Loss-of-Function Variants in Cytoskeletal Genes Are Associated with Early-Onset Atrial Fibrillation

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Populations
2.2. Genetic Sequencing
2.3. Selection of Candidate Genes
2.4. Evaluation of Identified Variants
2.5. Pathway Analysis
2.6. Statistical Analyses
3. Results
3.1. Clinical Characteristics
3.2. Genetic Variation
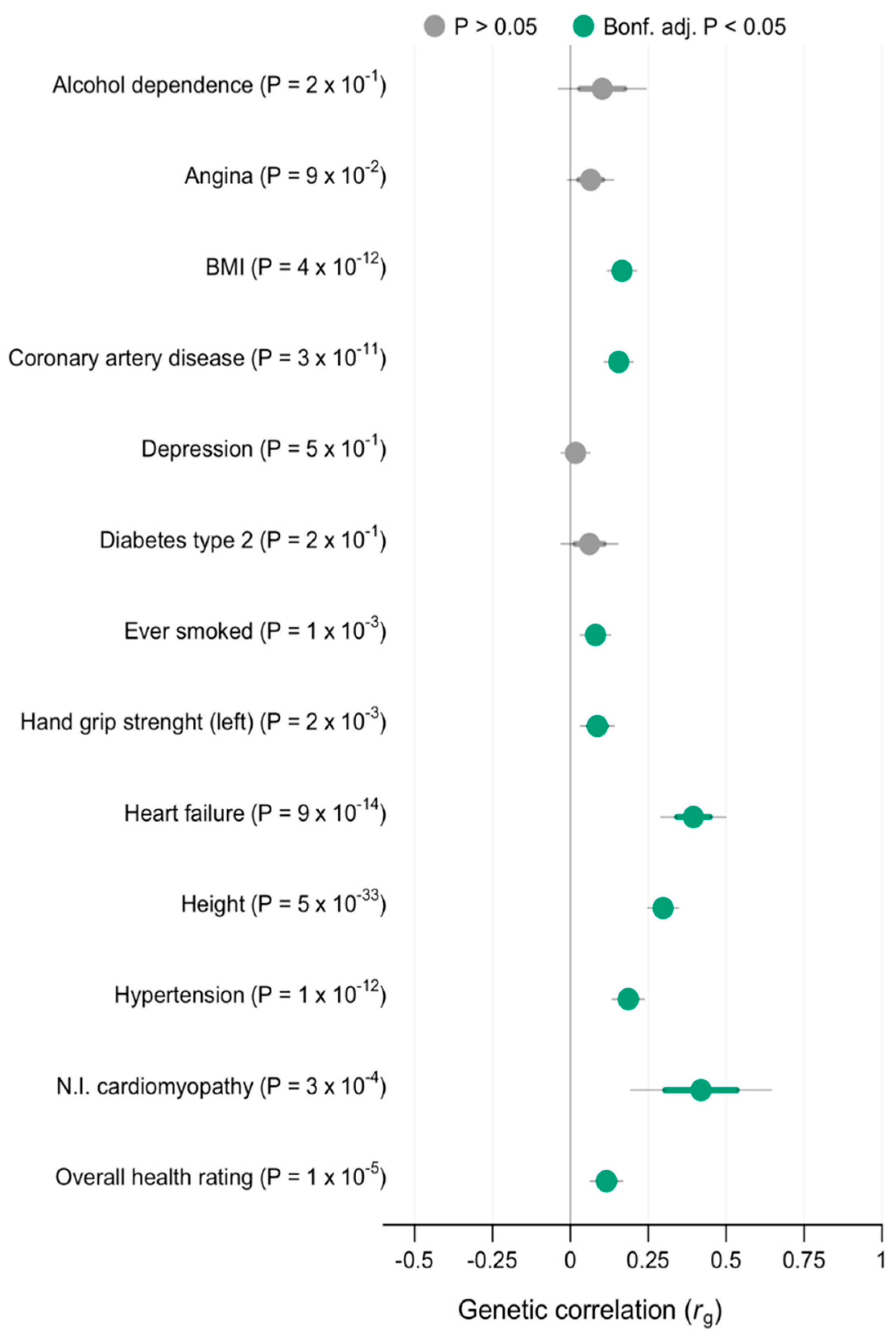
3.3. Genetic Correlation
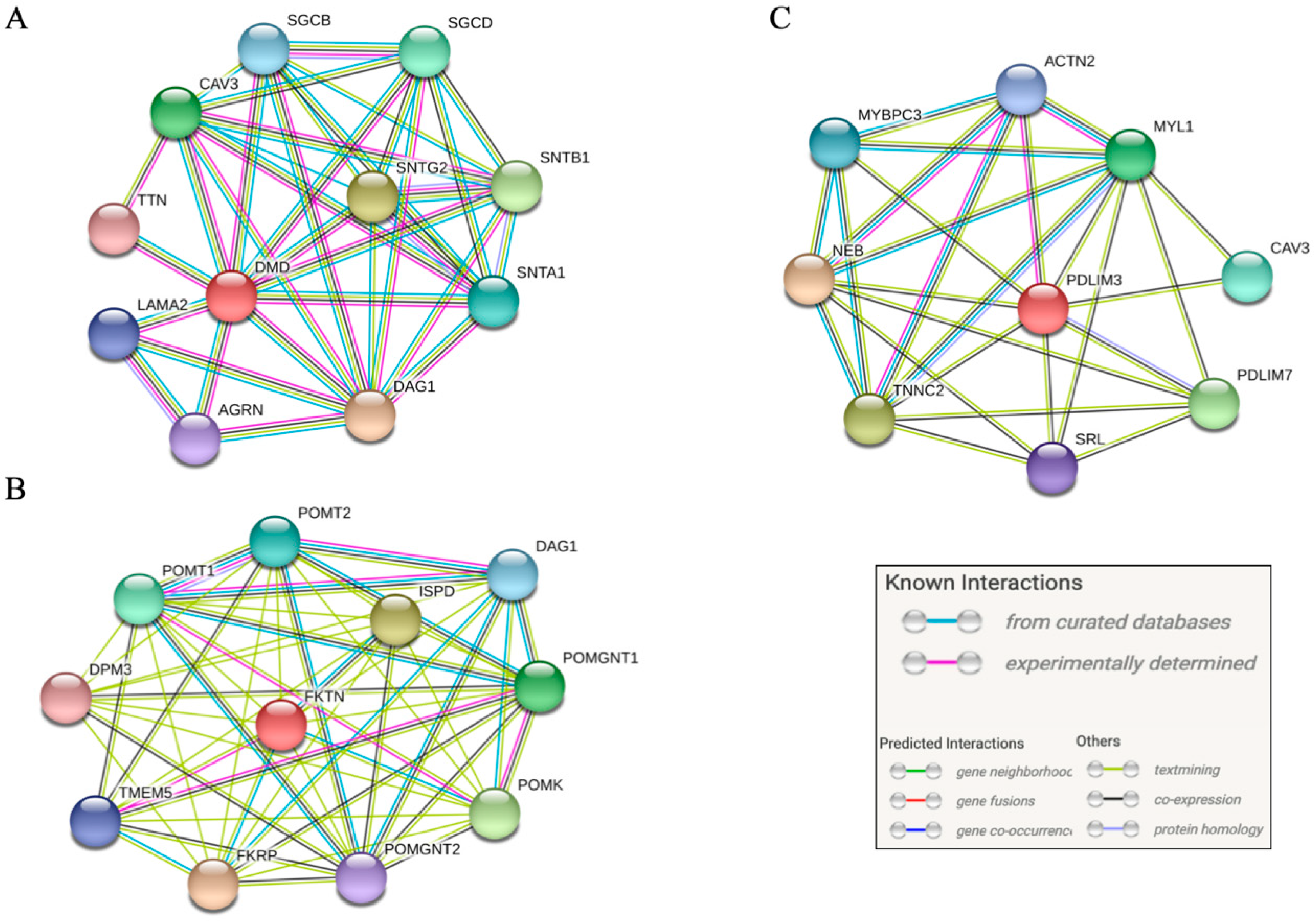
3.4. Pathway Analysis
4. Discussion
4.1. Variants in DMD
4.2. Variants in FKTN and PDLIM3
4.3. Atrial Cardiomyopathy
4.4. Treatment Consequences
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H.; Zheng, Z.J.; et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. Heart J. 2016, 37, 2893–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, S.; Murphy, N.F; Murphy, N.; Walker, A.; McGuire, A.; McMurray, J.J.V. Cost of an emerging epidemic: An economic analysis of atrial fibrillation in the UK. Heart 2004, 90, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Nattel, S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 2010, 375, 1212–1223. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Thorolfsdottir, R.B.; Fritsche, L.G.; Zhou, W.; Skov, M.W.; Graham, S.E.; Herron, T.J.; McCarthy, S.; Schmidt, E.M.; Sveinbjornsson, G.; et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 2018, 50, 1234–1239. [Google Scholar] [CrossRef]
