Retinoic Acids in the Treatment of Most Lethal Solid Cancers





{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
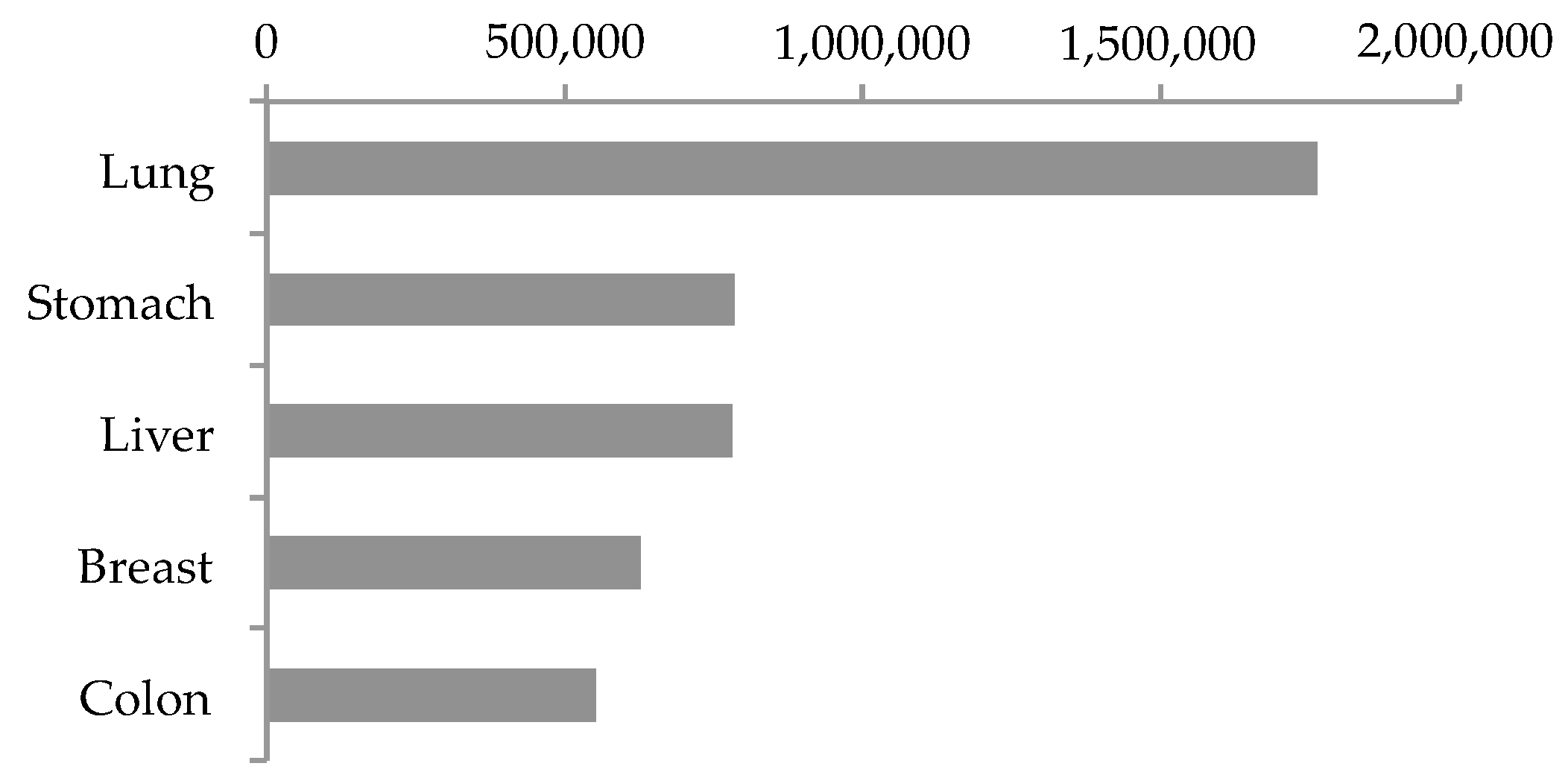
1.1. Top Five Lethal Solid Cancers
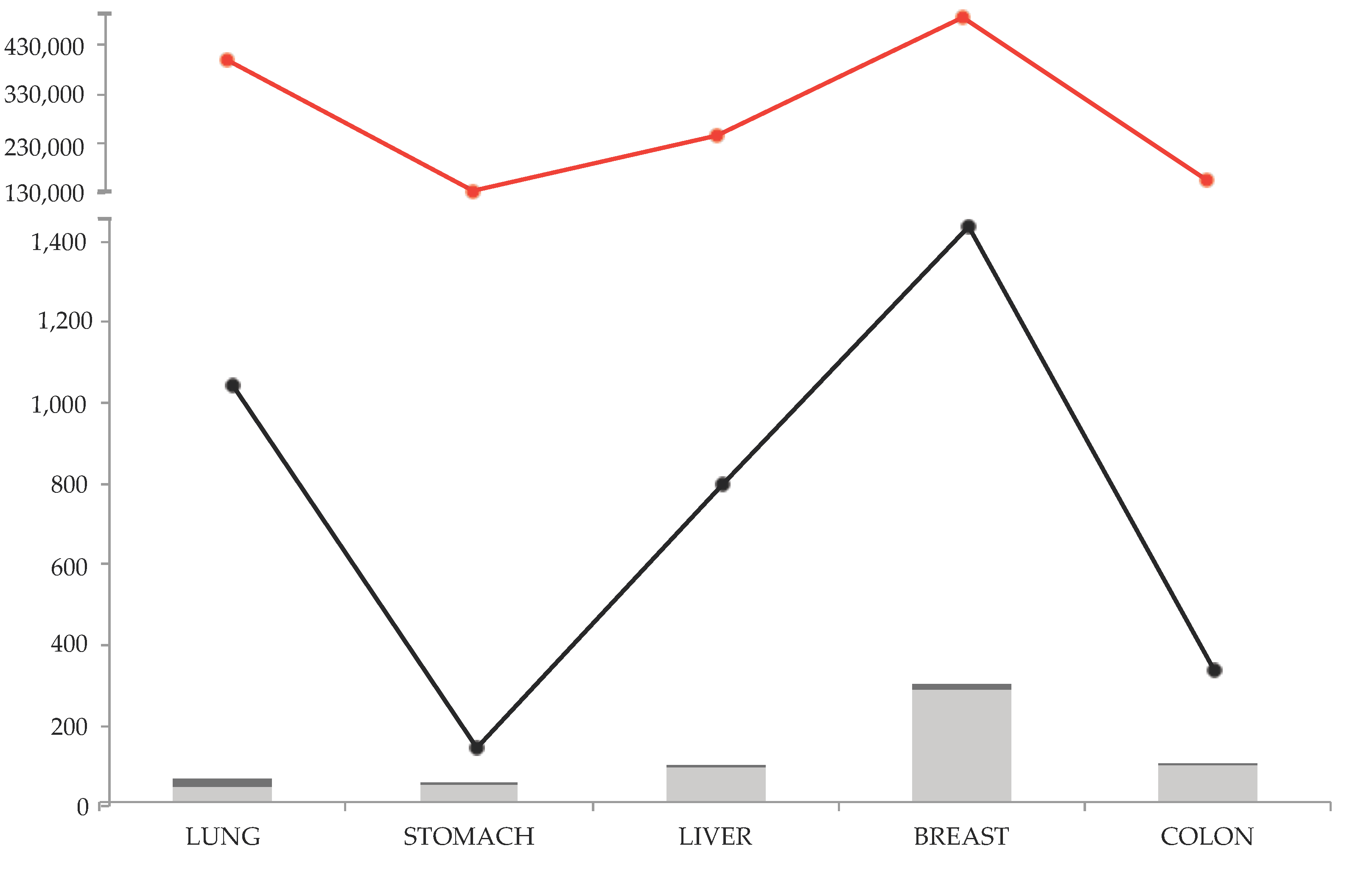
1.2. Revision Method and Results
2. Retinoic Acids’ Action as Single Agent in Solid Cancers: Clinical Trials
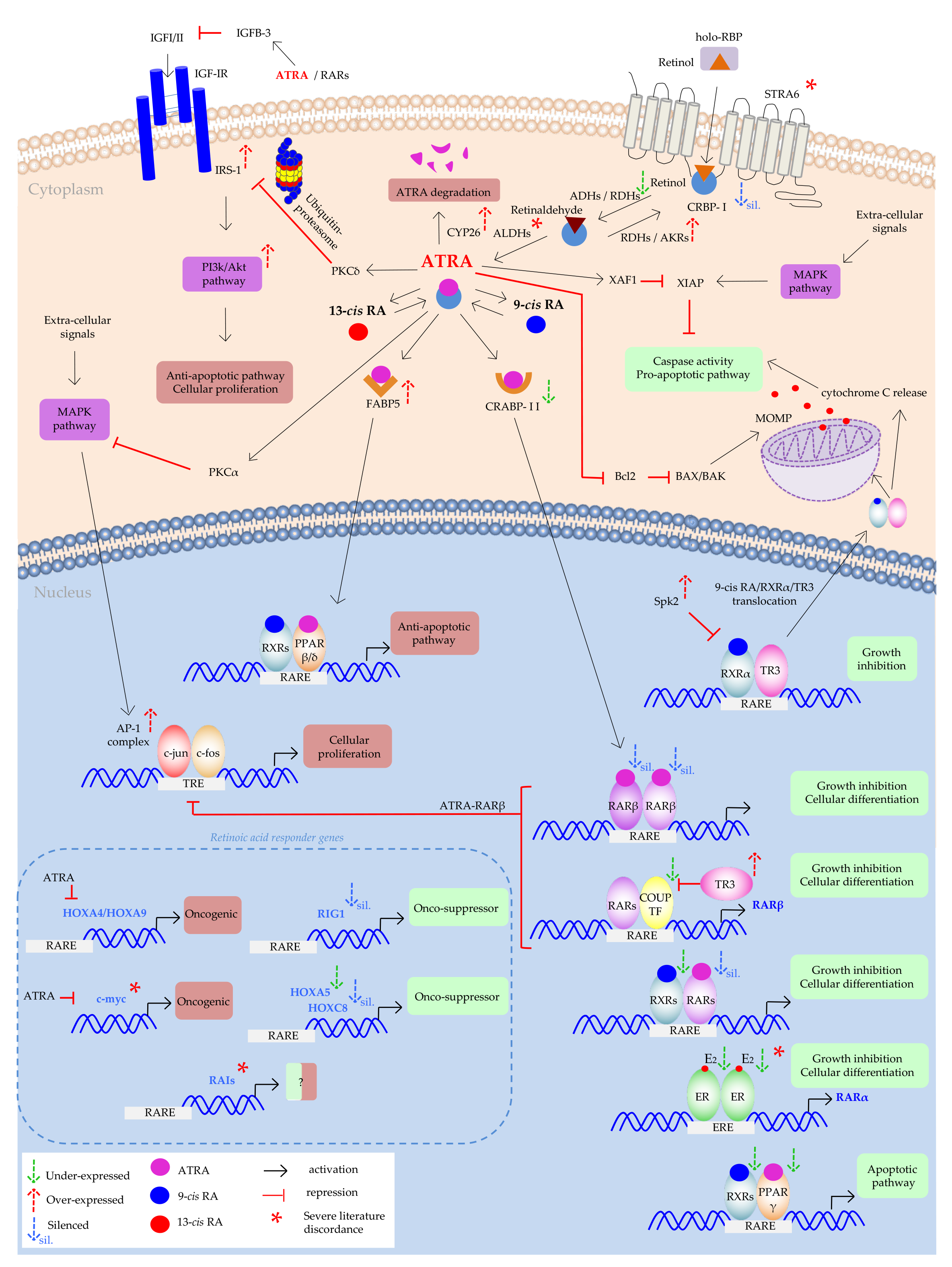
3. Pre-Clinical Studies
3.1. Defective Membrane Signaling
3.2. Impaired Cytoplasmic Signaling
3.2.1. Cytoplasmic Trafficking, Defective RBPs Expression
3.2.2. Metabolic Enzymes
3.2.3. Escape Routes from Apoptosis
Insulin-Like Growth Factors (IGFs) Pro-Survival Action
3.3. Impaired Nuclear Receptor Signaling
3.3.1. Loss of RARβ Expression
3.3.2. Other RARs and RXRs Defects
3.3.3. RARs vs. ER
3.3.4. PPARs Impaired Signaling as Retinoic Acids’ Resistance Mechanism
3.3.5. AP-1 Over-Expression as Retinoids’ Escape Route
3.4. Retinoid-Responsive Genes
3.4.1. Retinoid-Inducible Gene 1 (RIG1) Hypermethylation
3.4.2. Retinoic Acid-Induced (RAI) Genes: Oncogenes or Tumor Suppressors?
3.4.3. Homeobox (HOXs) Genes Impaired Expression
3.4.4. c-myc Oncogene Over-Expression
3.5. Tumor Progression: CSCs and Invasion
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Raghu, P.; Sivakumar, B. Interactions amongst Plasma Retinol-Binding Protein, Transthyretin and Their Ligands: Implications in Vitamin A Homeostasis and Transthyretin Amyloidosis. Biochim. Biophys. Acta BBA - Proteins Proteomics 2004, 1703, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of Retinoid Metabolism and Function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Urbach, J.; Rando, R.R. Isomerization of All-Trans-Retinoic Acid to 9-Cis-Retinoic Acid. Biochem. J. 1994, 299, 459–465. [Google Scholar] [CrossRef] [PubMed]
- di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic Acid Receptors: From Molecular Mechanisms to Cancer Therapy. Mol. Aspects Med. 2015, 41, 1–115. [Google Scholar] [CrossRef] [PubMed]
- Almeida, N.R.; Conda-Sheridan, M. A Review of the Molecular Design and Biological Activities of RXR Agonists. Med. Res. Rev. 2019, 39, 1372–1397. [Google Scholar] [CrossRef] [PubMed]
- Bastien, J.; Rochette-Egly, C. Nuclear Retinoid Receptors and the Transcription of Retinoid-Target Genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef]
- Idres, N.; Marill, J.; Flexor, M.A.; Chabot, G.G. Activation of Retinoic Acid Receptor-Dependent Transcription by All-Trans-Retinoic Acid Metabolites and Isomers. J. Biol. Chem. 2002, 277, 31491–31498. [Google Scholar] [CrossRef]
- Ruhl, R. Embryonic Subcellular Distribution of 13-Cis- and All-Trans-Retinoic Acid Indicates Differential Cytosolic/Nuclear Localization. Toxicol. Sci. 2001, 63, 82–89. [Google Scholar] [CrossRef][Green Version]
- Wolf, G. Retinoic Acid as Cause of Cell Proliferation or Cell Growth Inhibition Depending on Activation of One of Two Different Nuclear Receptors. Nutr. Rev. 2008, 66, 55–59. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Stark, R.; Holmes, K.A.; Schmidt, D.; Spyrou, C.; Russell, R.; Massie, C.E.; Vowler, S.L.; Eldridge, M.; Carroll, J.S. Cooperative Interaction between Retinoic Acid Receptor- and Estrogen Receptor in Breast Cancer. Genes Dev. 2010, 24, 171–182. [Google Scholar] [CrossRef]
- Lefebvre, P.; Martin, P.J.; Flajollet, S.; Dedieu, S.; Billaut, X.; Lefebvre, B. Transcriptional Activities of Retinoic Acid Receptors. In Vitamins & Hormones; 2005; Volume 70, pp. 199–264. [Google Scholar]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a Nuclear Receptor That Defines a Distinct Retinoid Response Pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lee, D.; Sysounthone, V.; Chandraratna, R.A.S.; Christakos, S.; Korah, R.; Wieder, R. 1,25-Dihydroxyvitamin D3 and Retinoic Acid Analogues Induce Differentiation in Breast Cancer Cells with Function- and Cell-Specific Additive Effects. Breast Cancer Res. Treat. 2001, 67, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing Effects of Retinoic Acid on Cell Growth Result from Alternate Activation of Two Different Nuclear Receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of Differentiation of the Human Promyelocytic Leukemia Cell Line (HL-60) by Retinoic Acid. Proc. Natl. Acad. Sci. 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.E.; Ye, Y.C.; Zhao, L. [Treatment of acute promyelocytic leukemia by retinoic acid with or without low dose cytosine arabinoside: Report of 4 cases]. Zhonghua Nei Ke Za Zhi 1987, 26, 330–332. [Google Scholar] [PubMed]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of Acute Promyelocytic Leukemia: Updated Recommendations from an Expert Panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academies Press (US): Washington, DC, USA, 2001. [Google Scholar]
- Jing, Y. The PML-RARα Fusion Protein and Targeted Therapy for Acute Promyelocytic Leukemia. Leuk. Lymphoma 2004, 45, 639–648. [Google Scholar] [CrossRef]
- Sokołowska-Wojdyło, M.; Ługowska-Umer, H.; Maciejewska-Radomska, A. Oral Retinoids and Rexinoids in Cutaneous T-Cell Lymphomas. Adv. Dermatol. Allergol. 2013, 1, 19–29. [Google Scholar] [CrossRef]
- Nakanishi, M.; Tomaru, Y.; Miura, H.; Hayashizaki, Y.; Suzuki, M. Identification of Transcriptional Regulatory Cascades in Retinoic Acid-Induced Growth Arrest of HepG2 Cells. Nucleic Acids Res. 2008, 36, 3443–3454. [Google Scholar] [CrossRef][Green Version]
