Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease


Abstract
1. Introduction
2. Neurobehavioral Abnormalities and Cognitive Decline as the Major Burden of MPS III
3. Animal Models of MPS III
4. Mechanisms of Neurodegeneration
4.1. Neuroinflammation
4.2. Mitochondrial Defects and Oxidative Stress
4.3. Autophagic Defects and Accumulation of Protein Aggregates
4.4. Specific Effects of Heparan Sulfate
4.5. Neuronal Death in MPS III
5. Biomarkers of MPS III Suitable for Diagnosis, Clinical Evaluation and Pharmacodynamics
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MPS | Mucopolysaccharidosis |
| LSD | lysosomal storage disorder |
| CNS | central nervous system |
| GAG | glycosaminoglycan |
| NAGLU | N-acetyl-α-D-glucosaminidase |
| SGSH | N-sulfoglucosamine sulfohydrolase |
| HGSNAT | acetyl-CoA:alpha-glucosaminide N-acetyltransferase |
| GNS | N-acetylglucosamine-6-sulfate sulfatase |
| IDUA | α-L-iduronidase |
| IDS | Iduronate-2-sulfatase |
| GALNS | Galactosamine-6-sulfatase |
| GLB1 | β-galactosidase |
| ARSB | Arylsulfatase B |
| CT-scan | computed tomography scan |
| MRI | magnetic resonance imaging |
| RP | rapid progressing |
| SP | slow progressing |
| OF | open field |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| SCMAS | subunit C of the mitochondrial ATP synthase |
| FGF | fibroblast growth factor |
| iPSC | induced pluripotent stem cell |
References
- Schultz, M.L.; Tecedor, L.; Chang, M.; Davidson, B.L. Clarifying lysosomal storage diseases. Trends Neurosci. 2011, 34, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D., Antonarakis, S., Ballabio, A., Beaudet, A., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Sanfilippo, S.J.; Podosin, R.; Langer, L.; Good, R.A. Mental retardation associated with acid mucopolysacchariduria (heparitin sulfate type). J. Pediatr. 1963, 63, 837–838. [Google Scholar] [CrossRef]
- Kresse, H.; Neufeld, E.F. The Sanfilippo A corrective factor. Purification and mode of action. J. Biol. Chem. 1972, 247, 2164–2170. [Google Scholar]
- von Figura, K.; Kresse, H. The sanfilippo B corrective factor: A N-acetyl-alpha-D-glucosamindiase. Biochem. Biophys. Res. Commun. 1972, 48, 262–269. [Google Scholar] [CrossRef]
- Klein, U.; Kresse, H.; von Figura, K. Sanfilippo syndrome type C: Deficiency of acetyl-CoA:alpha-glucosaminide N-acetyltransferase in skin fibroblasts. Proc. Natl. Acad. Sci. USA 1978, 75, 5185–5189. [Google Scholar] [CrossRef]
- Kresse, H.; Paschke, E.; von Figura, K.; Gilberg, W.; Fuchs, W. Sanfilippo disease type D: Deficiency of N-acetylglucosamine-6-sulfate sulfatase required for heparan sulfate degradation. Proc. Natl. Acad. Sci. USA 1980, 77, 6822–6826. [Google Scholar] [CrossRef]
- Feldhammer, M.; Durand, S.; Mrázová, L.; Boucher, R.-M.; Laframboise, R.; Steinfeld, R.; Wraith, J.E.; Michelakakis, H.; van Diggelen, O.P.; Hrebícek, M.; et al. Sanfilippo syndrome type C: Mutation spectrum in the heparan sulfate acetyl-CoA: Alpha-glucosaminide N-acetyltransferase (HGSNAT) gene. Hum. Mutat. 2009, 30, 918–925. [Google Scholar] [CrossRef]
- Hettiarachchi, D.; Nethikumara, N.; Pathirana, B.A.P.S.; Weththasigha, K.; Dissanayake, W.D.N.; Dissanayake, V.H.W. A novel mutation in the NAGLU gene associated with Sanfilippo syndrome type B (mucopolysaccharidosis III B). Clin. Case Rep. 2018, 6, 1051–1054. [Google Scholar] [CrossRef]
- Martins, C.; de Medeiros, P.F.V.; Leistner-Segal, S.; Dridi, L.; Elcioglu, N.; Wood, J.; Behnam, M.; Noyan, B.; Lacerda, L.; Geraghty, M.T.; et al. Molecular characterization of a large group of Mucopolysaccharidosis type IIIC patients reveals the evolutionary history of the disease. Hum. Mutat. 2019, 40, 1084–1100. [Google Scholar] [CrossRef]
- Tanwar, H.; Kumar, D.T.; Doss, C.G.P.; Zayed, H. Bioinformatics classification of mutations in patients with Mucopolysaccharidosis IIIA. Metab. Brain Dis. 2019, 34, 1577–1594. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Bertoli-Avella, A.M.; Wessels, M.W.; Ruijter, G.J.G.; de Graaf, B.; Olmer, R.; Elfferich, P.; Neijs, S.; Kariminejad, R.; Suheyl Ezgü, F.; et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum. Mutat. 2010, 31, E1348–E1360. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatol. (Oxford) 2011, 50 (Suppl. 5), v4–v12. [Google Scholar] [CrossRef]
- Cleary, M.A.; Wraith, J.E. Management of mucopolysaccharidosis type III. Arch. Dis. Child. 1993, 69, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Delgadillo, V.; O’Callaghan Mdel, M.; Gort, L.; Coll, M.J.; Pineda, M. Natural history of Sanfilippo syndrome in Spain. Orphanet J. Rare Dis. 2013, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Heron, B.; Mikaeloff, Y.; Froissart, R.; Caridade, G.; Maire, I.; Caillaud, C.; Levade, T.; Chabrol, B.; Feillet, F.; Ogier, H.; et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am. J. Med. Genet. A 2011, 155a, 58–68. [Google Scholar] [CrossRef]
- Ruijter, G.J.; Valstar, M.J.; van de Kamp, J.M.; van der Helm, R.M.; Durand, S.; van Diggelen, O.P.; Wevers, R.A.; Poorthuis, B.J.; Pshezhetsky, A.V.; Wijburg, F.A. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol. Genet. Metab. 2008, 93, 104–111. [Google Scholar] [CrossRef]
- Scott, H.S.; Anson, D.S.; Orsborn, A.M.; Nelson, P.V.; Clements, P.R.; Morris, C.P.; Hopwood, J.J. Human alpha-L-iduronidase: cDNA isolation and expression. Proc. Natl. Acad. Sci. USA 1991, 88, 9695–9699. [Google Scholar] [CrossRef]
- Wilson, P.J.; Morris, C.P.; Anson, D.S.; Occhiodoro, T.; Bielicki, J.; Clements, P.R.; Hopwood, J.J. Hunter syndrome: Isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 8531–8535. [Google Scholar] [CrossRef]
- Scott, H.S.; Blanch, L.; Guo, X.H.; Freeman, C.; Orsborn, A.; Baker, E.; Sutherland, G.R.; Morris, C.P.; Hopwood, J.J. Cloning of the sulphamidase gene and identification of mutations in Sanfilippo A syndrome. Nat. Genet. 1995, 11, 465–467. [Google Scholar] [CrossRef]
