peng, Q.; Madsen, S.J. Disruption of the blood-brain barrier following ALA-mediated photodynamic therapy. Lasers Surg. Med. 2008, 40, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, H.; Zhang, M.J.; Gach, H.M.; Uzal, F.A.; Peng, Q.; Sun, C.H.; Chighvinadze, D.; Madsen, S.J. Targeted delivery of bleomycin to the brain using photo-chemical internalization of Clostridium perfringens epsilon prototoxin. J. Neurooncol. 2009, 95, 317–329. [Google Scholar] [CrossRef]
- Doolittle, N.D.; Miner, M.E.; Hall, W.A.; Siegal, T.; Jerome, E.; Osztie, E.; McAllister, L.D.; Bubalo, J.S.; Kraemer, D.F.; Fortin, D.; et al. Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors. Cancer 2000, 88, 637–647. [Google Scholar] [CrossRef]
- Worthington, R.W.; Mulders, M.S. Effect of Clostridium perfringens epsilon toxin on the blood brain barrier of mice. Onderstepoort J. Vet. Res. 1975, 42, 25–27. [Google Scholar] [PubMed]
- Mathews, M.S.; Blickenstaff, J.W.; Shih, E.C.; Zamora, G.; Vo, V.; Sun, C.H.; Hirschberg, H.; Madsen, S.J. Photochemical internalization of bleomycin for glioma treatment. J. Biomed. Opt. 2012, 17, 058001. [Google Scholar] [CrossRef]
- Gederaas, O.A.; Hauge, A.; Ellingsen, P.G.; Berg, K.; Altin, D.; Bardal, T.; Hogset, A.; Lindgren, M. Photochemical internalization of bleomycin and temozolomide—In vitro studies on the glioma cell line F98. Photochem. Photobiol. Sci. 2015, 14, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Madsen, S.J.; Hirschberg, H. Macrophages as delivery vehicles for anticancer agents. Ther. Deliv. 2019, 10, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Christie, C.; Ju, D.; Nair, R.K.; Molina, S.; Berg, K.; Krasieva, T.B.; Madsen, S.J.; Hirschberg, H. Photochemical internalization enhanced macrophage delivered chemotherapy. Photodiagnosis Photodyn. Ther. 2018, 21, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Christie, C.E.; Zamora, G.; Kwon, Y.J.; Berg, K.; Madsen, S.J.; Hirschberg, H. Macrophage Mediated PCI Enhanced Gene-Directed Enzyme Prodrug Therapy; SPIE: San Francisco, CA, USA, 2015; Volume 9305. [Google Scholar]
- Zamora, G.; Wang, F.; Sun, C.H.; Trinidad, A.; Kwon, Y.J.; Cho, S.K.; Berg, K.; Madsen, S.J.; Hirschberg, H. Photochemical internalization-mediated nonviral gene transfection: Polyamine core-shell nanoparticles as gene carrier. J. Biomed. Opt. 2014, 19, 105009. [Google Scholar] [CrossRef]
- Christie, C.; Pomeroy, A.; Nair, R.; Berg, K.; Hirschberg, H. Photodynamic therapy enhances the efficacy of gene-directed enzyme prodrug therapy. Photodiagnosis Photodyn. Ther. 2017, 18, 140–148. [Google Scholar] [CrossRef]
- Johannesen, T.B.; Watne, K.; Lote, K.; Norum, J.; Hennig, R.; Tvera, K.; Hirschberg, H. Intracavity fractionated balloon brachytherapy in glioblastoma. Acta Neurochir. (Wien.) 1999, 141, 127–133. [Google Scholar] [CrossRef]
- Madsen, S.J.; Sun, C.H.; Tromberg, B.J.; Hirschberg, H. Development of a novel indwelling balloon applicator for optimizing light delivery in photodynamic therapy. Lasers Surg. Med. 2001, 29, 406–412. [Google Scholar] [CrossRef]
- Eljamel, M.S.; Goodman, C.; Moseley, H. ALA and Photofrin fluorescence-guided resection and repetitive PDT in glioblastoma multiforme: A single centre Phase III randomised controlled trial. Lasers Med. Sci. 2008, 23, 361–367. [Google Scholar] [CrossRef]
- Grigalavicius, M.; Mastrangelopoulou, M.; Berg, K.; Arous, D.; Ménard, M.; Raabe-Henriksen, T.; Brondz, E.; Siem, S.; Görgen, A.; Edin, N.F.J.; et al. Proton-dynamic therapy following photosensitiser activation by accelerated protons demonstrated through fluorescence and singlet oxygen production. Nat. Commun. 2019, 10, 3986. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, D.H. Wireless metronomic photodynamic therapy. Nat. Biomed. Eng. 2019, 3, 5–6. [Google Scholar] [CrossRef]
- Shin, D.; Nguyen, L.; Le, M.T.; Ju, D.; Le, J.N.; Berg, K.; Hirschberg, H. The effects of low irradiance long duration photochemical internalization on glioma spheroids. Photodiagnosis Photodyn. Ther. 2019, 26, 442–447. [Google Scholar] [CrossRef]
- Gonzales, J.; Nair, R.K.; Madsen, S.J.; Krasieva, T.; Hirschberg, H. Focused ultrasound-mediated sonochemical internalization: An alternative to light-based therapies. J. Biomed. Opt. 2016, 21, 78002. [Google Scholar] [CrossRef]
- Hirschberg, H.; Madsen, S.J. Synergistic efficacy of ultrasound, sonosensitizers and chemotherapy: A review. Ther. Deliv. 2017, 8, 331–342. [Google Scholar] [CrossRef]















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jerjes, W.; Theodossiou, T.A.; Hirschberg, H.; Høgset, A.; Weyergang, A.; Selbo, P.K.; Hamdoon, Z.; Hopper, C.; Berg, K. Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research. J. Clin. Med. 2020, 9, 528. https://doi.org/10.3390/jcm9020528
Jerjes W, Theodossiou TA, Hirschberg H, Høgset A, Weyergang A, Selbo PK, Hamdoon Z, Hopper C, Berg K. Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research. Journal of Clinical Medicine. 2020; 9(2):528. https://doi.org/10.3390/jcm9020528
Chicago/Turabian StyleJerjes, Waseem, Theodossis A. Theodossiou, Henry Hirschberg, Anders Høgset, Anette Weyergang, Pål Kristian Selbo, Zaid Hamdoon, Colin Hopper, and Kristian Berg. 2020. "Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research" Journal of Clinical Medicine 9, no. 2: 528. https://doi.org/10.3390/jcm9020528
APA StyleJerjes, W., Theodossiou, T. A., Hirschberg, H., Høgset, A., Weyergang, A., Selbo, P. K., Hamdoon, Z., Hopper, C., & Berg, K. (2020). Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research. Journal of Clinical Medicine, 9(2), 528. https://doi.org/10.3390/jcm9020528