- Muindi, J.; Frankel, S.R.; Miller, W.H.; Jakubowski, A.; Scheinberg, D.A.; Young, C.W.; Dmitrovsky, E.; Warrell, R.P. Continuous Treatment with All-Trans Retinoic Acid Causes a Progressive Reduction in Plasma Drug Concentrations: Implications for Relapse and Retinoid “Resistance” in Patients with Acute Promyelocytic Leukemia. Blood 1992, 79, 299–303. [Google Scholar] [CrossRef]
- Szuts, E.Z.; Harosi, F.I. Solubility of Retinoids in Water. Arch. Biochem. Biophys. 1991, 287, 297–304. [Google Scholar] [CrossRef]
- Adamson, P.C.; Widemann, B.C.; Reaman, G.H.; Seibel, N.L.; Murphy, R.F.; Gillespie, A.F.; Balis, F.M. A Phase I Trial and Pharmacokinetic Study of 9-Cis-Retinoic Acid (ALRT1057) in Pediatric Patients with Refractory Cancer: A Joint Pediatric Oncology Branch, National Cancer Institute, and Children’s Cancer Group Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 3034–3039. [Google Scholar]
- Sutton, L.M.; Warmuth, M.A.; Petros, W.P.; Winer, E.P. Pharmacokinetics and Clinical Impact of All- Trans Retinoic Acid in Metastatic Breast Cancer: A Phase II Trial. Cancer Chemother. Pharmacol. 1997, 40, 335–341. [Google Scholar] [CrossRef] [PubMed]
- GLOBOCAN 2018. Available online: http://gco.iarc.fr/ (accessed on 12 April 2019).
- Thun, M.J.; Hannan, L.M.; Adams-Campbell, L.L.; Boffetta, P.; Buring, J.E.; Feskanich, D.; Flanders, W.D.; Jee, S.H.; Katanoda, K.; Kolonel, L.N.; et al. Lung Cancer Occurrence in Never-Smokers: An Analysis of 13 Cohorts and 22 Cancer Registry Studies. PLoS Med. 2008, 5, e185. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung Cancer: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Vital Signs: Current Cigarette Smoking among Adults Aged >or=18 Years --- United States, 2009. MMWR Morb. Mortal. Wkly. Rep. 2010, 59, 1135–1140. [Google Scholar]
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung Cancer Stem Cells—Origin, Characteristics and Therapy. Stem Cell Investig. 2018, 5, 6. [Google Scholar] [CrossRef]
- Fu, Y.; Du, P.; Zhao, J.; Hu, C.; Qin, Y.; Huang, G. Gastric Cancer Stem Cells: Mechanisms and Therapeutic Approaches. Yonsei Med. J. 2018, 59, 1150. [Google Scholar] [CrossRef]
- Johnston, F.M.; Beckman, M. Updates on Management of Gastric Cancer. Curr. Oncol. Rep. 2019, 21. [Google Scholar] [CrossRef]
- Njei, B.; Rotman, Y.; Ditah, I.; Lim, J.K. Emerging Trends in Hepatocellular Carcinoma Incidence and Mortality: NJEI ET AL. Hepatology 2015, 61, 191–199. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of Hepatocellular Carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lai, E.C.H.; Zhang, C.; Yu, H.; Liu, Z.; Wan, B.; Liu, L.; Tian, Z.; Deng, H.; Sun, Q.; et al. The Strategies for Treating Primary Hepatocellular Carcinoma with Portal Vein Tumor Thrombus. Int. J. Surg. 2015, 20, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhu, Y. Targeting Liver Cancer Stem Cells for the Treatment of Hepatocellular Carcinoma. Ther. Adv. Gastroenterol. 2019, 12, 175628481882156. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast Cancer Development and Progression: Risk Factors, Cancer Stem Cells, Signaling Pathways, Genomics, and Molecular Pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Giovannelli, P.; Donato, M.D.; Galasso, G.; Zazzo, E.D.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast Cancer Stem Cells: The Role of Sex Steroid Receptors. World J. Stem Cells 2019, 11, 594–603. [Google Scholar] [CrossRef]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and Colorectal Cancer: Roles in Carcinogenesis and Clinical Potential. Mol. Aspects Med. 2019, 69, 93–106. [Google Scholar] [CrossRef]
- Gupta, R.; Bhatt, L.K.; Johnston, T.P.; Prabhavalkar, K.S. Colon Cancer Stem Cells: Potential Target for the Treatment of Colorectal Cancer. Cancer Biol. Ther. 2019, 20, 1068–1082. [Google Scholar] [CrossRef]
- Lee, J.S.; Newman, R.A.; Lippman, S.M.; Huber, M.H.; Minor, T.; Raber, M.N.; Krakoff, I.H.; Hong, W.K. Phase I Evaluation of All-Trans-Retinoic Acid in Adults with Solid Tumors. J. Clin. Oncol. 1993, 11, 959–966. [Google Scholar] [CrossRef]
- Iv, H.C.P.; Rubin, J.; Kovach, J.S.; Schutt, A.J.; Adamson, P.C. All-Trans Retinoic Acid: A Dose-Seeking Study in Solid Tumors. Ann. N. Y. Acad. Sci. 1993, 691, 250–252. [Google Scholar] [CrossRef]
- Smith, M.A.; Adamson, P.C.; Balis, F.M.; Feusner, J.; Aronson, L.; Murphy, R.F.; Horowitz, M.E.; Reaman, G.; Hammond, G.D.; Fenton, R.M. Phase I and Pharmacokinetic Evaluation of All-Trans-Retinoic Acid in Pediatric Patients with Cancer. J. Clin. Oncol. 1992, 10, 1666–1673. [Google Scholar] [CrossRef]
- Conley, B.A.; Egorin, M.J.; Sridhara, R.; Finley, R.; Hemady, R.; Wu, S.; Tait, N.S.; Echo, D.A.V.; Echo, D.A.V. Phase I Clinical Trial of All-Trans-Retinoic Acid with Correlation of Its Pharmacokinetics and Pharmacodynamics. Cancer Chemother. Pharmacol. 1997, 39, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Kurie, J.M.; Lee, J.S.; Griffin, T.; Lippman, S.M.; Drum, P.; Thomas, M.P.; Weber, C.; Bader, M.; Massimini, G.; Hong, W.K. Phase I Trial of 9-Cis Retinoic Acid in Adults with Solid Tumors. Clin. Cancer Res. 1996, 2, 287–293. [Google Scholar] [PubMed]
- Food and Drug Administration, FDA. Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. July 2005, Pharmacology and Toxicology. Available online: https://www.fda.gov/media/72309/download (accessed on 3 October 2019).