- Zhao, H.G.; Li, H.H.; Bach, G.; Schmidtchen, A.; Neufeld, E.F. The molecular basis of Sanfilippo syndrome type B. Proc. Natl. Acad. Sci. USA 1996, 93, 6101–6105. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, H.; Zhang, S.; Bagshaw, R.D.; Tropak, M.B.; Callahan, J.W.; Mahuran, D.J. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C). Am. J. Hum. Genet. 2006, 79, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Hrebicek, M.; Mrazova, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Noskova, L.; Hartmannova, H.; Ivanek, R.; Cizkova, A.; Poupetova, H.; et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.A.; Freeman, C.; Nelson, P.V.; Morris, C.P.; Hopwood, J.J. Human glucosamine-6-sulfatase cDNA reveals homology with steroid sulfatase. Biochem. Biophys. Res. Commun. 1988, 157, 218–224. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fukuda, S.; Masue, M.; Sukegawa, K.; Fukao, T.; Yamagishi, A.; Hori, T.; Iwata, H.; Ogawa, T.; Nakashima, Y.; et al. Morquio disease: Isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem. Biophys. Res. Commun. 1991, 181, 677–683. [Google Scholar] [CrossRef]
- Oshima, A.; Tsuji, A.; Nagao, Y.; Sakuraba, H.; Suzuki, Y. Cloning, sequencing, and expression of cDNA for human beta-galactosidase. Biochem. Biophys. Res. Commun. 1988, 157, 238–244. [Google Scholar] [CrossRef]
- Peters, C.; Schmidt, B.; Rommerskirch, W.; Rupp, K.; Zuhlsdorf, M.; Vingron, M.; Meyer, H.E.; Pohlmann, R.; von Figura, K. Phylogenetic conservation of arylsulfatases. cDNA cloning and expression of human arylsulfatase B. J. Biol. Chem. 1990, 265, 3374–3381. [Google Scholar]
- Oshima, A.; Kyle, J.W.; Miller, R.D.; Hoffmann, J.W.; Powell, P.P.; Grubb, J.H.; Sly, W.S.; Tropak, M.; Guise, K.S.; Gravel, R.A. Cloning, sequencing, and expression of cDNA for human beta-glucuronidase. Proc. Natl. Acad. Sci. USA 1987, 84, 685–689. [Google Scholar] [CrossRef]
- Frost, G.I.; Csóka, T.B.; Wong, T.; Stern, R. Purification, Cloning, and Expression of Human Plasma Hyaluronidase. Biochem. Biophys. Res. Commun. 1997, 236, 10–15. [Google Scholar] [CrossRef]
- Buhrman, D.; Thakkar, K.; Poe, M.; Escolar, M.L. Natural history of Sanfilippo syndrome type A. J. Inherit. Metab. Dis. 2014, 37, 431–437. [Google Scholar] [CrossRef]
- Meyer, A.; Kossow, K.; Gal, A.; Muhlhausen, C.; Ullrich, K.; Braulke, T.; Muschol, N. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics 2007, 120, e1255–e1261. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Bruggenwirth, H.T.; Olmer, R.; Wevers, R.A.; Verheijen, F.W.; Poorthuis, B.J.; Halley, D.J.; Wijburg, F.A. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J. Inherit. Metab. Dis. 2010, 33, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Nijmeijer, S.C.M.; van den Born, L.I.; Kievit, A.J.A.; Stepien, K.M.; Langendonk, J.; Marchal, J.P.; Roosing, S.; Wijburg, F.A.; Wagenmakers, M.A.E.M. The attenuated end of the phenotypic spectrum in MPS III: From late-onset stable cognitive impairment to a non-neuronopathic phenotype. Orphanet J. Rare Dis. 2019, 14, 249. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.C.; Cao, H.; Kaplan, P.; Silver, K.; Leonard, G.; De Meirleir, L.; Lissens, W.; Liebaers, I.; Veilleux, M.; Andermann, F.; et al. Sanfilippo syndrome type D: Natural history and identification of 3 novel mutations in the GNS Gene. Arch. Neurol. 2007, 64, 1629–1634. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chuang, C.K.; Lee, C.L.; Tu, R.Y.; Lo, Y.T.; Chiu, P.C.; Niu, D.M.; Fang, Y.Y.; Chen, T.L.; Tsai, F.J.; et al. Mucopolysaccharidosis III in Taiwan: Natural history, clinical and molecular characteristics of 28 patients diagnosed during a 21-year period. Am. J. Med. Genet. A 2018, 176, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Nestrasil, I.; Delaney, K.A.; Rudser, K.; Kovac, V.; Nair, N.; Richard, C.W.; Haslett, P.; Whitley, C.B. A Prospective Natural History Study of Mucopolysaccharidosis Type IIIA. J. Pediatr. 2016, 170, 278–287. [Google Scholar] [CrossRef]
- Truxal, K.V.; Fu, H.; McCarty, D.M.; McNally, K.A.; Kunkler, K.L.; Zumberge, N.A.; Martin, L.; Aylward, S.C.; Alfano, L.N.; Berry, K.M.; et al. A prospective one-year natural history study of mucopolysaccharidosis types IIIA and IIIB: Implications for clinical trial design. Mol. Genet. Metab. 2016, 119, 239–248. [Google Scholar] [CrossRef]
- Valstar, M.J.; Neijs, S.; Bruggenwirth, H.T.; Olmer, R.; Ruijter, G.J.; Wevers, R.A.; van Diggelen, O.P.; Poorthuis, B.J.; Halley, D.J.; Wijburg, F.A. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann. Neurol. 2010, 68, 876–887. [Google Scholar] [CrossRef]
- Velasco, H.M.; Sanchez, Y.; Martin, A.M.; Umana, L.A. Natural History of Sanfilippo Syndrome Type C in Boyaca, Colombia. J. Child. Neurol. 2017, 32, 177–183. [Google Scholar] [CrossRef]
- Muschol, N.M.; Pape, D.; Kossow, K.; Ullrich, K.; Arash-Kaps, L.; Hennermann, J.B.; Stucker, R.; Breyer, S.R. Growth charts for patients with Sanfilippo syndrome (Mucopolysaccharidosis type III). Orphanet J. Rare Dis. 2019, 14, 93. [Google Scholar] [CrossRef]
- Wilhelm, C.M.; Truxal, K.V.; McBride, K.L.; Kovalchin, J.P.; Flanigan, K.M. Natural history of echocardiographic abnormalities in mucopolysaccharidosis III. Mol. Genet. Metab. 2018, 124, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Ahmed, A.; Whitley, C.; Delaney, K. Observing the advanced disease course in mucopolysaccharidosis, type IIIA; a case series. Mol. Genet. Metab. 2018, 123, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Nestrasil, I.; Ahmed, A.; Wey, A.; Rudser, K.R.; Delaney, K.A.; Rumsey, R.K.; Haslett, P.A.; Whitley, C.B.; Potegal, M. Quantifying behaviors of children with Sanfilippo syndrome: The Sanfilippo Behavior Rating Scale. Mol. Genet. Metab. 2015, 114, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Marchal, J.P.; Grootenhuis, M.; Colland, V.; Wijburg, F.A. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J. Rare Dis. 2011, 6, 43. [Google Scholar] [CrossRef]