- Olesen, M.S.; Nielsen, M.W.; Haunsø, S.; Svendsen, J.H. Atrial fibrillation: The role of common and rare genetic variants. Eur. J. Hum. Genet. 2014, 22, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Ahlberg, G.; Refsgaard, L.; Lundegaard, P.R.; Andreasen, L.; Ranthe, M.F.; Linscheid, N.; Nielsen, J.B.; Melbye, M.; Haunsø, S.; Sajadieh, A.; et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Choi, S.H.; Weng, L.-C.; Roselli, C.; Lin, H.; Haggerty, C.M.; Shoemaker, M.B.; Barnard, J.; Arking, D.E.; Chasman, D.I.; Albert, C.M.; et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA 2018, 320, 2354–2364. [Google Scholar] [CrossRef]
- Roselli, C.; Chaffin, M.D.; Weng, L.-C.; Aeschbacher, S.; Ahlberg, G.; Albert, C.M.; Almgren, P.; Alonso, A.; Anderson, C.D.; Aragam, K.G.; et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Europace 2016, 18, 1455–1490. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Di Toro, A.; Giuliani, L.; Favalli, V.; Narula, N.; Grasso, M. Cardiac Phenotypes in Hereditary Muscle Disorders: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2485–2506. [Google Scholar] [CrossRef] [PubMed]
- McNally Elizabeth, M.; Mestroni, L. Dilated Cardiomyopathy. Circ. Res. 2017, 121, 731–748. [Google Scholar]