- Cassidy, J.; Lippman, M.; Lacroix, A.; Peck, G. Phase II Trial of 13-Cis-Retinoic Acid in Metastatic Breast Cancer. Eur. J. Cancer Clin. Oncol. 1982, 18, 925–928. [Google Scholar] [CrossRef]
- Lotan, R. Different Susceptibilities of Human Melanoma and Breast Carcinoma Cell Lines to Retinoic Acid-Induced Growth Inhibition. Cancer Res. 1979, 39, 1014–1019. [Google Scholar]
- Lacroix, A.; Lippman, M.E. Binding of Retinoids to Human Breast Cancer Cell Lines and Their Effects on Cell Growth. J. Clin. Investig. 1980, 65, 586–591. [Google Scholar] [CrossRef]
- Marth, C.; Mayer, I.; Daxenbichler, G. Effect of Retinoic Acid and 4-Hydroxytamoxifen on Human Breast Cancer Cell Lines. Biochem. Pharmacol. 1984, 33, 2217–2221. [Google Scholar] [CrossRef]
- Hoosein, N.M.; Brattain, D.E.; McKnight, M.K.; Childress, K.E.; Chakrabarty, S.; Brattain, M.G. Comparison of the Antiproliferative Effects of Transforming Growth Factor-β, N,N-Dimethylformamide and Retinoic Acid on a Human Colon Carcinoma Cell Line. Cancer Lett. 1988, 40, 219–232. [Google Scholar] [CrossRef]
- Kéri, G.; Balogh, A.; Teplén, I.; Csuka, O. Comparison of the Tyrosine Kinase Activity with the Proliferation Rate in Human Colon Solid Tumors and Tumor Cell Lines. Tumor Biol. 1988, 9, 315–322. [Google Scholar]
- Reynolds, S.; Rajagopal, S.; Chakrabarty, S. Differentiation-Inducing Effect of Retinoic Acid, Difluoromethylornithine, Sodium Butyrate and Sodium Suramin in Human Colon Cancer Cells. Cancer Lett. 1998, 134, 53–60. [Google Scholar] [CrossRef]
- Ai, Z.W. Reversing effect of retinoic acid on some phenotypes of human hepatocarcinoma cell line. Zhonghua Zhong Liu Za Zhi 1991, 13, 9–12. [Google Scholar]
- Jung, H.; Park, S.; Yoo, Y.; Kim, J.; Kim, Y. CDK2/4 Regulate Retinoic Acid-Induced G1 Arrest in Hepatocellular Carcinoma Cells. Hepatol. Res. 2005, 31, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Geradts, J.; Chen, J.Y.; Russell, E.K.; Yankaskas, J.R.; Nieves, L.; Minna, J.D. Human Lung Cancer Cell Lines Exhibit Resistance to Retinoic Acid Treatment. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1993, 4, 799–809. [Google Scholar]
- Wan, H.; Dawson, M.I.; Hong, W.K.; Lotan, R. Enhancement of Calu-1 Human Lung Carcinoma Cell Growth in Serum-Free Medium by Retinoids: Dependence on AP-1 Activation, but Not on Retinoid Response Element Activation. Oncogene 1997, 15, 2109–2118. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shyu, R.-Y.; Jiang, S.-Y.; Huang, S.-L.; Chang, T.-C.; Wu, K.-L.; Roffler, S.R.; Yeh, M.-Y. Growth Regulation by All-Trans-Retinoic Acid and Retinoic Acid Receptor Messenger Ribonucleic Acids Expression in Gastric Cancer Cells. Eur. J. Cancer 1995, 31, 237–243. [Google Scholar] [CrossRef]
- Naka, K.; Yokozaki, H.; Domen, T.; Hayashi, K.; Kuniyasu, H.; Yasui, W.; Tahara, E.; Lotan, R. Growth Inhibition of Cultured Human Gastric Cancer Cells by 9-Cis-Retinoic Acid with Induction of Cdk Inhibitor Waf1/Cip1/Sdi1/P21 Protein. Differentiation 1997, 61, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.-Y.; Xiao, S.-D. Effect of Trans-Retinoic Acid and Folic Acid on Apoptosis in Human Gastric Cancer Cell Lines MKN-45 and MKN-28. J. Gastroenterol. 1998, 33, 656–661. [Google Scholar] [CrossRef]
- Szeto, W.; Jiang, W.; Tice, D.A.; Rubinfeld, B.; Hollingshead, P.G.; Fong, S.E.; Dugger, D.L.; Pham, T.; Yansura, D.G.; Wong, T.A.; et al. Over-expression of the Retinoic Acid-Responsive Gene Stra6 in Human Cancers and Its Synergistic Induction by Wnt-1 and Retinoic Acid. Cancer Res. 2001, 61, 4197–4205. [Google Scholar]
- Berry, D.C.; Levi, L.; Noy, N. Holo-Retinol-Binding Protein and Its Receptor STRA6 Drive Oncogenic Transformation. Cancer Res. 2014, 74, 6341–6351. [Google Scholar] [CrossRef]
- Karunanithi, S.; Levi, L.; DeVecchio, J.; Karagkounis, G.; Reizes, O.; Lathia, J.D.; Kalady, M.F.; Noy, N. RBP4-STRA6 Pathway Drives Cancer Stem Cell Maintenance and Mediates High-Fat Diet-Induced Colon Carcinogenesis. Stem Cell Rep. 2017, 9, 438–450. [Google Scholar] [CrossRef]
- Lin, L.; Xiao, J.; Shi, L.; Chen, W.; Ge, Y.; Jiang, M.; Li, Z.; Fan, H.; Yang, L.; Xu, Z. STRA6 Exerts Oncogenic Role in Gastric Tumorigenesis by Acting as a Crucial Target of MiR-873. J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef]
- Carrera, S.; Cuadrado-Castano, S.; Samuel, J.; Jones, G.D.D.; Villar, E.; Lee, S.W.; Macip, S. Stra6, a Retinoic Acid-Responsive Gene, Participates in P53-Induced Apoptosis after DNA Damage. Cell Death Differ. 2013, 20, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Shutoh, M.; Oue, N.; Aung, P.P.; Noguchi, T.; Kuraoka, K.; Nakayama, H.; Kawahara, K.; Yasui, W. DNA Methylation of Genes Linked with Retinoid Signaling in Gastric Carcinoma: Expression of the Retinoid Acid Receptor β, Cellular Retinol-Binding Protein 1, and Tazarotene-Induced Gene 1 Genes Is Associated with DNA Methylation. Cancer 2005, 104, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Pierzchalski, K.; Yu, J.; Norman, V.; Kane, M.A. CrbpI Regulates Mammary Retinoic Acid Homeostasis and the Mammary Microenvironment. FASEB J. 2013, 27, 1904–1916. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Guo, M.; Moreno, V.; Peinado, M.A.; Capella, G.; Galm, O.; Baylin, S.B.; Herman, J.G. Hypermethylation-Associated Inactivation of the Cellular Retinol-Binding-Protein 1 Gene in Human Cancer. Cancer Res. 2002, 62, 5902–5905. [Google Scholar]
- He, D.; Zhang, Y.; Zhang, N.; Zhou, L.; Chen, J.; Jiang, Y.; Shao, C. Aberrant Gene Promoter Methylation of P16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein–Barr Virus-Associated Gastric Carcinomas. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef]
- Di Croce, L. Methyltransferase Recruitment and DNA Hypermethylation of Target Promoters by an Oncogenic Transcription Factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef]
- Jing, Y.; Waxman, S.; Mira-y-Lopez, R. The Cellular Retinoic Acid Binding Protein II Is a Positive Regulator of Retinoic Acid Signaling in Breast Cancer Cells. Cancer Res. 1997, 57, 1668–1672. [Google Scholar]
- Lu, M.; Mira-y-Lopez, R.; Nakajo, S.; Nakaya, K.; Jing, Y. Expression of Estrogen Receptor α, Retinoic Acid Receptor α and Cellular Retinoic Acid Binding Protein II Genes Is Coordinately Regulated in Human Breast Cancer Cells. Oncogene 2005, 24, 4362–4369. [Google Scholar] [CrossRef][Green Version]
- Budhu, A.S.; Noy, N. Direct Channeling of Retinoic Acid between Cellular Retinoic Acid-Binding Protein II and Retinoic Acid Receptor Sensitizes Mammary Carcinoma Cells to Retinoic Acid-Induced Growth Arrest. Mol. Cell. Biol. 2002, 22, 2632–2641. [Google Scholar] [CrossRef]
- Dong, D.; Ruuska, S.E.; Levinthal, D.J.; Noy, N. Distinct Roles for Cellular Retinoic Acid-Binding Proteins I and II in Regulating Signaling by Retinoic Acid. J. Biol. Chem. 1999, 274, 23695–23698. [Google Scholar] [CrossRef]
- Schug, T.T.; Berry, D.C.; Toshkov, I.A.; Cheng, L.; Nikitin, A.Y.; Noy, N. Overcoming Retinoic Acid-Resistance of Mammary Carcinomas by Diverting Retinoic Acid from PPAR / to RAR. Proc. Natl. Acad. Sci. 2008, 105, 7546–7551. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-Z.; Graham, K.; Glubrecht, D.D.; Germain, D.R.; Mackey, J.R.; Godbout, R. Association of FABP5 Expression With Poor Survival in Triple-Negative Breast Cancer. Am. J. Pathol. 2011, 178, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Levi, L.; Banerjee, P.; Jain, M.; Noy, N. Kruppel-like Factor 2 Suppresses Mammary Carcinoma Growth by Regulating Retinoic Acid Signaling. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Kannan-Thulasiraman, P.; Seachrist, D.D.; Mahabeleshwar, G.H.; Jain, M.K.; Noy, N. Fatty Acid-Binding Protein 5 and PPARβ/δ Are Critical Mediators of Epidermal Growth Factor Receptor-Induced Carcinoma Cell Growth. J. Biol. Chem. 2010, 285, 19106–19115. [Google Scholar] [CrossRef] [PubMed]
- Coyle, K.; Dean, C.; Thomas, M.; Vidovic, D.; Giacomantonio, C.; Helyer, L.; Marcato, P. DNA Methylation Predicts the Response of Triple-Negative Breast Cancers to All-Trans Retinoic Acid. Cancers 2018, 10, 397. [Google Scholar] [CrossRef]
- Okamoto, K.; Andreola, F.; Chiantore, M.V.; Dedrick, R.L.; De Luca, L.M. Differences in Uptake and Metabolism of Retinoic Acid between Estrogen Receptor-Positive and -Negative Human Breast Cancer Cells. Cancer Chemother. Pharmacol. 2000, 46, 128–134. [Google Scholar] [CrossRef]
- Hayden, L.J.; Satre, M.A. Alterations in Cellular Retinol Metabolism Contribute to Differential Retinoid Responsiveness in Normal Human Mammary Epithelial Cells Versus Breast Cancer Cells. Breast Cancer Res. Treat. 2002, 72, 95–105. [Google Scholar] [CrossRef]
- Kropotova, E.S.; Zinov’eva, O.L.; Zyryanova, A.F.; Choinzonov, E.L.; Afanas’ev, S.G.; Cherdyntseva, N.V.; Beresten’, S.F.; Oparina, N.Y.; Mashkova, T.D. Expression of Genes Involved in Retinoic Acid Biosynthesis in Human Gastric Cancer. Mol. Biol. 2013, 47, 280–292. [Google Scholar] [CrossRef]
- Kropotova, E.S.; Zinovieva, O.L.; Zyryanova, A.F.; Dybovaya, V.I.; Prasolov, V.S.; Beresten, S.F.; Oparina, N.Y.; Mashkova, T.D. Altered Expression of Multiple Genes Involved in Retinoic Acid Biosynthesis in Human Colorectal Cancer. Pathol. Oncol. Res. 2014, 20, 707–717. [Google Scholar] [CrossRef]
- Kuznetsova, E.S.; Zinovieva, O.L.; Oparina, N. Yu.; Prokofjeva, M.M.; Spirin, P.V.; Favorskaya, I.A.; Zborovskaya, I.B.; Lisitsyn, N.A.; Prassolov, V.S.; Mashkova, T.D. Abnormal Expression of Genes That Regulate Retinoid Metabolism and Signaling in Non-Small-Cell Lung Cancer. Mol. Biol. 2016, 50, 220–229. [Google Scholar] [CrossRef]
- Zeindl-Eberhart, E.; Haraida, S.; Liebmann, S.; Jungblut, P.R.; Lamer, S.; Mayer, D.; Jäger, G.; Chung, S.; Rabes, H.M. Detection and Identification of Tumor-Associated Protein Variants in Human Hepatocellular Carcinomas. Hepatol. 2004, 39, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, S.-I. Over-expression of the Aldo-Keto Reductase Family Protein AKR1B10 Is Highly Correlated with Smokers’ Non-Small Cell Lung Carcinomas. Clin. Cancer Res. 2005, 11, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Luo, D.-X.; Huang, C.; Shen, Y.; Bu, Y.; Markwell, S.; Gao, J.; Liu, J.; Zu, X.; Cao, Z.; et al. AKR1B10 Over-expression in Breast Cancer: Association with Tumor Size, Lymph Node Metastasis and Patient Survival and Its Potential as a Novel Serum Marker. Int. J. Cancer 2012, 131, E862–E871. [Google Scholar] [CrossRef] [PubMed]