- Whitley, C.B.; Cleary, M.; Eugen Mengel, K.; Harmatz, P.; Shapiro, E.; Nestrasil, I.; Haslett, P.; Whiteman, D.; Alexanderian, D. Observational Prospective Natural History of Patients with Sanfilippo Syndrome Type B. J. Pediatr. 2018, 197, 198–206. [Google Scholar] [CrossRef]
- Fraser, J.; Gason, A.A.; Wraith, J.E.; Delatycki, M.B. Sleep disturbance in Sanfilippo syndrome: A parental questionnaire study. Arch. Dis. Child. 2005, 90, 1239–1242. [Google Scholar] [CrossRef]
- Guerrero, J.M.; Pozo, D.; Diaz-Rodriguez, J.L.; Martinez-Cruz, F.; Vela-Campos, F. Impairment of the melatonin rhythm in children with Sanfilippo syndrome. J. Pineal Res. 2006, 40, 192–193. [Google Scholar] [CrossRef]
- Mahon, L.V.; Lomax, M.; Grant, S.; Cross, E.; Hare, D.J.; Wraith, J.E.; Jones, S.; Bigger, B.; Langford-Smith, K.; Canal, M. Assessment of sleep in children with mucopolysaccharidosis type III. PLoS ONE 2014, 9, e84128. [Google Scholar] [CrossRef]
- Rumsey, R.K.; Rudser, K.; Delaney, K.; Potegal, M.; Whitley, C.B.; Shapiro, E. Acquired autistic behaviors in children with mucopolysaccharidosis type IIIA. J. Pediatr. 2014, 164, 1147–1151. [Google Scholar] [CrossRef]
- Shapiro, E.; King, K.; Ahmed, A.; Rudser, K.; Rumsey, R.; Yund, B.; Delaney, K.; Nestrasil, I.; Whitley, C.; Potegal, M. The Neurobehavioral Phenotype in Mucopolysaccharidosis Type IIIB: An Exploratory Study. Mol. Genet. Metab. Rep. 2016, 6, 41–47. [Google Scholar] [CrossRef]
- Potegal, M.; Yund, B.; Rudser, K.; Ahmed, A.; Delaney, K.; Nestrasil, I.; Whitley, C.B.; Shapiro, E.G. Mucopolysaccharidosis Type IIIA presents as a variant of Kluver-Bucy syndrome. J. Clin. Exp. Neuropsychol. 2013, 35, 608–616. [Google Scholar] [CrossRef][Green Version]
- Lee, C.; Dineen, T.E.; Brack, M.; Kirsch, J.E.; Runge, V.M. The mucopolysaccharidoses: Characterization by cranial MR imaging. AJNR Am. J. Neuroradiol. 1993, 14, 1285–1292. [Google Scholar]
- Zafeiriou, D.I.; Savvopoulou-Augoustidou, P.A.; Sewell, A.; Papadopoulou, F.; Badouraki, M.; Vargiami, E.; Gombakis, N.P.; Katzos, G.S. Serial magnetic resonance imaging findings in mucopolysaccharidosis IIIB (Sanfilippo’s syndrome B). Brain Dev. 2001, 23, 385–389. [Google Scholar] [CrossRef]
- Jolly, R.D.; Johnstone, A.C.; Norman, E.J.; Hopwood, J.J.; Walkley, S.U. Pathology of mucopolysaccharidosis IIIA in Huntaway dogs. Vet. Pathol. 2007, 44, 569–578. [Google Scholar] [CrossRef]
- Fischer, A.; Carmichael, K.P.; Munnell, J.F.; Jhabvala, P.; Thompson, J.N.; Matalon, R.; Jezyk, P.F.; Wang, P.; Giger, U. Sulfamidase deficiency in a family of Dachshunds: A canine model of mucopolysaccharidosis IIIA (Sanfilippo A). Pediatr. Res. 1998, 44, 74–82. [Google Scholar] [CrossRef]
- Ellinwood, N.M.; Wang, P.; Skeen, T.; Sharp, N.J.; Cesta, M.; Decker, S.; Edwards, N.J.; Bublot, I.; Thompson, J.N.; Bush, W.; et al. A model of mucopolysaccharidosis IIIB (Sanfilippo syndrome type IIIB): N-acetyl-alpha-D-glucosaminidase deficiency in Schipperke dogs. J. Inherit. Metab. Dis. 2003, 26, 489–504. [Google Scholar] [CrossRef]
- Genger, S.C.; Mizukami, K.; Martin, M.P.; Applegate, J.R., Jr.; Barnes, H.J.; Giger, U. Mucopolysaccharidosis IIIB (Sanfilippo syndrome B) in a commercial emu (Dromaius novaehollandiae) flock. Avian Pathol. 2018, 47, 100–107. [Google Scholar] [CrossRef]
- Jones, M.Z.; Alroy, J.; Boyer, P.J.; Cavanagh, K.T.; Johnson, K.; Gage, D.; Vorro, J.; Render, J.A.; Common, R.S.; Leedle, R.A.; et al. Caprine mucopolysaccharidosis-IIID: Clinical, biochemical, morphological and immunohistochemical characteristics. J. Neuropathol. Exp. Neurol. 1998, 57, 148–157. [Google Scholar] [CrossRef]
- Webber, D.L.; Choo, A.; Hewson, L.J.; Trim, P.J.; Snel, M.F.; Hopwood, J.J.; Richards, R.I.; Hemsley, K.M.; O’Keefe, L.V. Neuronal-specific impairment of heparan sulfate degradation in Drosophila reveals pathogenic mechanisms for Mucopolysaccharidosis type IIIA. Exp. Neurol. 2018, 303, 38–47. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, X.; Xing, Y.; Jiang, C.; Jiang, K.; Xu, P.; Liu, W.; Ren, J.; Huang, L. A model of mucopolysaccharidosis type IIIB in pigs. Biol. Open 2018, 7. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Gliddon, B.; Beccari, T.; Hopwood, J.J.; Stanley, P. A novel missense mutation in lysosomal sulfamidase is the basis of MPS III A in a spontaneous mouse mutant. Glycobiology 2001, 11, 99–103. [Google Scholar] [CrossRef]
- Bhaumik, M.; Muller, V.J.; Rozaklis, T.; Johnson, L.; Dobrenis, K.; Bhattacharyya, R.; Wurzelmann, S.; Finamore, P.; Hopwood, J.J.; Walkley, S.U.; et al. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology 1999, 9, 1389–1396. [Google Scholar] [CrossRef]
- Crawley, A.C.; Gliddon, B.L.; Auclair, D.; Brodie, S.L.; Hirte, C.; King, B.M.; Fuller, M.; Hemsley, K.M.; Hopwood, J.J. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006, 1104, 1–17. [Google Scholar] [CrossRef]
- Li, H.H.; Yu, W.H.; Rozengurt, N.; Zhao, H.Z.; Lyons, K.M.; Anagnostaras, S.; Fanselow, M.S.; Suzuki, K.; Vanier, M.T.; Neufeld, E.F. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc. Natl. Acad. Sci. USA 1999, 96, 14505–14510. [Google Scholar] [CrossRef]
- Martins, C.; Hulkova, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef]
- Lau, A.A.; King, B.M.; Thorsen, C.L.; Hassiotis, S.; Beard, H.; Trim, P.J.; Whyte, L.S.; Tamang, S.J.; Duplock, S.K.; Snel, M.F.; et al. A novel conditional Sgsh knockout mouse model recapitulates phenotypic and neuropathic deficits of Sanfilippo syndrome. J. Inherit. Metab. Dis. 2017, 40, 715–724. [Google Scholar] [CrossRef]
- Marco, S.; Pujol, A.; Roca, C.; Motas, S.; Ribera, A.; Garcia, M.; Molas, M.; Villacampa, P.; Melia, C.S.; Sanchez, V.; et al. Progressive neurologic and somatic disease in a novel mouse model of human mucopolysaccharidosis type IIIC. Dis. Models Mech. 2016, 9, 999–1013. [Google Scholar] [CrossRef]