- Sajadieh, A.; Nielsen, O.W.; Rasmussen, V.; Hein, H.O.; Hansen, J.F. Prevalence and prognostic significance of daily-life silent myocardial ischaemia in middle-aged and elderly subjects with no apparent heart disease. Eur. Heart J. 2005, 26, 1402–1409. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Wilde, A.A.M.; Behr, E.R. Genetic testing for inherited cardiac disease. Nat. Rev. Cardiol. 2013, 10, 571–583. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, T.; Chong, Z.; Rohrdanz, M.A.; Melott, J.M.; Wakefield, C.; Zeng, J.; Weinstein, J.M.; Meric-Bernstam, F.; Mills, G.B.; et al. TransVar: A multilevel variant annotator for precision genomics. Nat. Methods 2015, 12, 1002. [Google Scholar] [CrossRef] [Green Version]
- GTEx Portal [Internet]. Available online: https://www.gtexportal.org/home/ (accessed on 28 November 2019).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids. Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef]
- Hadji-Turdeghal, K.; Andreasen, L.; Hagen, C.M.; Ahlberg, G.; Ghouse, J.; Bækvad-Hansen, M.; Bybjerg-Grauholm, J.; Hougaard, D.M.; Hedley, P.; Haunsø, S.; et al. Genome-wide association study identifies locus at chromosome 2q32.1 associated with syncope and collapse. Cardiovasc. Res. 2020, 116, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Dystrophin Isoforms and Their Expression [Internet]. Available online: https://www.dmd.nl/isoforms.html (accessed on 28 November 2019).