- Selga, E.; Noé, V.; Ciudad, C.J. Transcriptional Regulation of Aldo-Keto Reductase 1C1 in HT29 Human Colon Cancer Cells Resistant to Methotrexate: Role in the Cell Cycle and Apoptosis. Biochem. Pharmacol. 2008, 75, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Torres-Mena, J.E.; Salazar-Villegas, K.N.; Sánchez-Rodríguez, R.; López-Gabiño, B.; Del Pozo-Yauner, L.; Arellanes-Robledo, J.; Villa-Treviño, S.; Gutiérrez-Nava, M.A.; Pérez-Carreón, J.I. Aldo-Keto Reductases as Early Biomarkers of Hepatocellular Carcinoma: A Comparison Between Animal Models and Human HCC. Dig. Dis. Sci. 2018, 63, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.A.; Kumar, P.U.; Srinivasulu, M.; Triveni, B.; Sharada, K.; Ismail, A.; Reddy, G.B. Over-expression and Enhanced Specific Activity of Aldoketo Reductases (AKR1B1 & AKR1B10) in Human Breast Cancers. Breast 2017, 31, 137–143. [Google Scholar]
- Ohashi, T.; Idogawa, M.; Sasaki, Y.; Suzuki, H.; Tokino, T. AKR1B10, a Transcriptional Target of P53, Is Downregulated in Colorectal Cancers Associated with Poor Prognosis. Mol. Cancer Res. 2013, 11, 1554–1563. [Google Scholar] [CrossRef]
- Yao, H.-B.; Xu, Y.; Chen, L.-G.; Guan, T.-P.; Ma, Y.-Y.; He, X.-J.; Xia, Y.-J.; Tao, H.-Q.; Shao, Q.-S. AKR1B10, a Good Prognostic Indicator in Gastric Cancer. Eur. J. Surg. Oncol. EJSO 2014, 40, 318–324. [Google Scholar] [CrossRef]
- Rexer, B.N.; Zheng, W.L.; Ong, D.E. Retinoic Acid Biosynthesis by Normal Human Breast Epithelium Is via Aldehyde Dehydrogenase 6, Absent in MCF-7 Cells. Cancer Res. 2001, 61, 7065–7070. [Google Scholar]
- Moreb, J.S.; Gabr, A.; Vartikar, G.R.; Gowda, S.; Zucali, J.R.; Mohuczy, D. Retinoic Acid Down-Regulates Aldehyde Dehydrogenase and Increases Cytotoxicity of 4-Hydroperoxycyclophosphamide and Acetaldehyde. J. Pharmacol. Exp. Ther. 2005, 312, 339–345. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Giraud, J.; Staedel, C.; Chambonnier, L.; Dubus, P.; Chevret, E.; Bœuf, H.; Gauthereau, X.; Rousseau, B.; Fevre, M.; et al. All-Trans Retinoic Acid Targets Gastric Cancer Stem Cells and Inhibits Patient-Derived Gastric Carcinoma Tumor Growth. Oncogene 2016, 35, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, Z.; Xu, X.; Chen, C.; Wei, W.; Fan, M.; Chen, X.; Li, J.J.; Wang, Y.; Huang, J. All-Trans Retinoic Acids Induce Differentiation and Sensitize a Radioresistant Breast Cancer Cells to Chemotherapy. BMC Complement. Altern. Med. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Coyle, K.M.; Maxwell, S.; Thomas, M.L.; Marcato, P. Profiling of the Transcriptional Response to All-Trans Retinoic Acid in Breast Cancer Cells Reveals RARE-Independent Mechanisms of Gene Expression. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Shelton, D.N.; Sandoval, I.T.; Eisinger, A.; Chidester, S.; Ratnayake, A.; Ireland, C.M.; Jones, D.A. Up-Regulation of CYP26A1 in Adenomatous Polyposis Coli–Deficient Vertebrates via a WNT-Dependent Mechanism: Implications for Intestinal Cell Differentiation and Colon Tumor Development. Cancer Res. 2006, 66, 7571–7577. [Google Scholar] [CrossRef]
- Osanai, M.; Sawada, N.; Lee, G.-H. Oncogenic and Cell Survival Properties of the Retinoic Acid Metabolizing Enzyme, CYP26A1. Oncogene 2010, 29, 1135–1144. [Google Scholar] [CrossRef]
- Wang, W.; Xu, G.; Ding, C.-L.; Zhao, L.-J.; Zhao, P.; Ren, H.; Qi, Z.-T. All- Trans Retinoic Acid Protects Hepatocellular Carcinoma Cells against Serum-Starvation-Induced Cell Death by Upregulating Collagen 8A2. FEBS J. 2013, 280, 1308–1319. [Google Scholar] [CrossRef]
- Osanai, M.; Lee, G.-H. Elevated Expression of the Retinoic Acid-Metabolizing Enzyme CYP26C1 in Primary Breast Carcinomas. Med. Mol. Morphol. 2016, 49, 22–27. [Google Scholar] [CrossRef]
- Jiang, M.; Zhu, K.; Grenet, J.; Lahti, J.M. Retinoic Acid Induces Caspase-8 Transcription via Phospho-CREB and Increases Apoptotic Responses to Death Stimuli in Neuroblastoma Cells. Biochim. Biophys. Acta (BBA) – Mol. Cell Res. 2008, 1783, 1055–1067. [Google Scholar] [CrossRef]
- Engedal, N.; Auberger, P.; Blomhoff, H.K. Retinoic Acid Regulates Fas-Induced Apoptosis in Jurkat T Cells: Reversal of Mitogen-Mediated Repression of Fas DISC Assembly. J. Leukoc. Biol. 2009, 85, 469–480. [Google Scholar] [CrossRef]
- Dhandapani, L.; Yue, P.; Ramalingam, S.S.; Khuri, F.R.; Sun, S.-Y. Retinoic Acid Enhances TRAIL-Induced Apoptosis in Cancer Cells by Upregulating TRAIL Receptor 1 Expression. Cancer Res. 2011, 71, 5245–5254. [Google Scholar] [CrossRef]
- Teixeira, C.; Pratt, M.A.C. CDK2 Is a Target for Retinoic Acid-Mediated Growth Inhibition in MCF-7 Human Breast Cancer Cells. Mol. Endocrinol. 1997, 11, 1191–1202. [Google Scholar] [CrossRef]
- Christine Pratt, M.A.; Niu, M.; White, D. Differential Regulation of Protein Expression, Growth and Apoptosis by Natural and Synthetic Retinoids. J. Cell. Biochem. 2003, 90, 692–708. [Google Scholar] [CrossRef]
- Wang, J.; Peng, Y.; Sun, Y.W.; He, H.; Zhu, S.; An, X.; Li, M.; Lin, M.C.M.; Zou, B.; Xia, H.H.; et al. All-Trans Retinoic Acid Induces XAF1 Expression Through an Interferon Regulatory Factor-1 Element in Colon Cancer. Gastroenterology 2006, 130, 747–758. [Google Scholar] [CrossRef]
- Donato, L.J.; Noy, N. Suppression of Mammary Carcinoma Growth by Retinoic Acid: Proapoptotic Genes Are Targets for Retinoic Acid Receptor and Cellular Retinoic Acid–Binding Protein II Signaling. Cancer Res. 2005, 65, 8193–8199. [Google Scholar] [CrossRef]
- Seewaldt, V.L.; Kim, J.-H.; Parker, M.B.; Dietze, E.C.; Srinivasan, K.V.; Caldwell, L.E. Dysregulated Expression of Cyclin D1 in Normal Human Mammary Epithelial Cells Inhibits All-Trans-Retinoic Acid-Mediated G0/G1-Phase Arrest and Differentiation in Vitro. Exp. Cell Res. 1999, 249, 70–85. [Google Scholar] [CrossRef]
- Dillard, A.C.; Lane, M.A. Retinol Decreases β-Catenin Protein Levels in Retinoic Acid-Resistant Colon Cancer Cell Lines. Mol. Carcinog. 2007, 46, 315–329. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, W.; Zheng, M.; Liao, X.; Wang, J.; Yang, D.; Lu, W.; Wang, L.; Zhang, S.; Liu, H.; et al. Pin1 Inhibition Potently Suppresses Gastric Cancer Growth and Blocks PI3K/AKT and Wnt/Β-catenin Oncogenic Pathways. Mol. Carcinog. 2019, 58, 1450–1464. [Google Scholar] [CrossRef]
- Seewaldt, V.L.; Dietze, E.C.; Johnson, B.S.; Collins, S.J.; Parker, M.B. Retinoic Acid-Mediated G1-S-Phase Arrest of Normal Human Mammary Epithelial Cells Is Independent of the Level of P53 Protein Expression. Cell Growth Differ. 1999, 10, 49–59. [Google Scholar]
- Heo, S.-H.; Kwak, J.; Jang, K.L. All-Trans Retinoic Acid Induces P53-Depenent Apoptosis in Human Hepatocytes by Activating P14 Expression via Promoter Hypomethylation. Cancer Lett. 2015, 362, 139–148. [Google Scholar] [CrossRef]
- Lee, H.Y.; Dohi, D.F.; Kim, Y.H.; Walsh, G.L.; Consoli, U.; Andreeff, M.; Dawson, M.I.; Hong, W.K.; Kurie, J.M. All-Trans Retinoic Acid Converts E2F into a Transcriptional Suppressor and Inhibits the Growth of Normal Human Bronchial Epithelial Cells through a Retinoic Acid Receptor- Dependent Signaling Pathway. J. Clin. Invest. 1998, 101, 1012–1019. [Google Scholar] [CrossRef][Green Version]
- Sueoka, N.; Lee, H.Y.; Walsh, G.L.; Hong, W.K.; Kurie, J.M. Posttranslational Mechanisms Contribute to the Suppression of Specific Cyclin: CDK Complexes by All-Trans Retinoic Acid in Human Bronchial Epithelial Cells. Cancer Res. 1999, 59, 3838–3844. [Google Scholar] [PubMed]
- García-Regalado, A.; Vargas, M.; García-Carrancá, A.; Aréchaga-Ocampo, E.; González-De la Rosa, C.H. Activation of Akt Pathway by Transcription-Independent Mechanisms of Retinoic Acid Promotes Survival and Invasion in Lung Cancer Cells. Mol. Cancer 2013, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Quintero Barceinas, R.S.; García-Regalado, A.; Aréchaga-Ocampo, E.; Villegas-Sepúlveda, N.; González-De la Rosa, C.H. All-Trans Retinoic Acid Induces Proliferation, Survival, and Migration in A549 Lung Cancer Cells by Activating the ERK Signaling Pathway through a Transcription-Independent Mechanism. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, H.A.N.; Schuller, H.M. Cyclic Adenosine Monophosphate-Dependent Cell Type-Specific Modulation of Mitogenic Signaling by Retinoids in Normal and Neoplastic Lung Cells. Cancer Detect. Prev. 2006, 30, 403–411. [Google Scholar] [CrossRef][Green Version]
- Cho, Y.; Tighe, A.P.; Talmage, D.A. Retinoic Acid Induced Growth Arrest of Human Breast Carcinoma Cells Requires Protein Kinase Cα Expression and Activity. J. Cell. Physiol. 1997, 172, 306–313. [Google Scholar] [CrossRef]
- Cho, Y.; Talmage, D.A. Protein Kinase Cα Expression Confers Retinoic Acid Sensitivity on MDA-MB-231 Human Breast Cancer Cells. Exp. Cell Res. 2001, 269, 97–108. [Google Scholar] [CrossRef]
- Kambhampati, S.; Li, Y.; Verma, A.; Sassano, A.; Majchrzak, B.; Deb, D.K.; Parmar, S.; Giafis, N.; Kalvakolanu, D.V.; Rahman, A.; et al. Activation of Protein Kinase Cδ by All- Trans -Retinoic Acid. J. Biol. Chem. 2003, 278, 32544–32551. [Google Scholar] [CrossRef]
- Berardi, D.E.; Bessone, M.I.D.; Motter, A.; Bal de Kier Joffé, E.D.; Urtreger, A.J.; Todaro, L.B. Involvement of Protein Kinase C α and δ Activities on the Induction of the Retinoic Acid System in Mammary Cancer Cells: PKC/RETINOIDS CROSSTALK AS THERAPEUTIC TARGET. Mol. Carcinog. 2015, 54, 1110–1121. [Google Scholar] [CrossRef]
- Lin, X.-F. RXR Acts as a Carrier for TR3 Nuclear Export in a 9-Cis Retinoic Acid-Dependent Manner in Gastric Cancer Cells. J. Cell Sci. 2004, 117, 5609–5621. [Google Scholar] [CrossRef]
- Ye, X.; Wu, Q.; Liu, S.; Lin, X.; Zhang, B.; Wu, J.; Cai, J.; Zhang, M.; Su, W. Distinct Role and Functional Mode of TR3 and RARα in Mediating ATRA-Induced Signaling Pathway in Breast and Gastric Cancer Cells. Int. J. Biochem. Cell Biol. 2004, 36, 98–113. [Google Scholar] [CrossRef]