- Roca, C.; Motas, S.; Marco, S.; Ribera, A.; Sanchez, V.; Sanchez, X.; Bertolin, J.; Leon, X.; Perez, J.; Garcia, M.; et al. Disease correction by AAV-mediated gene therapy in a new mouse model of mucopolysaccharidosis type IIID. Hum. Mol. Genet. 2017, 26, 1535–1551. [Google Scholar] [CrossRef]
- Hemsley, K.M.; Hopwood, J.J. Development of motor deficits in a murine model of mucopolysaccharidosis type IIIA (MPS-IIIA). Behav. Brain Res. 2005, 158, 191–199. [Google Scholar] [CrossRef]
- Heldermon, C.D.; Hennig, A.K.; Ohlemiller, K.K.; Ogilvie, J.M.; Herzog, E.D.; Breidenbach, A.; Vogler, C.; Wozniak, D.F.; Sands, M.S. Development of sensory, motor and behavioral deficits in the murine model of Sanfilippo syndrome type B. PLoS ONE 2007, 2, e772. [Google Scholar] [CrossRef]
- Fu, H.; Bartz, J.D.; Stephens, R.L., Jr.; McCarty, D.M. Peripheral nervous system neuropathology and progressive sensory impairments in a mouse model of Mucopolysaccharidosis IIIB. PLoS ONE 2012, 7, e45992. [Google Scholar] [CrossRef] [PubMed]
- Mader, K.M.; Beard, H.; King, B.M.; Hopwood, J.J. Effect of high dose, repeated intra-cerebrospinal fluid injection of sulphamidase on neuropathology in mucopolysaccharidosis type IIIA mice. Genes Brain Behav. 2008, 7, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Kaidonis, X.; Byers, S.; Ranieri, E.; Sharp, P.; Fletcher, J.; Derrick-Roberts, A. N-butyldeoxynojirimycin treatment restores the innate fear response and improves learning in mucopolysaccharidosis IIIA mice. Mol. Genet. Metab. 2016, 118, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; Hannouche, H.; Rozaklis, T.; Hassiotis, S.; Hopwood, J.J.; Hemsley, K.M. Allogeneic stem cell transplantation does not improve neurological deficits in mucopolysaccharidosis type IIIA mice. Exp. Neurol. 2010, 225, 445–454. [Google Scholar] [CrossRef]
- Holley, R.J.; Ellison, S.M.; Fil, D.; O’Leary, C.; McDermott, J.; Senthivel, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; D’Souza, Z.; Parker, H.; et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain 2018, 141, 99–116. [Google Scholar] [CrossRef]
- Kan, S.-H.; Aoyagi-Scharber, M.; Le, S.Q.; Vincelette, J.; Ohmi, K.; Bullens, S.; Wendt, D.J.; Christianson, T.M.; Tiger, P.M.N.; Brown, J.R.; et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. USA 2014, 111, 14870–14875. [Google Scholar] [CrossRef]
- Langford-Smith, A.; Wilkinson, F.L.; Langford-Smith, K.J.; Holley, R.J.; Sergijenko, A.; Howe, S.J.; Bennett, W.R.; Jones, S.A.; Wraith, J.; Merry, C.L.; et al. Hematopoietic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1610–1621. [Google Scholar] [CrossRef]
- Malinowska, M.; Wilkinson, F.L.; Langford-Smith, K.J.; Langford-Smith, A.; Brown, J.R.; Crawford, B.E.; Vanier, M.T.; Grynkiewicz, G.; Wynn, R.F.; Wraith, J.E.; et al. Genistein improves neuropathology and corrects behaviour in a mouse model of neurodegenerative metabolic disease. PLoS ONE 2010, 5, e14192. [Google Scholar] [CrossRef]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Zheng, Y.; Ryazantsev, S.; Ohmi, K.; Zhao, H.-Z.; Rozengurt, N.; Kohn, D.B.; Neufeld, E.F. Retrovirally transduced bone marrow has a therapeutic effect on brain in the mouse model of mucopolysaccharidosis IIIB. Mol. Genet. Metab. 2004, 82, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Beard, H.; Hassiotis, S.; Gai, W.P.; Parkinson-Lawrence, E.; Hopwood, J.J.; Hemsley, K.M. Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp. Neurol. 2017, 295, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, K.; Kudo, L.C.; Ryazantsev, S.; Zhao, H.Z.; Karsten, S.L.; Neufeld, E.F. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 8332–8337. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, A.; Langford-Smith, K.J.; Jones, S.A.; Wynn, R.F.; Wraith, J.E.; Wilkinson, F.L.; Bigger, B.W. Female mucopolysaccharidosis IIIA mice exhibit hyperactivity and a reduced sense of danger in the open field test. PLoS ONE 2011, 6, e25717. [Google Scholar] [CrossRef]
- Sorrentino, N.C.; D’Orsi, L.; Sambri, I.; Nusco, E.; Monaco, C.; Spampanato, C.; Polishchuk, E.; Saccone, P.; De Leonibus, E.; Ballabio, A.; et al. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol. Med. 2013, 5, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Cressant, A.; Desmaris, N.; Verot, L.; Bréjot, T.; Froissart, R.; Vanier, M.-T.; Maire, I.; Heard, J.M. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 10229–10239. [Google Scholar] [CrossRef]
- Lotfi, P.; Tse, D.Y.; Di Ronza, A.; Seymour, M.L.; Martano, G.; Cooper, J.D.; Pereira, F.A.; Passafaro, M.; Wu, S.M.; Sardiello, M. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy 2018, 14, 1419–1434. [Google Scholar] [CrossRef]
- Willing, A.E.; Garbuzova-Davis, S.N.; Zayko, O.; Derasari, H.M.; Rawls, A.E.; James, C.R.; Mervis, R.F.; Sanberg, C.D.; Kuzmin-Nichols, N.; Sanberg, P.R. Repeated Administrations of Human Umbilical Cord Blood Cells Improve Disease Outcomes in a Mouse Model of Sanfilippo Syndrome Type III B. Cell Transplant. 2014, 23, 1613–1630. [Google Scholar] [CrossRef]
- Lau, A.A.; Crawley, A.C.; Hopwood, J.J.; Hemsley, K.M. Open field locomotor activity and anxiety-related behaviors in mucopolysaccharidosis type IIIA mice. Behav. Brain Res. 2008, 191, 130–136. [Google Scholar] [CrossRef]
- Gliddon, B.L.; Hopwood, J.J. Enzyme-Replacement Therapy from Birth Delays the Development of Behavior and Learning Problems in Mucopolysaccharidosis Type IIIA Mice. Pediatr. Res. 2004, 56, 65–72. [Google Scholar] [CrossRef]