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.; Swift, M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, S.A.; Duncan, N.M.; Rash, S.M.; Sadeghi, A.; Dewan, A.K.; Pillers, D.A. Redefinition of dystrophin isoform distribution in mouse tissue by RT-PCR implies role in nonmuscle manifestations of duchenne muscular dystrophy. Mol. Genet. Metab. 1998, 65, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Scheiper, S.; Ramos-Luis, E.; Blanco-Verea, A.; Niess, C.; Beckmann, B.-M.; Schmidt, U.; Kettner, M.; Geisen, C.; Verhoff, M.A.; Brion, M.; et al. Sudden unexpected death in the young—Value of massive parallel sequencing in postmortem genetic analyses. Forensic Sci. Int. 2018, 293, 70–76. [Google Scholar] [CrossRef]
- Andreasen, C.; Nielsen, J.B.; Refsgaard, L.; Holst, A.G.; Christensen, A.H.; Andreasen, L.; Sajadieh, A.; Haunsø, S.; Svendsen, J.H.; Olesen, M.S. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur. J. Hum. Genet. 2013, 21, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Paludan-Müller, C.; Ghouse, J.; Vad, O.B.; Herfelt, C.B.; Lundegaard, P.; Ahlberg, G.; Schmitt, N.; Svendsen, J.H.; Haunsø, S.; Bundgaard, H.; et al. Reappraisal of variants previously linked with sudden infant death syndrome: Results from three population-based cohorts. Eur. J. Hum. Genet. 2019, 27, 1427–1435. [Google Scholar] [CrossRef]
- Andreasen, L.; Nielsen, J.B.; Olesen, M.S. Genetic aspects of lone atrial fibrillation: What do we know? Curr. Pharm. Des. 2015, 21, 667–678. [Google Scholar] [CrossRef]
- Saito, K. Fukuyama Congenital Muscular Dystrophy; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; 2006 Jan 26 [Updated 2019 Jul 3]; GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993–2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1206/ (accessed on 2 January 2020).
- Murakami, T.; Hayashi, Y.K.; Noguchi, S.; Ogawa, M.; Nonaka, I.; Tanabe, Y.; Ogino, M.; Takada, F.; Eriguchi, M.; Kotooka, N.; et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann. Neurol. 2006, 60, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Brockington, M.; Straub, V.; Quijano-Roy, S.; Yuva, Y.; Herrmann, R.; Brown, S.C.; Torelli, S.; Dubowitz, V.; Blake, D.J.; et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann. Neurol. 2003, 53, 537–542. [Google Scholar] [CrossRef]
- Wang, D.; Fang, J.; Lv, J.; Pan, Z.; Yin, X.; Cheng, H.; Gou, X. Novel polymorphisms in PDLIM3 and PDLIM5 gene encoding Z-line proteins increase risk of idiopathic dilated cardiomyopathy. J. Cell Mol. Med. 2019, 23, 7054–7062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsawa, N.; Koebis, M.; Suo, S.; Nishino, I.; Ishiura, S. Alternative splicing of PDLIM3/ALP, for α-actinin-associated LIM protein 3, is aberrant in persons with myotonic dystrophy. Biochem. Biophys. Res. Commun. 2011, 409, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Marrouche, N.F.; Wilber, D.; Hindricks, G.; Jais, P.; Akoum, N.; Marchlinski, F.; Kholmovski, E.; Burgon, N.; Hu, N.; Mont, L.; et al. Association of Atrial Tissue Fibrosis Identified by Delayed Enhancement MRI and Atrial Fibrillation Catheter Ablation: The DECAAF Study. JAMA 2014, 311, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Dobrev, D. Controversies About Atrial Fibrillation Mechanisms. Circ. Res. 2017, 120, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Siebermair, J.; Kholmovski, E.G.; Marrouche, N. Assessment of Left Atrial Fibrosis by Late Gadolinium Enhancement Magnetic Resonance Imaging: Methodology and Clinical Implications. JACC Clin. Electrophysiol. 2017, 3, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodder, E.M.; Scicluna, B.P.; Beekman, L.; Arends, D.; Moerland, P.D.; Tanck, M.W.T.; Adriaens, M.E.; Bezzina, C.R. Integrative genomic approach identifies multiple genes involved in cardiac collagen deposition. Circ. Cardiovasc. Genet. 2014, 7, 790–798. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Early-Onset AF Cohort (n = 527) | Control Cohort (n = 383) | |