- Lin, B.; Kolluri, S.K.; Lin, F.; Liu, W.; Han, Y.-H.; Cao, X.; Dawson, M.I.; Reed, J.C.; Zhang, X. Conversion of Bcl-2 from Protector to Killer by Interaction with Nuclear Orphan Receptor Nur77/TR3. Cell 2004, 116, 527–540. [Google Scholar] [CrossRef]
- Sun, D.-F.; Gao, Z.-H.; Liu, H.-P.; Yuan, Y.; Qu, X.-J. Sphingosine 1-Phosphate Antagonizes the Effect of All-Trans Retinoic Acid (ATRA) in a Human Colon Cancer Cell Line by Modulation of RARβ Expression. Cancer Lett. 2012, 319, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.-N.; Cui, S.-X.; Song, Z.-Y.; Wang, S.-Q.; Sun, S.-Y.; Yu, X.-F.; Li, Y.; Zhang, Y.-H.; Gao, Z.-H.; Qu, X.-J. Over-expression of SphK2 Contributes to ATRA Resistance in Colon Cancer through Rapid Degradation of Cytoplasmic RXRα; by K48/K63-Linked Polyubiquitination. Oncotarget 2017, 8. [Google Scholar]
- Fontana, J.A.; Burrows-Mezu, A.; Clemmons, D.R.; Leroith, D. Retinoid Modulation of Insulin-Like Growth Factor-Binding Proteins and Inhibition of Breast Carcinoma Proliferation*. Endocrinology 1991, 128, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Bentel, J.M.; Lebwohl, D.E.; Cullen, K.J.; Rubin, M.S.; Rosen, N.; Mendelsohn, J.; Miller, W.H. Insulin-like Growth Factors Modulate the Growth Inhibitory Effects of Retinoic Acid on MCF-7 Breast Cancer Cells. J. Cell. Phys. 1995, 165, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Coverley, J.A.; Pattison, S.T.; Baxter, R.C. Insulin-like Growth Factor-Binding Protein-3 Production by MCF-7 Breast Cancer Cells: Stimulation by Retinoic Acid and Cyclic Adenosine Monophosphate and Differential Effects of Estradiol. Endocrinology 1995, 136, 1219–1226. [Google Scholar] [CrossRef]
- Gucev, Z.S.; Oh, Y.; Kelley, K.M.; Rosenfeld, R.G. Insulin-like Growth Factor Binding Protein 3 Mediates Retinoic Acid- and Transforming Growth Factor Beta2-Induced Growth Inhibition in Human Breast Cancer Cells. Cancer Res. 1996, 56, 1545–1550. [Google Scholar]
- Shang, Y.; Baumrucker, C.R.; Green, M.H. Signal Relay by Retinoic Acid Receptors α and β in the Retinoic Acid-Induced Expression of Insulin-like Growth Factor-Binding Protein-3 in Breast Cancer Cells. J. Biol. Chem. 1999, 274, 18005–18010. [Google Scholar] [CrossRef]
- Murakami, K.; Matsuura, T.; Hasumura, S.; Nagamori, S.; Yamada, Y.; Saiki, I. Involvement of Insulin-like Growth Factor Binding Protein-3 in the Retinoic Acid Receptor-α-Mediated Inhibition of Hepatocellular Carcinoma Cell Proliferation. Cancer Lett. 2000, 151, 63–70. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Chang, B.-D.; Fang, J.; Roninson, I.B. Retinoid-Induced Growth Arrest of Breast Carcinoma Cells Involves Co-Activation of Multiple Growth-Inhibitory Genes. Cancer Biol. Ther. 2002, 1, 24–27. [Google Scholar] [CrossRef]
- del Rincón, S.V.; Rousseau, C.; Samanta, R.; Miller, W.H. Retinoic Acid-Induced Growth Arrest of MCF-7 Cells Involves the Selective Regulation of the IRS-1/PI 3-Kinase/AKT Pathway. Oncogene 2003, 22, 3353–3360. [Google Scholar] [CrossRef][Green Version]
- Oh, Y.-I.; Kim, J.-H.; Kang, C.-W. Involvement of Insulin-Like Growth Factor-I Secretion and All-Trans-Retinoic Acid-Induced Decrement in Viability in MCF-7 Cells. Chemotherapy 2011, 57, 17–26. [Google Scholar] [CrossRef]
- del Rincón, S.V.; Guo, Q.; Morelli, C.; Shiu, H.-Y.; Surmacz, E.; Miller, W.H. Retinoic Acid Mediates Degradation of IRS-1 by the Ubiquitin–Proteasome Pathway, via a PKC-Dependant Mechanism. Oncogene 2004, 23, 9269–9279. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schedlich, L.J.; O’Han, M.K.; Leong, G.M.; Baxter, R.C. Insulin-like Growth Factor Binding Protein-3 Prevents Retinoid Receptor Heterodimerization: Implications for Retinoic Acid-Sensitivity in Human Breast Cancer Cells. Biochem. Biophys. Res. Comm. 2004, 314, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Ping, S.; Wang, S.; Zhang, J.; Peng, X. Effect of All-Trans-Retinoic Acid on MRNA Binding Protein P62 in Human Gastric Cancer Cells. Int. J. Biochem. Cell Biol. 2005, 37, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kang, Y.-H.; Schaffer, B.S.; Bach, L.A.; MacDonald, R.G.; Park, J.H.Y. Inhibition of Caco-2 Cell Proliferation by All-Trans Retinoic Acid: Role of Insulin-like Growth Factor Binding Protein-6. J. Cell. Phys. 2002, 190, 92–100. [Google Scholar] [CrossRef]
- Carney, D.N.; De Leij, L. Lung Cancer Biology. Semin. Oncol. 1988, 15, 199–214. [Google Scholar]
- Xu, X.-C. Tumor-Suppressive Activity of Retinoic Acid Receptor-β in Cancer. Cancer Lett. 2007, 253, 14–24. [Google Scholar] [CrossRef]
- Houle, B.; Rochette-Egly, C.; Bradley, W.E. Tumor-Suppressive Effect of the Retinoic Acid Receptor Beta in Human Epidermoid Lung Cancer Cells. Proc. Natl. Acad. Sci. 1993, 90, 985–989. [Google Scholar] [CrossRef]
- Sever, C.E.; Locker, J. Expression of Retinoic Acid Alpha and Beta Receptor Genes in Liver and Hepatocellular Carcinoma. Mol. Carcinog. 1991, 4, 138–144. [Google Scholar] [CrossRef]
- Yang, B.; Guo, M.; Herman, J.G.; Clark, D.P. Aberrant Promoter Methylation Profiles of Tumor Suppressor Genes in Hepatocellular Carcinoma. Am. J. Pathol. 2003, 163, 1101–1107. [Google Scholar] [CrossRef]
- Sano, K.; Takayama, T.; Murakami, K.; Saiki, I.; Makuuchi, M. Over-expression of Retinoic Acid Receptor Alpha in Hepatocellular Carcinoma. Clin. Cancer Res. 2003, 9, 3679–3683. [Google Scholar] [PubMed]
- Jung, J.K.; Park, S.-H.; Jang, K.L. Hepatitis B Virus X Protein Overcomes the Growth-Inhibitory Potential of Retinoic Acid by Downregulating Retinoic Acid Receptor- 2 Expression via DNA Methylation. J. Gen. Virol. 2010, 91, 493–500. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Rice, A.; Chronopoulos, A.; Robinson, B.; Thorpe, S.; Lee, D.A.; Possamai, L.A.; Wang, H.; Pinato, D.J.; et al. Retinoic Acid Receptor-β Is Downregulated in Hepatocellular Carcinoma and Cirrhosis and Its Expression Inhibits Myosin-Driven Activation and Durotaxis in Hepatic Stellate Cells. Hepatology 2019, 69, 785–802. [Google Scholar] [CrossRef] [PubMed]
- Swisshelm, K.; Ryan, K.; Lee, X.; Tsou, H.C.; Peacocke, M.; Sager, R. Down-Regulation of Retinoic Acid Receptor Beta in Mammary Carcinoma Cell Lines and Its up-Regulation in Senescing Normal Mammary Epithelial Cells. Cell Growth Differ. 1994, 5, 133–141. [Google Scholar] [PubMed]
- Deng, G.; Lu, Y.; Zlotnikov, G.; Thor, A.D.; Smith, H.S. Loss of Heterozygosity in Normal Tissue Adjacent to Breast Carcinomas. Science 1996, 274, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Berger, J.; Daxenbichler, G.; Müller-Holzner, E.; Widschwendter, A.; Mayr, A.; Marth, C.; Zeimet, A.G. Loss of Retinoic Acid Receptor Beta Expression in Breast Cancer and Morphologically Normal Adjacent Tissue but Not in the Normal Breast Tissue Distant from the Cancer. Cancer Res. 1997, 57, 4158–4161. [Google Scholar]
- Peng, X.; Yun, D.; Christov, K. Breast Cancer Progression in MCF10A Series of Cell Lines Is Associated with Alterations in Retinoic Acid and Retinoid X Receptors and with Differential Response to Retinoids. Int. J. Oncol. 2004, 25, 961–971. [Google Scholar]
- Ravi, R.K.; Knudsen, E.S.; Williams, J.R.; Dillehay, L.E.; Nelkin, B.D.; Kalemkerian, G.P.; Feramisco, J.R.; Mabry, M. Retinoic Acid-Mediated Growth Inhibition of Small Cell Lung Cancer Cells Is Associated with Reduced Myc and Increased P27Kip1 Expression. Int. J. Cancer 1999, 80, 935–943. [Google Scholar]
- Li, Y.; Dawson, M.I.; Agadir, A.; Lee, M.O.; Jong, L.; Hobbs, P.D.; Zhang, X.K. Regulation of RAR Beta Expression by RAR- and RXR-Selective Retinoids in Human Lung Cancer Cell Lines: Effect on Growth Inhibition and Apoptosis Induction. Int. J. Cancer 1998, 75, 88–95. [Google Scholar] [CrossRef]
- Wan, H.; Hong, W.K.; Lotan, R. Increased Retinoic Acid Responsiveness in Lung Carcinoma Cells That Are Nonresponsive despite the Presence of Endogenous Retinoic Acid Receptor (RAR) Beta by Expression of Exogenous Retinoid Receptors Retinoid X Receptor Alpha, RAR Alpha, and RAR Gamma. Cancer Res. 2001, 61, 556–564. [Google Scholar] [PubMed]
- Choi, E.J.; Whang, Y.M.; Kim, S.J.; Kim, H.J.; Kim, Y.H. Combinational Treatment with Retinoic Acid Derivatives in Non-Small Cell Lung Carcinoma In Vitro. J. Korean Med. Sci. 2007, 22, S52. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Choi, Y.-K.; Kwon, H.-J.; Yang, H.-K.; Choi, J.-H.; Kim, D.-Y. Downregulation of Gelsolin and Retinoic Acid Receptor Beta Expression in Gastric Cancer Tissues through Histone Deacetylase 1. J. Gastroenterol. Hepatol. 2004, 19, 218–224. [Google Scholar] [CrossRef]
- Hu, K.-W.; Chen, F.-H.; Ge, J.-F.; Cao, L.-Y.; Li, H. Retinoid Receptors in Gastric Cancer: Expression and Influence on Prognosis. Asian Pac. J. Cancer Prev. 2012, 13, 1809–1817. [Google Scholar] [CrossRef]
- Nicke, B.; Riecken, E.-O.; Rosewicz, S. Induction of Retinoic Acid Receptor Beta Mediates Growth Inhibition in Retinoid Resistant Human Colon Carcinoma Cells. Gut 1999, 45, 51–57. [Google Scholar] [CrossRef][Green Version]
- Lee, M.-O.; Han, S.-Y.; Jiang, S.; Han Park, J.; Kim, S.J. Differential Effects of Retinoic Acid on Growth and Apoptosis in Human Colon Cancer Cell Lines Associated with the Induction of Retinoic Acid Receptor β. Biochem. Pharmacol. 2000, 59, 485–496. [Google Scholar] [CrossRef]
- Côté, S.; Momparler, R.L. Antineoplastic action of all-trans retinoic acid and 5-Aza-2'deoxycytidine on human DLD-1 colon carcinoma cells. Cell Pharmacol. 1995, 2, 221–228. [Google Scholar]
- Côté, S.; Momparler, R.L. Activation of the Retinoic Acid Receptor β Gene by 5-Aza-2'-Deoxycytidine in Human DLD-1 Colon Carcinoma Cells. Anti-Cancer Drugs 1997, 8, 56–61. [Google Scholar]
- Côté, S.; Sinnett, D.; Momparler, R.L. Demethylation by 5-Aza-2'-Deoxycytidine of Specific 5-Methylcytosine Sites in the Promoter Region of the Retinoic Acid Receptor β Gene in Human Colon Carcinoma Cells. Anti-Cancer Drugs 1998, 9, 743–750. [Google Scholar]