- Roberts, A.L.K.; Rees, M.H.; Klebe, S.; Fletcher, J.M.; Byers, S. Improvement in behaviour after substrate deprivation therapy with rhodamine B in a mouse model of MPS IIIA. Mol. Genet. Metab. 2007, 92, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, B.; Lamanna, W.C.; Lawrence, R.; Damme, M.; Stroobants, S.; Padva, M.; Kalus, I.; Frese, M.A.; Lubke, T.; Lullmann-Rauch, R.; et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10310–10315. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, B.; Heimann, P.; Ortkras, T.; Lullmann-Rauch, R.; Sawada, T.; Walkley, S.U.; Dierks, T.; Damme, M. Ataxia is the major neuropathological finding in arylsulfatase G-deficient mice: Similarities and dissimilarities to Sanfilippo disease (mucopolysaccharidosis type III). Hum. Mol. Genet. 2015, 24, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Ruzo, A.; Marco, S.; Garcia, M.; Villacampa, P.; Ribera, A.; Ayuso, E.; Maggioni, L.; Mingozzi, F.; Haurigot, V.; Bosch, F. Correction of pathological accumulation of glycosaminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum. Gene Ther. 2012, 23, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- DiRosario, J.; Divers, E.; Wang, C.; Etter, J.; Charrier, A.; Jukkola, P.; Auer, H.; Best, V.; Newsom, D.L.; McCarty, D.M.; et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J. Neurosci. Res. 2009, 87, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Arfi, A.; Richard, M.; Gandolphe, C.; Bonnefont-Rousselot, D.; Therond, P.; Scherman, D. Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: The positive effect of long-term aspirin treatment. Mol. Genet. Metab. 2011, 103, 18–25. [Google Scholar] [CrossRef]
- Killedar, S.; Dirosario, J.; Divers, E.; Popovich, P.G.; McCarty, D.M.; Fu, H. Mucopolysaccharidosis IIIB, a lysosomal storage disease, triggers a pathogenic CNS autoimmune response. J. Neuroinflamm. 2010, 7, 39. [Google Scholar] [CrossRef]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef]
- Ali, S.; Palmer, A.C.; Banerjee, B.; Fritchley, S.J.; Kirby, J.A. Examination of the function of RANTES, MIP-1alpha, and MIP-1beta following interaction with heparin-like glycosaminoglycans. J. Biol. Chem. 2000, 275, 11721–11727. [Google Scholar] [CrossRef]
- Billings, P.C.; Pacifici, M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: Mechanisms and mysteries. Connect. Tissue Res. 2015, 56, 272–280. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, Y.; Platt, J.L. Modification of antigen-presenting cell functions by heparan sulfate. Transplant. Proc. 2000, 32, 947. [Google Scholar] [CrossRef]
- Wrenshall, L.E.; Cerra, F.B.; Carlson, A.; Bach, F.H.; Platt, J.L. Regulation of murine splenocyte responses by heparan sulfate. J. Immunol. 1991, 147, 455–459. [Google Scholar] [PubMed]
- Wrenshall, L.E.; Cerra, F.B.; Singh, R.K.; Platt, J.L. Heparan sulfate initiates signals in murine macrophages leading to divergent biologic outcomes. J. Immunol. 1995, 154, 871–880. [Google Scholar] [PubMed]
- Wrenshall, L.E.; Stevens, R.B.; Cerra, F.B.; Platt, J.L. Modulation of macrophage and B cell function by glycosaminoglycans. J. Leukoc. Biol. 1999, 66, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; van der Spoel, A.C.; d’Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef]
- Wu, Y.P.; Proia, R.L. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc. Natl. Acad. Sci. USA 2004, 101, 8425–8430. [Google Scholar] [CrossRef]
- Dawson, G.; Fuller, M.; Helmsley, K.M.; Hopwood, J.J. Abnormal gangliosides are localized in lipid rafts in Sanfilippo (MPS3a) mouse brain. Neurochem. Res. 2012, 37, 1372–1380. [Google Scholar] [CrossRef]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef]
- Villani, G.R.; Gargiulo, N.; Faraonio, R.; Castaldo, S.; Gonzalez, Y.R.E.; Di Natale, P. Cytokines, neurotrophins, and oxidative stress in brain disease from mucopolysaccharidosis IIIB. J. Neurosci. Res. 2007, 85, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.R.; Di Domenico, C.; Musella, A.; Cecere, F.; Di Napoli, D.; Di Natale, P. Mucopolysaccharidosis IIIB: Oxidative damage and cytotoxic cell involvement in the neuronal pathogenesis. Brain Res. 2009, 1279, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Trecherel, E.; Gomila, C.; Peltier, M.; Aubignat, M.; Gubler, B.; Morliere, P.; Heard, J.M.; Ausseil, J. Oxidative stress is independent of inflammation in the neurodegenerative Sanfilippo syndrome type B. J. Neurosci. Res. 2015, 93, 424–432. [Google Scholar] [CrossRef]
- Ryazantsev, S.; Yu, W.H.; Zhao, H.Z.; Neufeld, E.F.; Ohmi, K. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol. Genet. Metab. 2007, 90, 393–401. [Google Scholar] [CrossRef]
- Kida, E.; Wisniewski, K.E.; Golabek, A.A. Increased expression of subunit c of mitochondrial ATP synthase in brain tissue from neuronal ceroid lipofuscinoses and mucopolysaccharidosis cases but not in long-term fibroblast cultures. Neurosci. Lett. 1993, 164, 121–124. [Google Scholar] [CrossRef]
- de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef]
- Woś, M.; Szczepanowska, J.; Pikuła, S.; Tylki-Szymańska, A.; Zabłocki, K.; Bandorowicz-Pikuła, J. Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch. Biochem. Biophys. 2016, 593, 50–59. [Google Scholar] [CrossRef]
- Suzuki, K.; Yamaguchi, A.; Yamanaka, S.; Kanzaki, S.; Kawashima, M.; Togo, T.; Katsuse, O.; Koumitsu, N.; Aoki, N.; Iseki, E.; et al. Accumulated α-synuclein affects the progression of GM2 gangliosidoses. Exp. Neurol. 2016, 284, 38–49. [Google Scholar] [CrossRef]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; Hoek, G.v.d.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Galvin, J.E.; Lee, V.M.; Rorke, L.B.; Dickson, D.W.; Wolfe, J.H.; Jones, M.Z.; Trojanowski, J.Q. Accumulation of intracellular amyloid-beta peptide (A beta 1-40) in mucopolysaccharidosis brains. J. Neuropathol. Exp. Neurol. 1999, 58, 815–824. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Garcia-Reitbock, P.; Ban, M.; Evans, J.R.; Jacques, T.S.; Kemppinen, A.; Foltynie, T.; Williams-Gray, C.H.; Chinnery, P.F.; Hudson, G.; et al. Genetic and pathological links between Parkinson’s disease and the lysosomal disorder Sanfilippo syndrome. Mov. Disord. 2012, 27, 312–315. [Google Scholar] [CrossRef]