|---|---|---|
| Sex, male, n (%) | 441 (83.6) | 257 (67) |
| Age, years, median (IQR) | 30 (24–36) a | 71 (66–76) b |
| Height, cm, mean (SD) | 183 (±9) | 172 (±9) |
| Weight, kg, mean (SD) | 89 (±17) | 77 (±15) |
| BMI, kg/m2, mean (SD) | 26 (±5) | 26 (±5) |
| Comorbidities: | ||
| Hypertension, n (%) | 0 (0) | 246 (64.2) |
| Diabetes, n (%) | 0 (0) | 39 (10.2) |
| Heart failure, n (%) | 0 (0) | 0 (0) |
| Ischemic heart disease, n (%) | 0 (0) | 0 (0) |
| Valvular heart disease, n (%) | 0 (0) | 0 (0) |
| Patient | Gene | Variant | RefSNP | Gender | Genotype | Onset of AF (age in years) | AF Type | LVEF (%) | Family History of AF (self-reported) |
|---|---|---|---|---|---|---|---|---|---|
| I | DMD | p.D615Efs*6 | rs752332058 | Male | Hemizygote | 28 | Persistent | >55 | No |
| II | DMD | p.D615Efs*6 | rs752332058 | Male | Hemizygote | 25 | Persistent | >55 | Yes |
| III | DMD | c.10262+1G > A | rs145603325 | Male | Hemizygote | 21 | Persistent | >55 | Yes |
| IV | DMD | c.10262+1G > A | rs145603325 | Male | Hemizygote | 28 | Persistent | >55 | No |
| V | FKTN | Chr9:108358933C > T | NA | Male | Heterozygote | 31 | Paroxysmal | NA | No |
| VI | PDLIM3 | Chr4:186425651_186425652del | NA | Female | Heterozygote | 40 | Paroxysmal | >55 | Yes |
| Gene | Genomic Position | RefSNP | Transcript | Consequence | Effect | GnomAD MAF (%) |
|---|---|---|---|---|---|---|
| DMD | ChrX:31140001_31140013del | rs752332058 | ENST00000378723 | p.D615Efs*6 | Frameshift variant | 0.02491 |
| DMD | ChrX:31196048C>T | rs145603325 | ENST00000357033 | c.10262+1G>A | Splice donor | 0.02689 |
| FKTN | Chr9:108358933C>T | NA | ENST00000223528 | p.Q54* | Nonsense variant | NA |
| PDLIM3 | Chr4:186425651_186425652del | NA | ENST00000284771 | p.C246*fs*1 | Frameshift variant | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vad, O.B.; Paludan-Müller, C.; Ahlberg, G.; Kalstø, S.M.; Ghouse, J.; Andreasen, L.; Haunsø, S.; Tveit, A.; Sajadieh, A.; Christophersen, I.E.; et al. Loss-of-Function Variants in Cytoskeletal Genes Are Associated with Early-Onset Atrial Fibrillation. J. Clin. Med. 2020, 9, 372. https://doi.org/10.3390/jcm9020372
Vad OB, Paludan-Müller C, Ahlberg G, Kalstø SM, Ghouse J, Andreasen L, Haunsø S, Tveit A, Sajadieh A, Christophersen IE, et al. Loss-of-Function Variants in Cytoskeletal Genes Are Associated with Early-Onset Atrial Fibrillation. Journal of Clinical Medicine. 2020; 9(2):372. https://doi.org/10.3390/jcm9020372
Chicago/Turabian StyleVad, Oliver Bundgaard, Christian Paludan-Müller, Gustav Ahlberg, Silje Madeleine Kalstø, Jonas Ghouse, Laura Andreasen, Stig Haunsø, Arnljot Tveit, Ahmad Sajadieh, Ingrid Elisabeth Christophersen, and et al. 2020. "Loss-of-Function Variants in Cytoskeletal Genes Are Associated with Early-Onset Atrial Fibrillation" Journal of Clinical Medicine 9, no. 2: 372. https://doi.org/10.3390/jcm9020372
APA StyleVad, O. B., Paludan-Müller, C., Ahlberg, G., Kalstø, S. M., Ghouse, J., Andreasen, L., Haunsø, S., Tveit, A., Sajadieh, A., Christophersen, I. E., Svendsen, J. H., & Olesen, M. S. (2020). Loss-of-Function Variants in Cytoskeletal Genes Are Associated with Early-Onset Atrial Fibrillation. Journal of Clinical Medicine, 9(2), 372. https://doi.org/10.3390/jcm9020372