- Youssef, E.M.; Estecio, M.R.H.; Issa, J.-P.J. Methylation and Regulation of Expression of Different Retinoic Acid Receptor Beta Isoforms in Human Colon Cancer. Cancer Biol. Ther. 2004, 3, 82–86. [Google Scholar] [CrossRef][Green Version]
- Sirchia, S.M.; Ferguson, A.T.; Sironi, E.; Subramanyan, S.; Orlandi, R.; Sukumar, S.; Sacchi, N. Evidence of Epigenetic Changes Affecting the Chromatin State of the Retinoic Acid Receptor Β2 Promoter in Breast Cancer Cells. Oncogene 2000, 19, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Berger, J.; Hermann, M.; Muller, H.M.; Amberger, A.; Zeschnigk, M.; Widschwendter, A.; Abendstein, B.; Zeimet, A.G.; Daxenbichler, G.; et al. Methylation and Silencing of the Retinoic Acid Receptor- 2 Gene in Breast Cancer. JNCI J. Natl. Cancer Inst. 2000, 92, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Mori, I.; Shan, L.; Nakamura, M.; Nakamura, Y.; Utsunomiya, H.; Yoshimura, G.; Suzuma, T.; Tamaki, T.; Umemura, T.; et al. Biallelic Inactivation of Retinoic Acid Receptor Beta2 Gene by Epigenetic Change in Breast Cancer. Am. J. Pathol. 2001, 158, 299–303. [Google Scholar] [CrossRef]
- Suh, Y.-A.; Lee, H.-Y.; Virmani, A.; Wong, J.; Mann, K.K.; Miller, W.H.; Gazdar, A.; Kurie, J.M. Loss of Retinoic Acid Receptor Beta Gene Expression Is Linked to Aberrant Histone H3 Acetylation in Lung Cancer Cell Lines. Cancer Res. 2002, 62, 3945–3949. [Google Scholar]
- Zöchbauer-Müller, S.; Lam, S.; Toyooka, S.; Virmani, A.K.; Toyooka, K.O.; Seidl, S.; Minna, J.D.; Gazdar, A.F. Aberrant Methylation of Multiple Genes in the Upper Aerodigestive Tract Epithelium of Heavy Smokers: Methylation in Smokers. Int. J. Cancer 2003, 107, 612–616. [Google Scholar] [CrossRef]
- Hayashi, K.; Yokozaki, H.; Goodison, S.; Oue, N.; Suzuki, T.; Lotan, R.; Yasui, W.; Tahara, E. Inactivation of Retinoic Acid Receptor Beta by Promoter CpG Hypermethylation in Gastric Cancer. Differentiation 2001, 68, 13–21. [Google Scholar] [CrossRef]
- Oue, N.; Motoshita, J.; Yokozaki, H.; Hayashi, K.; Tahara, E.; Taniyama, K.; Matsusaki, K.; Yasui, W. Distinct Promoter Hypermethylation Ofp16INK4a,CDH1, AndRAR-Beta in Intestinal, Diffuse-Adherent, and Diffuse-Scattered Type Gastric Carcinomas. J. Pathol. 2002, 198, 55–59. [Google Scholar] [CrossRef]
- Oue, N.; Oshimo, Y.; Nakayama, H.; Ito, R.; Yoshida, K.; Matsusaki, K.; Yasui, W. DNA Methylation of Multiple Genes in Gastric Carcinoma: Association with Histological Type and CpG Island Methylator Phenotype. Cancer Sci. 2003, 94, 901–905. [Google Scholar] [CrossRef]
- Swellam, M.; Abdelmaksoud, M.D.E.; Sayed Mahmoud, M.; Ramadan, A.; Abdel-Moneem, W.; Hefny, M.M. Aberrant Methylation of APC and RARβ2 Genes in Breast Cancer Patients: Aberrant Methylated Genes in Breast Cancer. IUBMB Life 2015, 67, 61–68. [Google Scholar] [CrossRef]
- Fang, C.; Jian, Z.-Y.; Shen, X.-F.; Wei, X.-M.; Yu, G.-Z.; Zeng, X.-T. Promoter Methylation of the Retinoic Acid Receptor Beta2 (RARβ2) Is Associated with Increased Risk of Breast Cancer: A PRISMA Compliant Meta-Analysis. PLoS ONE 2015, 10, e0140329. [Google Scholar] [CrossRef]
- Sun, J.; Xu, X.; Liu, J.; Liu, H.; Fu, L.; Gu, L. Epigenetic Regulation of Retinoic Acid Receptor Β2 Gene in the Initiation of Breast Cancer. Med. Oncol. 2011, 28, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Barnicle, A.; Seoighe, C.; Greally, J.M.; Golden, A.; Egan, L.J. Inflammation-Associated DNA Methylation Patterns in Epithelium of Ulcerative Colitis. Epigenetics 2017, 12, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q. Modulation of Retinoic Acid Sensitivity in Lung Cancer Cells through Dynamic Balance of Orphan Receptors Nur77 and COUP-TF and Their Heterodimerization. EMBO J. 1997, 16, 1656–1669. [Google Scholar] [CrossRef]
- Chen, G.; Lin, B.; Dawson, M.I.; Zhang, X. Nicotine Modulates the Effects of Retinoids on Growth Inhibition and RARβ Expression in Lung Cancer Cells. Int. J. Cancer 2002, 99, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Chen, G.-Q.; Xiao, D.; Kolluri, S.K.; Cao, X.; Su, H.; Zhang, X.-K. Orphan Receptor COUP-TF Is Required for Induction of Retinoic Acid Receptor Beta, Growth Inhibition, and Apoptosis by Retinoic Acid in Cancer Cells. Mol. Cell. Biol. 2000, 20, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, L.M.; Riggs, K.A.; Hockenberry, A.M.; Oliver, L.D.; Barnhart, K.G.; Cai, J.; Pierce, W.M.; Ivanova, M.M.; Bates, P.J.; Appana, S.N.; et al. Identification and Characterization of Nucleolin as a COUP-TFII Coactivator of Retinoic Acid Receptor β Transcription in Breast Cancer Cells. PLoS ONE 2012, 7, e38278. [Google Scholar] [CrossRef]
- Swift, M.E.; Wallden, B.; Wayner, E.A.; Swisshelm, K. Truncated RAR Beta Isoform Enhances Proliferation and Retinoid Resistance. J. Cell. Physiol. 2006, 209, 718–725. [Google Scholar] [CrossRef]
- Liu, S.; Wu, Q.; Chen, Z.M.; Su, W.J. The Effect Pathway of Retinoic Acid through Regulation of Retinoic Acid Receptor α in Gastric Cancer Cells. World J. Gastroenterol. 2001, 7, 662. [Google Scholar] [CrossRef]
- Centritto, F.; Paroni, G.; Bolis, M.; Garattini, S.K.; Kurosaki, M.; Barzago, M.M.; Zanetti, A.; Fisher, J.N.; Scott, M.F.; Pattini, L.; et al. Cellular and Molecular Determinants of All- Trans Retinoic Acid Sensitivity in Breast Cancer: Luminal Phenotype and RAR α Expression. EMBO Mol. Med. 2015, 7, 950–972. [Google Scholar] [CrossRef]
- Alsafadi, S.; Even, C.; Falet, C.; Goubar, A.; Commo, F.; Scott, V.; Quidville, V.; Albiges, L.; Dieci, M.-V.; Guegan, J.; et al. Retinoic Acid Receptor Alpha Amplifications and Retinoic Acid Sensitivity in Breast Cancers. Clin. Breast Cancer 2013, 13, 401–408. [Google Scholar] [CrossRef]
- Farias, E.F.; Arapshian, A.; Bleiweiss, I.J.; Waxman, S.; Zelent, A.; Mira-Y-Lopez, R. Retinoic Acid Receptor Alpha2 Is a Growth Suppressor Epigenetically Silenced in MCF-7 Human Breast Cancer Cells. Cell Growth Differ. 2002, 13, 335–341. [Google Scholar] [PubMed]
- Yan, T.-D.; Wu, H.; Zhang, H.-P.; Lu, N.; Ye, P.; Yu, F.-H.; Zhou, H.; Li, W.-G.; Cao, X.; Lin, Y.-Y.; et al. Oncogenic Potential of Retinoic Acid Receptor- in Hepatocellular Carcinoma. Cancer Res. 2010, 70, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Nass, S.J.; Herman, J.G.; Gabrielson, E.; Iversen, P.W.; Parl, F.F.; Davidson, N.E.; Graff, J.R. Aberrant Methylation of the Estrogen Receptor and E-Cadherin 5’ CpG Islands Increases with Malignant Progression in Human Breast Cancer. Cancer Res. 2000, 60, 4346–4348. [Google Scholar] [PubMed]
- Duffy, M.J. Estrogen Receptors: Role in Breast Cancer. Crit. Rev. Clin. Lab. Sci. 2006, 43, 325–347. [Google Scholar] [CrossRef]
- Roman, S.D.; Clarke, C.L.; Hall, R.E.; Alexander, I.E.; Sutherland, R.L. Expression and Regulation of Retinoic Acid Receptors in Human Breast Cancer Cells. Cancer Res. 1992, 52, 2236–2242. [Google Scholar]
- van der Burg, B.; van der Leede, B.M.; Kwakkenbos-Isbrücker, L.; Salverda, S.; de Laat, S.W.; van der Saag, P.T. Retinoic Acid Resistance of Estradiol-Independent Breast Cancer Cells Coincides with Diminished Retinoic Acid Receptor Function. Mol. Cell. Endocrinol. 1993, 91, 149–157. [Google Scholar] [CrossRef]
- Rubin, M.; Fenig, E.; Rosenauer, A.; Menendez-Botet, C.; Achkar, C.; Bentel, J.M.; Yahalom, J.; Mendelsohn, J.; Miller, W.H. 9-Cis Retinoic Acid Inhibits Growth of Breast Cancer Cells and down-Regulates Estrogen Receptor RNA and Protein. Cancer Res. 1994, 54, 6549–6556. [Google Scholar]
- Zhao, Z.; Zhang, Z.; Soprano, D.R.; Soprano, K.J. Effect of 9-Cis-Retinoic Acid on Growth and RXR Expression in Human Breast Cancer Cells. Exp. Cell Res. 1995, 219, 555–561. [Google Scholar] [CrossRef]
- Liu, Y.; Lee, M.O.; Wang, H.G.; Li, Y.; Hashimoto, Y.; Klaus, M.; Reed, J.C.; Zhang, X. Retinoic Acid Receptor Beta Mediates the Growth-Inhibitory Effect of Retinoic Acid by Promoting Apoptosis in Human Breast Cancer Cells. Mol. Cell. Biol. 1996, 16, 1138–1149. [Google Scholar] [CrossRef]
- Fitzgerald, P.; Teng, M.; Chandraratna, R.A.; Heyman, R.A.; Allegretto, E.A. Retinoic Acid Receptor Alpha Expression Correlates with Retinoid-Induced Growth Inhibition of Human Breast Cancer Cells Regardless of Estrogen Receptor Status. Cancer Res. 1997, 57, 2642–2650. [Google Scholar]
- Schneider, S.M.; Offterdinger, M.; Huber, H.; Grunt, T.W. Activation of Retinoic Acid Receptor Alpha Is Sufficient for Full Induction of Retinoid Responses in SK-BR-3 and T47D Human Breast Cancer Cells. Cancer Res. 2000, 60, 5479–5487. [Google Scholar]
- Sheikh, M.S.; Shao, Z.-M.; Chen, J.-C.; Hussain, A.; Jetten, A.M.; Fontana, J.A. Estrogen Receptor-Negative Breast Cancer Cells Transfected with the Estrogen Receptor Exhibit Increased RARα Gene Expression and Sensitivity to Growth Inhibition by Retinoic Acid. J. Cell. Biochem. 1993, 53, 394–404. [Google Scholar] [CrossRef]
- Toma, S.; Isnardi, L.; Raffo, P.; Dastoli, G.; De Francisci, E.; Riccardi, L.; Palumbo, R.; Bollag, W. Effects of ALL-Trans-Retinoic Acid and 13-Cis-Retinoic Acid on Breast-Cancer Cell Lines: Growth Inhibition and Apoptosis Induction. Int. J. Cancer 1997, 70, 619–627. [Google Scholar] [CrossRef]
- Phipps, S.M.O.; Love, W.K.; White, T.; Andrews, L.G.; Tollefsbol, T.O. Retinoid-Induced Histone Deacetylation Inhibits Telomerase Activity in Estrogen Receptor-Negative Breast Cancer Cells. Anticancer Res. 2009, 29, 4959–4964. [Google Scholar]
- Hua, S.; Kittler, R.; White, K.P. Genomic Antagonism between Retinoic Acid and Estrogen Signaling in Breast Cancer. Cell 2009, 137, 1259–1271. [Google Scholar] [CrossRef]
- Salvatori, L.; Ravenna, L.; Caporuscio, F.; Principessa, L.; Coroniti, G.; Frati, L.; Russo, M.A.; Petrangeli, E. Action of Retinoic Acid Receptor on EGFR Gene Transactivation and Breast Cancer Cell Proliferation: Interplay with the Estrogen Receptor. Biomed. Pharmacother. 2011, 65, 307–312. [Google Scholar] [CrossRef]
- Miro Estruch, I.; de Haan, L.H.J.; Melchers, D.; Houtman, R.; Louisse, J.; Groten, J.P.; Rietjens, I.M.C.M. The Effects of All-Trans Retinoic Acid on Estrogen Receptor Signaling in the Estrogen-Sensitive MCF/BUS Subline. J. Recept. Signal Transduct. 2018, 38, 112–121. [Google Scholar] [CrossRef]