- Ohmi, K.; Zhao, H.Z.; Neufeld, E.F. Defects in the medial entorhinal cortex and dentate gyrus in the mouse model of Sanfilippo syndrome type B. PLoS ONE 2011, 6, e27461. [Google Scholar] [CrossRef]
- Sambri, I.; D’Alessio, R.; Ezhova, Y.; Giuliano, T.; Sorrentino, N.C.; Cacace, V.; De Risi, M.; Cataldi, M.; Annunziato, L.; De Leonibus, E.; et al. Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol. Med. 2017, 9, 112–132. [Google Scholar] [CrossRef]
- Soe, K.; Beard, H.; Neumann, D.; Trim, P.J.; Duplock, S.; Snel, M.F.; Hopwood, J.J.; Hemsley, K.M. Early disease course is unaltered in mucopolysaccharidosis type IIIA (MPS IIIA) mice lacking alpha-synuclein. Neuropathol. Appl. Neurobiol. 2019, 45, 715–731. [Google Scholar] [CrossRef]
- Abdelmotilib, H.; Maltbie, T.; Delic, V.; Liu, Z.; Hu, X.; Fraser, K.B.; Moehle, M.S.; Stoyka, L.; Anabtawi, N.; Krendelchtchikova, V.; et al. α-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol. Dis. 2017, 105, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H., 3rd; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against α-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 5144–5159. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, L.; Tammewar, G.; Ayakta, N.; Baker, S.L.; Bejanin, A.; Boxer, A.L.; Gorno-Tempini, M.L.; Janabi, M.; Kramer, J.H.; Lazaris, A.; et al. Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer’s Disease. Neuroimage Clin. 2017, 17, 452–464. [Google Scholar] [CrossRef]
- De Pasquale, V.; Pavone, L.M. Heparan sulfate proteoglycans: The sweet side of development turns sour in mucopolysaccharidoses. Biochim. Biophys. Acta Mol. Basis Dis. 2019. [Google Scholar] [CrossRef]
- Tordo, J.; O’Leary, C.; Antunes, A.S.L.M.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain A J. Neurol. 2018, 141, 2014–2031. [Google Scholar] [CrossRef]
- Hocquemiller, M.; Vitry, S.; Bigou, S.; Bruyere, J.; Ausseil, J.; Heard, J.M. GAP43 overexpression and enhanced neurite outgrowth in mucopolysaccharidosis type IIIB cortical neuron cultures. J. Neurosci. Res. 2010, 88, 202–213. [Google Scholar] [CrossRef]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef]
- Gómez-Pinilla, F.; Vu, L.; Cotman, C.W. Regulation of astrocyte proliferation by FGF-2 and heparan sulfate in vivo. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 2021–2029. [Google Scholar] [CrossRef]
- Li, H.H.; Zhao, H.Z.; Neufeld, E.F.; Cai, Y.; Gomez-Pinilla, F. Attenuated plasticity in neurons and astrocytes in the mouse model of Sanfilippo syndrome type B. J. Neurosci. Res. 2002, 69, 30–38. [Google Scholar] [CrossRef]
- Bruyere, J.; Roy, E.; Ausseil, J.; Lemonnier, T.; Teyre, G.; Bohl, D.; Etienne-Manneville, S.; Lortat-Jacob, H.; Heard, J.M.; Vitry, S. Heparan sulfate saccharides modify focal adhesions: Implication in mucopolysaccharidosis neuropathophysiology. J. Mol. Biol. 2015, 427, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Canals, I.; Soriano, J.; Orlandi, J.G.; Torrent, R.; Richaud-Patin, Y.; Jimenez-Delgado, S.; Merlin, S.; Follenzi, A.; Consiglio, A.; Vilageliu, L.; et al. Activity and High-Order Effective Connectivity Alterations in Sanfilippo C Patient-Specific Neuronal Networks. Stem Cell Rep. 2015, 5, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.D.; Yu, J.S.; Shiah, S.G.; Huang, J.J. Protein kinase FA/glycogen synthase kinase-3 alpha after heparin potentiation phosphorylates tau on sites abnormally phosphorylated in Alzheimer’s disease brain. J. Neurochem. 1994, 63, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Paudel, H.K.; Li, W. Heparin-induced conformational change in microtubule-associated protein Tau as detected by chemical cross-linking and phosphopeptide mapping. J. Biol. Chem. 1999, 274, 8029–8038. [Google Scholar] [CrossRef]
- Maccari, F.; Sorrentino, N.C.; Mantovani, V.; Galeotti, F.; Fraldi, A.; Volpi, N. Glycosaminoglycan levels and structure in a mucopolysaccharidosis IIIA mice and the effect of a highly secreted sulfamidase engineered to cross the blood-brain barrier. Metab. Brain Dis. 2017, 32, 203–210. [Google Scholar] [CrossRef]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem. 1997, 69, 2452–2465. [Google Scholar] [CrossRef]
- Barone, R.; Nigro, F.; Triulzi, F.; Musumeci, S.; Fiumara, A.; Pavone, L. Clinical and neuroradiological follow-up in mucopolysaccharidosis type III (Sanfilippo syndrome). Neuropediatrics 1999, 30, 270–274. [Google Scholar] [CrossRef]
- Calleja Gero, M.L.; Gonzalez Gutierrez-Solana, L.; Lopez Marin, L.; Lopez Pino, M.A.; Fournier Del Castillo, C.; Duat Rodriguez, A. Neuroimaging findings in patient series with mucopolysaccharidosis. Neurologia 2012, 27, 407–413. [Google Scholar] [CrossRef]
- Hadfield, M.G.; Ghatak, N.R.; Nakoneczna, I.; Lippman, H.R.; Myer, E.C.; Constantopoulos, G.; Bradley, R.M. Pathologic findings in mucopolysaccharidosis type IIIB (Sanfilippo’s sydnrome B). Arch. Neurol. 1980, 37, 645–650. [Google Scholar] [CrossRef]
- Wallace, B.J.; Kaplan, D.; Adachi, M.; Schneck, L.; Volk, B.W. Mucopolysaccharidosis type 3. Morphologic and biochemical studies of two siblings with Sanfilippo syndrome. Arch. Pathol. 1966, 82, 462–473. [Google Scholar]
- Vitry, S.; Ausseil, J.; Hocquemiller, M.; Bigou, S.; Dos Santos Coura, R.; Heard, J.M. Enhanced degradation of synaptophysin by the proteasome in mucopolysaccharidosis type IIIB. Mol. Cell. Neurosci. 2009, 41, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Moog, U.; van Mierlo, I.; van Schrojenstein Lantman-de Valk, H.M.; Spaapen, L.; Maaskant, M.A.; Curfs, L.M. Is Sanfilippo type B in your mind when you see adults with mental retardation and behavioral problems? Am. J. Med. Genet. Part C Semin. Med. Genet. 2007, 145c, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.; Trehan, A.; Landis, D.; Toro, C. Mucopolysaccharidosis type IIIB (MPS IIIB) masquerading as a behavioural disorder. BMJ Case Rep. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Colmenares-Bonilla, D.; Colin-Gonzalez, C.; Gonzalez-Segoviano, A.; Esquivel Garcia, E.; Vela-Huerta, M.M.; Lopez-Gomez, F.G. Diagnosis of Mucopolysaccharidosis Based on History and Clinical Features: Evidence from the Bajio Region of Mexico. Cureus 2018, 10, e3617. [Google Scholar] [CrossRef] [PubMed]