- Hsu, L.-H.; Chu, N.-M.; Kao, S.-H. Estrogen, Estrogen Receptor and Lung Cancer. Int. J. Mol. Sci. 2017, 18, 1713. [Google Scholar] [CrossRef]
- Sukocheva, O.A. Estrogen, Estrogen Receptors, and Hepatocellular Carcinoma: Are We There Yet? World J. Gastroenterol. 2018, 24, 1–4. [Google Scholar] [CrossRef]
- Ge, H.; Yan, Y.; Tian, F.; Wu, D.; Huang, Y. Prognostic Value of Estrogen Receptor α and Estrogen Receptor β in Gastric Cancer Based on a Meta-Analysis and The Cancer Genome Atlas (TCGA) Datasets. Int. J. Surg. 2018, 53, 24–31. [Google Scholar] [CrossRef]
- Caiazza, F.; Ryan, E.J.; Doherty, G.; Winter, D.C.; Sheahan, K. Estrogen Receptors and Their Implications in Colorectal Carcinogenesis. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-Cis Retinoic Acid and Peroxisome Proliferator Signaling Pathways through Heterodimer Formation of Their Receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Davies, P.J.A.; Crombie, D.L.; Bischoff, E.D.; Cesario, R.M.; Jow, L.; Hamann, L.G.; Boehm, M.F.; Mondon, C.E.; Nadzan, A.M.; et al. Sensitization of Diabetic and Obese Mice to Insulin by Retinoid X Receptor Agonists. Nature 1997, 386, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Miyazaki, Y.; Shinomura, Y.; Kondo, S.; Kanayama, S.; Matsuzawa, Y. Peroxisome Proliferator-Activated Receptor γ Induces Growth Arrest and Differentiation Markers of Human Colon Cancer Cells. Jpn. J. Cancer Res. 1999, 90, 75–80. [Google Scholar] [CrossRef]
- Elstner, E.; Muller, C.; Koshizuka, K.; Williamson, E.A.; Park, D.; Asou, H.; Shintaku, P.; Said, J.W.; Heber, D.; Koeffler, H.P. Ligands for Peroxisome Proliferator-Activated Receptor and Retinoic Acid Receptor Inhibit Growth and Induce Apoptosis of Human Breast Cancer Cells in Vitro and in BNX Mice. Proc. Natl. Acad. Sci. 1998, 95, 8806–8811. [Google Scholar] [CrossRef]
- Sato, H.; Ishihara, S.; Kawashima, K.; Moriyama, N.; Suetsugu, H.; Kazumori, H.; Okuyama, T.; Rumi, M.A.K.; Fukuda, R.; Nagasue, N.; et al. Expression of Peroxisome Proliferator-Activated Receptor (PPAR)γ in Gastric Cancer and Inhibitory Effects of PPARγ Agonists. Br. J. Cancer 2000, 83, 1394–1400. [Google Scholar] [CrossRef]
- Yang, W.-L. Activation of the PPAR Pathway Induces Apoptosis and COX-2 Inhibition in HT-29 Human Colon Cancer Cells. Carcinogenesis 2001, 22, 1379–1383. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Cione, E.; Qi, H.; Pingitore, A.; Perri, M.; Catalano, S.; Vizza, D.; Panno, M.L.; Genchi, G.; Fuqua, S.A.W.; et al. Combined Low Doses of PPARγ and RXR Ligands Trigger an Intrinsic Apoptotic Pathway in Human Breast Cancer Cells. Am. J. Pathol. 2009, 175, 1270–1280. [Google Scholar] [CrossRef]
- Allred, C.D.; Kilgore, M.W. Selective Activation of PPARγ in Breast, Colon, and Lung Cancer Cell Lines. Mol. Cell. Endocrinol. 2005, 235, 21–29. [Google Scholar] [CrossRef]
- Matsushima-Nishiwaki, R.; Okuno, M.; Adachi, S.; Sano, T.; Akita, K.; Moriwaki, H.; Friedman, S.L.; Kojima, S. Phosphorylation of Retinoid X Receptor Alpha at Serine 260 Impairs Its Metabolism and Function in Human Hepatocellular Carcinoma. Cancer Res. 2001, 61, 7675–7682. [Google Scholar]
- Yamazaki, K.; Shimizu, M.; Okuno, M.; Matsushima-Nishiwaki, R.; Kanemura, N.; Araki, H.; Tsurumi, H.; Kojima, S.; Weinstein, I.B.; Moriwaki, H. Synergistic Effects of RXR and PPAR Ligands to Inhibit Growth in Human Colon Cancer Cells Phosphorylated RXR Is a Critical Target for Colon Cancer Management. Gut 2007, 56, 1557–1563. [Google Scholar] [CrossRef]
- Shaw, N.; Elholm, M.; Noy, N. Retinoic Acid Is a High Affinity Selective Ligand for the Peroxisome Proliferator-Activated Receptor β/δ. J. Biol. Chem. 2003, 278, 41589–41592. [Google Scholar] [CrossRef]
- Wagner, E.F. AP-1 – Introductory Remarks. Oncogene 2001, 20, 2334–2335. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ma, W.-Y.; Dawson, M.I.; Rincon, M.; Flavell, R.A.; Dong, Z. Blocking Activator Protein-1 Activity, but Not Activating Retinoic Acid Response Element, Is Required for the Antitumor Promotion Effect of Retinoic Acid. Proc. Natl. Acad. Sci. 1997, 94, 5826–5830. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, B.; Slager-Davidov, R.; van der Leede, B.M.; de Laat, S.W.; van der Saag, P.T. Differential Regulation of AP1 Activity by Retinoic Acid in Hormone-Dependent and -Independent Breast Cancer Cells. Mol. Cell. Endocrinol. 1995, 112, 143–152. [Google Scholar] [CrossRef]
- Yang, L.; Kim, H.T.; Munoz-Medellin, D.; Reddy, P.; Brown, P.H. Induction of Retinoid Resistance in Breast Cancer Cells by Over-expression of CJun. Cancer Res. 1997, 57, 4652–4661. [Google Scholar]
- Sapi, E.; Flick, M.B.; Tartaro, K.; Kim, S.; Rakhlin, Y.; Rodov, S.; Kacinski, B.M. Effect of All-Trans-Retinoic Acid on c-Fms Proto-Oncogene [Colony-Stimulating Factor 1 (CSF-1) Receptor] Expression and CSF-1-Induced Invasion and Anchorage-Independent Growth of Human Breast Carcinoma Cells. Cancer Res. 1999, 59, 5578–5585. [Google Scholar]
- Lin, F.; Xiao, D.; Kolluri, S.K.; Zhang, X. Unique Anti-Activator Protein-1 Activity of Retinoic Acid Receptor Beta. Cancer Res. 2000, 60, 3271–3280. [Google Scholar]
- Dedieu, S.; Lefebvre, P. Retinoids Interfere with the AP1 Signaling Pathway in Human Breast Cancer Cells. Cell. Signal. 2006, 18, 889–898. [Google Scholar] [CrossRef]
- Lee, H.Y.; Dawson, M.I.; Claret, F.X.; Chen, J.D.; Walsh, G.L.; Hong, W.K.; Kurie, J.M. Evidence of a Retinoid Signaling Alteration Involving the Activator Protein 1 Complex in Tumorigenic Human Bronchial Epithelial Cells and Non-Small Cell Lung Cancer Cells. Cell Growth Differ. 1997, 8, 283–291. [Google Scholar]
- Wu, Q.; Chen, Z.; Su, W. Anticancer Effect of Retinoic Acid via AP-1 Activity Repression Is Mediated by Retinoic Acid Receptor α and β in Gastric Cancer Cells. Int. J. Biochem. Cell Biol. 2002, 34, 1102–1114. [Google Scholar] [CrossRef]
- Huang, S.-L.; Shyu, R.-Y.; Yeh, M.-Y.; Jiang, S.-Y. Cloning and Characterization of a Novel Retinoid-Inducible Gene 1(RIG1) Deriving from Human Gastric Cancer Cells. Mol. Cell. Endocrinol. 2000, 159, 15–24. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Wu, M.-S.; Chen, L.-M.; Hung, M.-W.; Lin, H.-E.; Chang, G.-G.; Chang, T.-C. Identification and Characterization of the Retinoic Acid Response Elements in the Human RIG1 Gene Promoter. Biochem. Biophys. Res. Comm. 2005, 331, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Shyu, R.-Y.; Jiang, S.-Y.; Chou, J.-M.; Shih, Y.-L.; Lee, M.-S.; Yu, J.-C.; Chao, P.-C.; Hsu, Y.-J.; Jao, S.-W. RARRES3 Expression Positively Correlated to Tumor Differentiation in Tissues of Colorectal Adenocarcinoma. Br. J. Cancer 2003, 89, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhou, Y.; Zheng, Y.; Fan, J.; Zhou, W.; Ng, I.O.L.; Sun, H.; Qin, L.; Qiu, S.; Lee, J.M.F.; et al. Hepatic RIG-I Predicts Survival and Interferon-α Therapeutic Response in Hepatocellular Carcinoma. Cancer Cell 2014, 25, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Shyu, R.-Y.; Chang, S.-C.; Yu, J.-C.; Hsu, S.-J.; Chou, J.-M.; Lee, M.-S.; Jiang, S.-Y. Expression and Regulation of Retinoid-Inducible Gene 1 (RIG1) in Breast Cancer. Anticancer Res. 2005, 25, 2453–2460. [Google Scholar]
- Liu, Z.; Dou, C.; Jia, Y.; Li, Q.; Zheng, X.; Yao, Y.; Liu, Q.; Song, T. RIG-I Suppresses the Migration and Invasion of Hepatocellular Carcinoma Cells by Regulating MMP9. Int. J. Oncol. 2015, 46, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, E.; Chandraratna, R.A.S.; Hong, W.K.; Lotan, R. Induction of TIG3, a Putative Class II Tumor Suppressor Gene, by Retinoic Acid in Head and Neck and Lung Carcinoma Cells and Its Association with Suppression of the Transformed Phenotype. Oncogene 2003, 22, 4627–4635. [Google Scholar] [CrossRef] [PubMed]
- Youssef, E.M.; Chen, X.; Higuchi, E.; Kondo, Y.; Garcia-Manero, G.; Lotan, R.; Issa, J.-P.J. Hypermethylation and Silencing of the Putative Tumor Suppressor Tazarotene-Induced Gene 1 in Human Cancers. Cancer Res. 2004, 64, 2411–2417. [Google Scholar] [CrossRef] [PubMed]
- Son, M.S.; Kang, M.-J.; Park, H.C.; Chi, S.-G.; Kim, Y.H. Expression and Mutation Analysis of TIG1 (Tazarotene-Induced Gene 1) in Human Gastric Cancer. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2009, 17, 571–580. [Google Scholar] [CrossRef] [PubMed]
- So, K.; Tamura, G.; Honda, T.; Homma, N.; Waki, T.; Togawa, N.; Nishizuka, S.; Motoyama, T. Multiple Tumor Suppressor Genes Are Increasingly Methylated with Age in Non-Neoplastic Gastric Epithelia. Cancer Sci. 2006, 97, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-H.; Wu, W.-G.; Ding, J. Aberrant TIG1 Methylation Associated with Its Decreased Expression and Clinicopathological Significance in Hepatocellular Carcinoma. Tumor Biol. 2014, 35, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-Y. Decreased Expression of Type II Tumor Suppressor Gene RARRES3 in Tissues of Hepatocellular Carcinoma and Cholangiocarcinoma. World J. Gastroenterol. 2005, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chiu, D.K.-C.; Tsang, F.H.-C.; Law, C.-T.; Cheng, C.L.-H.; Au, S.L.-K.; Lee, J.M.-F.; Wong, C.C.-L.; Ng, I.O.-L.; Wong, C.-M. Histone Methyltransferase G9a Promotes Liver Cancer Development by Epigenetic Silencing of Tumor Suppressor Gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Suzuki, Y.; Matsui, T.; Tohyama, M.; Wanaka, A.; Takagi, T. Cloning of a Retinoic Acid-Induced Gene, GT1, in the Embryonal Carcinoma Cell Line P19: Neuron-Specific Expression in the Mouse Brain. Mol. Brain Res. 1995, 31, 1–9. [Google Scholar] [CrossRef]
- Laperriere, D.; Wang, T.-T.; White, J.H.; Mader, S. Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genomics 2007, 8, 23. [Google Scholar] [CrossRef]
- Werner, S.; Brors, B.; Eick, J.; Marques, E.; Pogenberg, V.; Parret, A.; Kemming, D.; Wood, A.W.; Edgren, H.; Neubauer, H.; et al. Suppression of Early Hematogenous Dissemination of Human Breast Cancer Cells to Bone Marrow by Retinoic Acid-Induced 2. Cancer Discov. 2015, 5, 506–519. [Google Scholar] [CrossRef]
- Yan, W.; Wu, K.; Herman, J.G.; Xu, X.; Yang, Y.; Dai, G.; Guo, M. Retinoic Acid-Induced 2 (RAI2) Is a Novel Tumor Suppressor, and Promoter Region Methylation of RAI2 Is a Poor Prognostic Marker in Colorectal Cancer. Clin. Epigenetics 2018, 10. [Google Scholar] [CrossRef]
- Cheng, Y.; Lotan, R. Molecular Cloning and Characterization of a Novel Retinoic Acid-Inducible Gene That Encodes a Putative G Protein-Coupled Receptor. J. Biol. Chem. 1998, 273, 35008–35015. [Google Scholar] [CrossRef]
- Nagahata, T. Identification of RAI3 as a Therapeutic Target for Breast Cancer. Endocr. Relat. Cancer 2005, 12, 65–73. [Google Scholar] [CrossRef]
- Wu, Q.; Ding, W.; Mirza, A.; Van Arsdale, T.; Wei, I.; Bishop, W.R.; Basso, A.; McClanahan, T.; Luo, L.; Kirschmeier, P.; et al. Integrative Genomics Revealed RAI3 Is a Cell Growth-Promoting Gene and a Novel P53 Transcriptional Target. J. Biol. Chem. 2005, 280, 12935–12943. [Google Scholar] [CrossRef] [PubMed]
- Zougman, A.; Hutchins, G.G.; Cairns, D.A.; Verghese, E.; Perry, S.L.; Jayne, D.G.; Selby, P.J.; Banks, R.E. Retinoic Acid-Induced Protein 3: Identification and Characterisation of a Novel Prognostic Colon Cancer Biomarker. Eur. J. Cancer 2013, 49, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Fujimoto, J.; Men, T.; Ye, X.; Deng, J.; Lacroix, L.; Clifford, J.L.; Mao, L.; Van Pelt, C.S.; Lee, J.J.; et al. Identification of the Retinoic Acid-Inducible Gprc5a As a New Lung Tumor Suppressor Gene. JNCI J. Natl. Cancer Inst. 2007, 99, 1668–1682. [Google Scholar] [CrossRef] [PubMed]
- Kadara, H.; Fujimoto, J.; Men, T.; Ye, X.; Lotan, D.; Lee, J.-S.; Lotan, R. A Gprc5a Tumor Suppressor Loss of Expression Signature Is Conserved, Prevalent, and Associated with Survival in Human Lung Adenocarcinomas. Neoplasia 2010, 12, 499-IN8. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Hu, H.; Kuang, M.; Chen, Z.; Tao, X.; Fang, S.; Sun, Y.; Zhang, Y.; Chen, H. Super Enhancer Associated RAI14 Is a New Potential Biomarker in Lung Adenocarcinoma. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yong, W.-P.; Yap, C.S.; Vijayaraghavan, A.; Sinha, R.A.; Singh, B.K.; Xiu, S.; Manesh, S.; Ngo, A.; Lim, A.; et al. An Integrative Approach Identified Genes Associated with Drug Response in Gastric Cancer. Carcinogenesis 2015, 36, 441–451. [Google Scholar] [CrossRef] [PubMed]
- He, X.-Y.; Zhao, J.; Chen, Z.-Q.; Jin, R.; Liu, C.-Y. High Expression of Retinoic Acid Induced 14 (RAI14) in Gastric Cancer and Its Prognostic Value. Med. Sci. Monit. 2018, 24, 2244–2251. [Google Scholar] [CrossRef]
- Chen, C.; Maimaiti, A.; Zhang, X.; Qu, H.; Sun, Q.; He, Q.; Yu, W. Knockdown of RAI14 Suppresses the Progression of Gastric Cancer. OncoTargets Ther. 2018, 11, 6693–6703. [Google Scholar] [CrossRef]
- Nolte, C.; De Kumar, B.; Krumlauf, R. Hox Genes: Downstream “Effectors” of Retinoic Acid Signaling in Vertebrate Embryogenesis. Genes 2019, e23306. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, L.-J.; Yang, Y.; Wang, R.-Y.; Ren, H.; Zhao, P.; Zhou, W.-P.; Qi, Z.-T. Retinoic Acid Induced 16 Enhances Tumorigenesis and Serves as a Novel Tumor Marker for Hepatocellular Carcinoma. Carcinogenesis 2012, 33, 2578–2585. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, H.; Lee, J.; Liang, X.; Wu, X.; Zhu, T.; Lo, P.; Zhang, X.; Sukumar, S. HOXA5 Acts Directly Downstream of Retinoic Acid Receptor β and Contributes to Retinoic Acid–Induced Apoptosis and Growth Inhibition. Cancer Res. 2007, 67, 8007–8013. [Google Scholar] [CrossRef] [PubMed]
- Teo, W.W.; Merino, V.F.; Cho, S.; Korangath, P.; Liang, X.; Wu, R.; Neumann, N.M.; Ewald, A.J.; Sukumar, S. HOXA5 Determines Cell Fate Transition and Impedes Tumor Initiation and Progression in Breast Cancer through Regulation of E-Cadherin and CD24. Oncogene 2016, 35, 5539–5551. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Cardenas, R.; Wang, B.; Persson, J.; Mongan, N.P.; Grabowska, A.; Allegrucci, C. HOXC8 Regulates Self-Renewal, Differentiation and Transformation of Breast Cancer Stem Cells. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Bhatlekar, S.; Viswanathan, V.; Fields, J.Z.; Boman, B.M. Over-expression of HOXA4 and HOXA9 Genes Promotes Self-Renewal and Contributes to Colon Cancer Stem Cell Overpopulation. J. Cell. Physiol. 2018, 233, 727–735. [Google Scholar] [CrossRef]
- Kalemkerian, G.P.; Jasti, R.K.; Celano, P.; Nelkin, B.D.; Mabry, M. All-Trans-Retinoic Acid Alters Myc Gene Expression and Inhibits in Vitro Progression in Small Cell Lung Cancer. Cell Growth Differ. 1994, 5, 55–60. [Google Scholar] [PubMed]
- Saunders, D.E.; Christensen, C.; Wappler, N.L.; Schultz, J.F.; Lawrence, W.D.; Malviya, V.K.; Malone, J.M.; Deppe, G. Inhibition of C-Myc in Breast and Ovarian Carcinoma Cells by 1,25-Dihydroxyvitamin D3, Retinoic Acid and Dexamethasone. Anti-Cancer Drugs 1993, 4, 201–208. [Google Scholar] [CrossRef]
- Stopera, S.A.; Bird, R.P. Effects of All-Trans Retinoic Acid as a Potential Chemopreventive Agent on the Formation of Azoxymethane-Induced Aberrant Crypt Foci: Differential Expression of c-Myc and c-Fos Mrna and Protein. Int. J. Cancer 1993, 53, 798–803. [Google Scholar] [CrossRef]
- Akie, K.; Dosaka-Akita, H.; Murakami, A.; Kawakami, Y. A Combination Treatment of C-Myc Antisense DNA with All-Trans-Retinoic Acid Inhibits Cell Proliferation by Downregulating c-Myc Expression in Small Cell Lung Cancer. Antisense Nucleic Acid Drug Dev. 2000, 10, 243–249. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Inhibition of Aldehyde Dehydrogenase (ALDH) Activity Reduces Chemotherapy and Radiation Resistance of Stem-like ALDHhiCD44+ Human Breast Cancer Cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef]
- Modarai, S.R.; Gupta, A.; Opdenaker, L.M.; Kowash, R.; Masters, G.; Viswanathan, V.; Zhang, T.; Fields, J.Z.; Boman, B.M. The Anti-Cancer Effect of Retinoic Acid Signaling in CRC Occurs via Decreased Growth of ALDH+ Colon Cancer Stem Cells and Increased Differentiation of Stem Cells. Oncotarget 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, W.; Zhang, X.; Bai, J.; Chen, G.; Li, L.; Li, M. All-Trans Retinoic Acid-Induced Deficiency of the Wnt/β-Catenin Pathway Enhances Hepatic Carcinoma Stem Cell Differentiation. PLoS ONE 2015, 10, e0143255. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-J.; Kim, M.R.; Chen, Y.-S.; Yang, J.-Y.; Chang, C.-J. Retinoic Acid Directs Breast Cancer Cell State Changes through Regulation of TET2-PKCζ Pathway. Oncogene 2017, 36, 3193–3206. [Google Scholar] [CrossRef]
- Shi, G.; Zheng, X.; Wu, X.; Wang, S.; Wang, Y.; Xing, F. All- Trans Retinoic Acid Reverses Epithelial-Mesenchymal Transition in Paclitaxel-Resistant Cells by Inhibiting Nuclear Factor Kappa B and Upregulating Gap Junctions. Cancer Sci. 2019, 110, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Benbow, U.; Schoenermark, M.P.; Orndorff, K.A.; Givan, A.L.; Brinckerhoff, C.E. Human Breast Cancer Cells Activate Procollagenase-1 and Invade Type I Collagen: Invasion Is Inhibited by All-Trans Retinoic Acid. Clin. Exp. Metastasis 1999, 17, 231–238. [Google Scholar] [CrossRef]
- Liu, H.; Zang, C.; Fenner, M.H.; Possinger, K.; Elstner, E. PPARgamma Ligands and ATRA Inhibit the Invasion of Human Breast Cancer Cells in Vitro. Breast Cancer Res. Treat. 2003, 79, 63–74. [Google Scholar] [CrossRef]
- Dutta, A.; Sen, T.; Chatterjee, A. All-Trans Retinoic Acid (ATRA) Downregulated MMP-9 by Modulating Its Regulatory Molecules. Cell Adhes. Migr. 2010, 4, 409–418. [Google Scholar] [CrossRef][Green Version]
- Dutta, A.; Sen, T.; Banerji, A.; Das, S.; Chatterjee, A. Studies on Multifunctional Effect of All-Trans Retinoic Acid (ATRA) on Matrix Metalloproteinase-2 (MMP-2) and Its Regulatory Molecules in Human Breast Cancer Cells (MCF-7). J. Oncol. 2009, 2009, 1–13. [Google Scholar] [CrossRef][Green Version]
- Adachi, Y.; Itoh, F.; Yamamoto, H.; Iku, S.; Matsuno, K.; Arimura, Y.; Imai, K. Retinoic Acids Reduce Matrilysin (Matrix Metalloproteinase 7) and Inhibit Tumor Cell Invasion in Human Colon Cancer. Tumor Biol. 2001, 22, 247–253. [Google Scholar] [CrossRef]
- Park, E.Y.; Wilder, E.T.; Lane, M.A. Retinol Inhibits the Invasion of Retinoic Acid–Resistant Colon Cancer Cells In Vitro and Decreases Matrix Metalloproteinase MRNA, Protein, and Activity Levels. Nutr. Cancer 2007, 57, 66–77. [Google Scholar] [CrossRef]
- Vermeulen, S.; Bruyneel, E.; van Roy, F.; Mareel, M.; Bracke, M. Activation of the E-Cadherin/Catenin Complex in Human MCF-7 Breast Cancer Cells by All-Trans-Retinoic Acid. Br. J. Cancer 1995, 72, 1447–1453. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ara, C.; Devirgiliis, L.C.; Massimi, M. Influence of Retinoic Acid on Adhesion Complexes in Human Hepatoma Cells: A Clue to Its Antiproliferative Effects. Cell Commun. Adhes. 2004, 11, 13–23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, W.Y.; Jones, C.S.; Amin, S.; Matsukuma, K.; Haque, M.; Vuligonda, V.; Chandraratna, R.A.; De Luca, L.M. Retinoic Acid Increases Tyrosine Phosphorylation of Focal Adhesion Kinase and Paxillin in MCF-7 Human Breast Cancer Cells. Cancer Res. 1999, 59, 85–90. [Google Scholar] [PubMed]
- Mezquita, B.; Mezquita, P.; Pau, M.; Gasa, L.; Navarro, L.; Samitier, M.; Pons, M.; Mezquita, C. All-Trans-Retinoic Acid Activates the pro-Invasive Src-YAP-Interleukin 6 Axis in Triple-Negative MDA-MB-231 Breast Cancer Cells While Cerivastatin Reverses This Action. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. J. Clin. Med. 2020, 9, 360. https://doi.org/10.3390/jcm9020360
Costantini L, Molinari R, Farinon B, Merendino N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. Journal of Clinical Medicine. 2020; 9(2):360. https://doi.org/10.3390/jcm9020360
Chicago/Turabian StyleCostantini, Lara, Romina Molinari, Barbara Farinon, and Nicolò Merendino. 2020. "Retinoic Acids in the Treatment of Most Lethal Solid Cancers" Journal of Clinical Medicine 9, no. 2: 360. https://doi.org/10.3390/jcm9020360
APA StyleCostantini, L., Molinari, R., Farinon, B., & Merendino, N. (2020). Retinoic Acids in the Treatment of Most Lethal Solid Cancers. Journal of Clinical Medicine, 9(2), 360. https://doi.org/10.3390/jcm9020360