- Rezayi, A.; Feshangchi-Bonab, M.; Taherian, R. An Uncommon Presentation of Mucopolysaccharidosis Type IIIb. Iran. J. Child. Neurol. 2019, 13, 105–111. [Google Scholar]
- Saini, A.G.; Singhi, P.; Sahu, J.K.; Ganesan, S.L.; Vyas, S.; Rao, S.; Sachdeva, M.U. Hyperactivity, unexplained speech delay, and coarse facies--is it Sanfilippo syndrome? J. Child. Neurol. 2014, 29, Np9-12. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Wegrzyn, G.; Burton, B.K.; Tylki-Szymanska, A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013, 102, 462–470. [Google Scholar] [CrossRef]
- Kuiper, G.A.; Meijer, O.L.M.; Langereis, E.J.; Wijburg, F.A. Failure to shorten the diagnostic delay in two ultra-orphan diseases (mucopolysaccharidosis types I and III): Potential causes and implications. Orphanet J. Rare Dis. 2018, 13, 2. [Google Scholar] [CrossRef]
- Vieira, T.; Schwartz, I.; Munoz, V.; Pinto, L.; Steiner, C.; Ribeiro, M.; Boy, R.; Ferraz, V.; de Paula, A.; Kim, C.; et al. Mucopolysaccharidoses in Brazil: What happens from birth to biochemical diagnosis? Am. J. Med. Genet. A 2008, 146a, 1741–1747. [Google Scholar] [CrossRef]
- Yodoshi, T.; Hurt, T.L. Avoiding diagnostic delay for mucopolysaccharidosis IIIB: Do not overlook common clues such as wheezing and otitis media. BMJ Case Rep. 2018, 2018, bcr-2018. [Google Scholar] [CrossRef]
- Boehringer, S.; Vollmar, T.; Tasse, C.; Wurtz, R.P.; Gillessen-Kaesbach, G.; Horsthemke, B.; Wieczorek, D. Syndrome identification based on 2D analysis software. Eur. J. Hum. Genet. 2006, 14, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Giugliani, R.; Wood, T. The laboratory diagnosis of mucopolysaccharidosis III (Sanfilippo syndrome): A changing landscape. Mol. Genet. Metab. 2014, 113, 34–41. [Google Scholar] [CrossRef]
- Filocamo, M.; Tomanin, R.; Bertola, F.; Morrone, A. Biochemical and molecular analysis in mucopolysaccharidoses: What a paediatrician must know. Ital. J. Pediatr. 2018, 44, 129. [Google Scholar] [CrossRef] [PubMed]
- Berry, H.K. Screening for mucopolysaccharide disorders with the Berry spot test. Clin. Biochem. 1987, 20, 365–371. [Google Scholar] [CrossRef]
- Chih-Kuang, C.; Shuan-Pei, L.; Shyue-Jye, L.; Tuen-Jen, W. MPS screening methods, the Berry spot and acid turbidity tests, cause a high incidence of false-negative results in sanfilippo and morquio syndromes. J. Clin. Lab. Anal. 2002, 16, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Sukegawa, K.; Orii, T. Screening test for urinary glycosaminoglycans and differentiation of various mucopolysaccharidoses. Clin. Chim. Acta 1985, 151, 147–156. [Google Scholar] [CrossRef]
- Tomatsu, S.; Gutierrez, M.A.; Ishimaru, T.; Pena, O.M.; Montano, A.M.; Maeda, H.; Velez-Castrillon, S.; Nishioka, T.; Fachel, A.A.; Cooper, A.; et al. Heparan sulfate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 743–757. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Lavoie, P.; Tomatsu, S.; Valayannopoulos, V.; Mitchell, J.J.; Raiman, J.; Beaudoin, M.; Maranda, B.; Clarke, J.T. UPLC-MS/MS detection of disaccharides derived from glycosaminoglycans as biomarkers of mucopolysaccharidoses. Anal. Chim. Acta 2016, 936, 139–148. [Google Scholar] [CrossRef]
- Khan, A.; Sergi, C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagn. (Basel) 2018, 8, 29. [Google Scholar] [CrossRef]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef]
- Kubaski, F.; Osago, H.; Mason, R.W.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol. Genet. Metab. 2017, 120, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Naimy, H.; Powell, K.D.; Moriarity, J.R.; Wu, J.; McCauley, T.G.; Haslett, P.A.; Barbier, A.J.; Qiu, Y. A novel LC-MS/MS assay for heparan sulfate screening in the cerebrospinal fluid of mucopolysaccharidosis IIIA patients. Bioanalysis 2016, 8, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Saville, J.T.; Flanigan, K.M.; Truxal, K.V.; McBride, K.L.; Fuller, M. Evaluation of biomarkers for Sanfilippo syndrome. Mol. Genet. Metab. 2019, 128, 68–74. [Google Scholar] [CrossRef]
- Saville, J.T.; McDermott, B.K.; Fletcher, J.M.; Fuller, M. Disease and subtype specific signatures enable precise diagnosis of the mucopolysaccharidoses. Genet. Med. 2019, 21, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Kubaski, F.; Sawamoto, K.; Mason, R.W.; Yasuda, E.; Shimada, T.; Montano, A.M.; Yamaguchi, S.; Suzuki, Y.; Orii, T. Newborn screening and diagnosis of mucopolysaccharidoses: Application of tandem mass spectrometry. Nihon Masu Sukuriningu Gakkai Shi 2014, 24, 19–37. [Google Scholar] [PubMed]
- Yi, F.; Hong, X.; Kumar, A.B.; Zong, C.; Boons, G.J.; Scott, C.R.; Turecek, F.; Robinson, B.H.; Gelb, M.H. Detection of mucopolysaccharidosis III-A (Sanfilippo Syndrome-A) in dried blood spots (DBS) by tandem mass spectrometry. Mol. Genet. Metab. 2018, 125, 59–63. [Google Scholar] [CrossRef]
- Zhang, H.; Young, S.P.; Auray-Blais, C.; Orchard, P.J.; Tolar, J.; Millington, D.S. Analysis of glycosaminoglycans in cerebrospinal fluid from patients with mucopolysaccharidoses by isotope-dilution ultra-performance liquid chromatography-tandem mass spectrometry. Clin. Chem. 2011, 57, 1005–1012. [Google Scholar] [CrossRef]
- Gelb, M.H.; Scott, C.R.; Turecek, F. Newborn screening for lysosomal storage diseases. Clin. Chem. 2015, 61, 335–346. [Google Scholar] [CrossRef]
- Zeng, Q.; Fan, Y.; Wang, L.; Huang, Z.; Gu, X.; Yu, Y. Molecular defects identified by whole exome sequencing in a child with atypical mucopolysaccharidosis IIIB. J. Pediatr. Endocrinol. Metab. 2017, 30, 463–469. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, J.J.; Park, S.I.; Cho, S.Y.; Jin, D.K.; Cho, Y.S.; Chung, W.H.; Hong, S.H.; Moon, I.J. Auditory Characteristics in Patients With Mucopolysaccharidosis. Otol. Neurotol. 2019, 40, e955–e961. [Google Scholar] [CrossRef]
- Ghosh, A.; Shapiro, E.; Rust, S.; Delaney, K.; Parker, S.; Shaywitz, A.J.; Morte, A.; Bubb, G.; Cleary, M.; Bo, T.; et al. Recommendations on clinical trial design for treatment of Mucopolysaccharidosis Type III. Orphanet J. Rare Dis. 2017, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, J.; Valstar, M.J.; Narajczyk, M.; Wegrzyn, G.; Kulik, W.; Ijlst, L.; Wagemans, T.; van der Wal, W.M.; Wijburg, F.A. Genistein in Sanfilippo disease: A randomized controlled crossover trial. Ann. Neurol. 2012, 71, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Hemsley, K.M.; King, B.; Hopwood, J.J. Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol. Genet. Metab. 2007, 90, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.S.; Hemsley, K.M.; Hopwood, J.J. Intracerebral injection of sulfamidase delays neuropathology in murine MPS-IIIA. Mol. Genet. Metab. 2004, 82, 273–285. [Google Scholar] [CrossRef]
- Tardieu, M.; Zerah, M.; Gougeon, M.L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: An uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017, 16, 712–720. [Google Scholar] [CrossRef]
- Tardieu, M.; Zerah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef]
- Clarke, L.A.; Winchester, B.; Giugliani, R.; Tylki-Szymanska, A.; Amartino, H. Biomarkers for the mucopolysaccharidoses: Discovery and clinical utility. Mol. Genet. Metab. 2012, 106, 395–402. [Google Scholar] [CrossRef]
- de Ruijter, J.; Ijlst, L.; Kulik, W.; van Lenthe, H.; Wagemans, T.; van Vlies, N.; Wijburg, F.A. Heparan sulfate derived disaccharides in plasma and total urinary excretion of glycosaminoglycans correlate with disease severity in Sanfilippo disease. J. Inherit. Metab. Dis. 2013, 36, 271–279. [Google Scholar] [CrossRef]
- Hopwood, J.J.; Muller, V.; Harrison, J.R.; Carey, W.F.; Elliott, H.; Robertson, E.F.; Pollard, A.C. Enzymatic diagnosis of the mucopolysaccharidoses: Experience of 96 cases diagnosed in a five-year period. Med. J. Aust. 1982, 1, 257–260. [Google Scholar] [CrossRef]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Tylki-Szymanska, A.; Czartoryska, B.; Wegrzyn, A.; Wegrzyn, G. Correlation between severity of mucopolysaccharidoses and combination of the residual enzyme activity and efficiency of glycosaminoglycan synthesis. Acta Paediatr. 2009, 98, 743–749. [Google Scholar] [CrossRef]
- Vance, J.M.; Conneally, P.M.; Wappner, R.S.; Yu, P.L.; Brandt, I.K.; Pericak-Vance, M.A. Carrier detection in Sanfilippo syndrome type B: Report of six families. Clin. Genet. 1981, 20, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Knottnerus, S.J.G.; Nijmeijer, S.C.M.; IJlst, L.; Te Brinke, H.; van Vlies, N.; Wijburg, F.A. Prediction of phenotypic severity in mucopolysaccharidosis type IIIA. Ann. Neurol. 2017, 82, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Meadows, A.S.; Pineda, R.J.; Mohney, R.P.; Stirdivant, S.; McCarty, D.M. Serum global metabolomics profiling reveals profound metabolic impairments in patients with MPS IIIA and MPS IIIB. Metab. Brain Dis. 2017, 32, 1403–1415. [Google Scholar] [CrossRef]
- Tebani, A.; Abily-Donval, L.; Schmitz-Afonso, I.; Heron, B.; Piraud, M.; Ausseil, J.; Zerimech, F.; Gonzalez, B.; Marret, S.; Afonso, C.; et al. Unveiling metabolic remodeling in mucopolysaccharidosis type III through integrative metabolomics and pathway analysis. J. Transl. Med. 2018, 16, 248. [Google Scholar] [CrossRef] [PubMed]


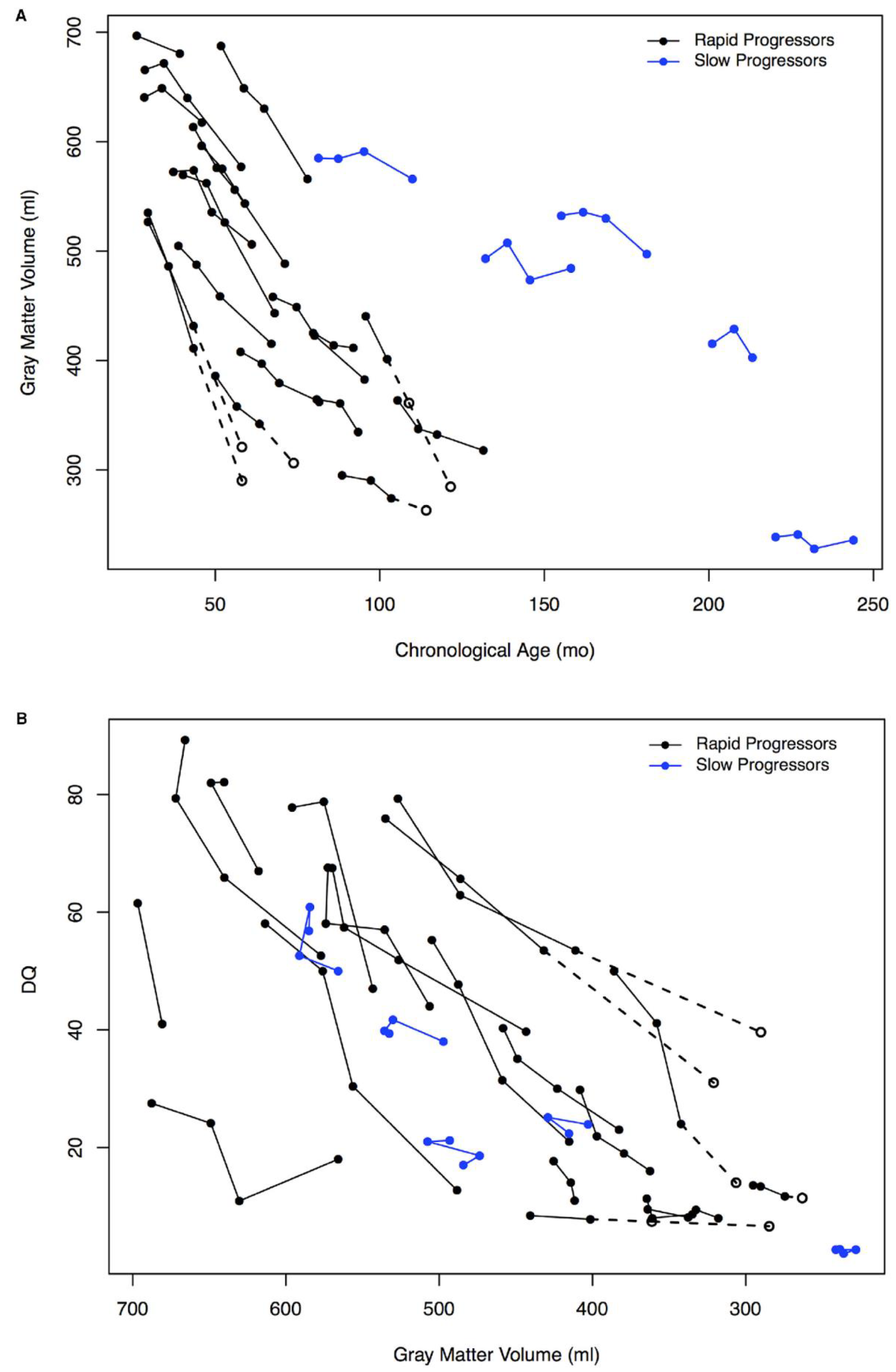{kind=link}
| MPS Subtype | Genetic Locus [14] | Enzyme Defect | Primary Storage Molecule | Somatic Disease [14] | CNS Disease [14] |
|---|---|---|---|---|---|
| MPS I (Hurler, Hurler-Scheie and Scheie syndrome) | IDUA [19] 4p16.3 | α-L-iduronidase | Dermatan sulfate, heparan sulfate | Severe | IS, IHS: None to mild IH: Severe |
| MPS II (Hunter syndrome) | IDS [20] Xq28 | Iduronate-2-sulfatase | Dermatan sulfate, heparan sulfate | Severe | None to severe |
| MPS IIIA-D (Sanfilippo syndrome) | SGSH [21] 17q25.3 | IIIA: N-sulfoglucosamine sulfohydrolase | Heparan sulfate | Mild | Severe |
| NAGLU [22] 17q21 | IIIB: N-acetyl-α-D-glucosaminidase | ||||
| HGSNAT [23,24] 8p11.1 | IIIC: acetyl-CoA:alpha-glucosaminide N-acetyltransferase | ||||
| GNS [25] 12q14 | IIID: N-acetylglucosamine-6-sulfate sulfatase | ||||
| MPS IV (Morquio syndrome) | GALNS [26] 16q24.3 | IVA: Galactosamine-6-sulfatase | Keratan sulfate, chondroitin sulfate Keratan sulfate | Severe | None |
| GLB1 [27] 3p21.33 | IVB: β-galactosidase | ||||
| MPS VI (Maroteaux-Lamy syndrome) | ARSB [28] 5q11-13 | Arylsulfatase B | Dermatan sulfate, chondroitin sulfate | Severe | None |
| MPS VII (Sly syndrome) | GUSB [29] 7q21.11 | β-glucuronidase | Dermatan sulfate, heparan sulfate, chondroitin sulfate | Severe | Severe |
| MPS IX | HYAL1 [30] 3p21.3-21.2 | Hyaluronidase | Hyaluronan | Mild to moderate | None |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin. Med. 2020, 9, 344. https://doi.org/10.3390/jcm9020344
Heon-Roberts R, Nguyen ALA, Pshezhetsky AV. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. Journal of Clinical Medicine. 2020; 9(2):344. https://doi.org/10.3390/jcm9020344
Chicago/Turabian StyleHeon-Roberts, Rachel, Annie L. A. Nguyen, and Alexey V. Pshezhetsky. 2020. "Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease" Journal of Clinical Medicine 9, no. 2: 344. https://doi.org/10.3390/jcm9020344
APA StyleHeon-Roberts, R., Nguyen, A. L. A., & Pshezhetsky, A. V. (2020). Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. Journal of Clinical Medicine, 9(2), 344. https://doi.org/10.3390/jcm9020344